
Attenuation of Ventilation-Enhanced Epithelial–Mesenchymal Transition through the Phosphoinositide 3-Kinase-γ in a Murine Bleomycin-Induced Acute Lung Injury Model

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
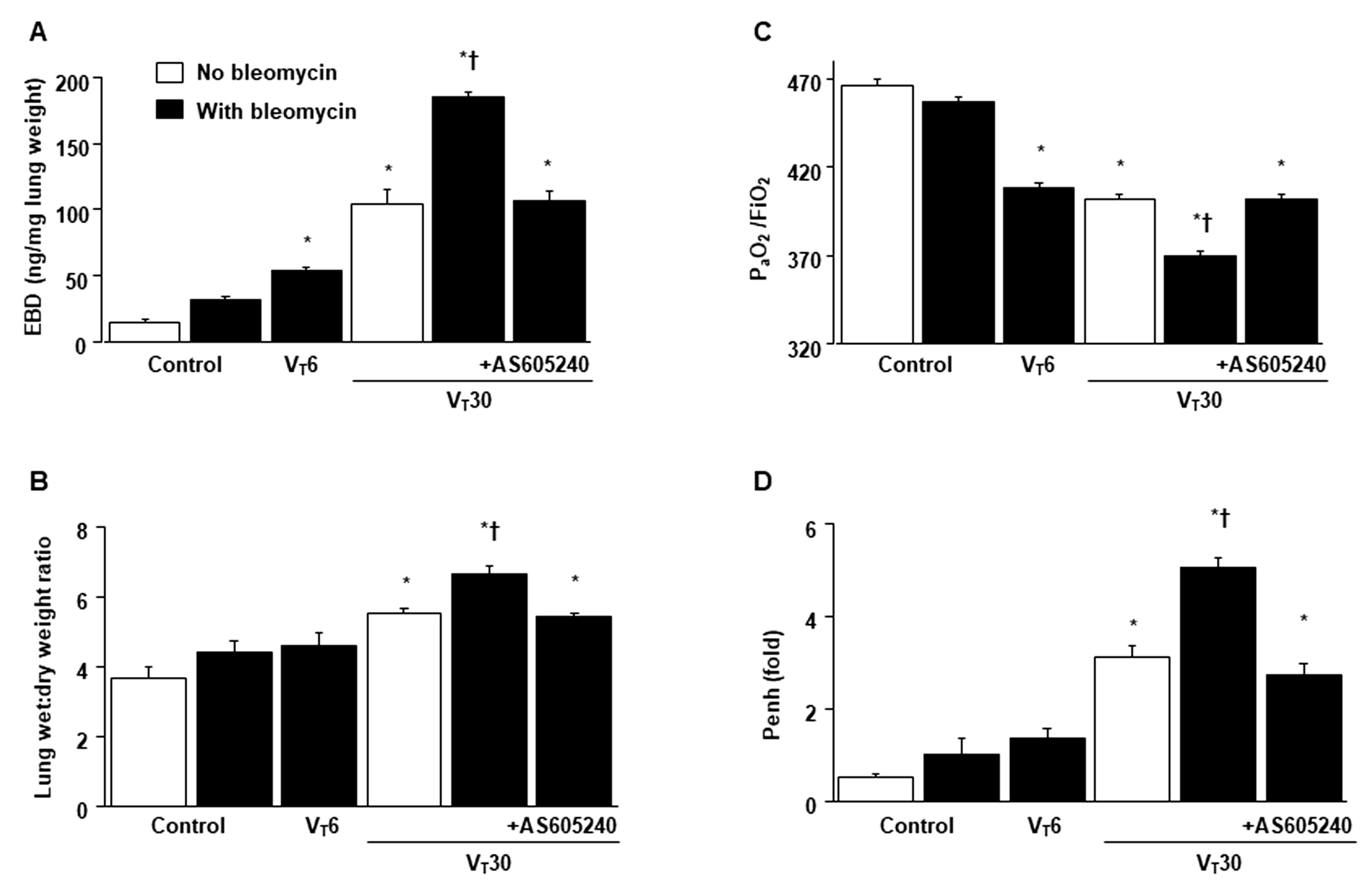
2.1. Inhibition of Bleomycin-Stimulated MV-Induced VILI through AS605240
2.2. Reduction in the Effects of MV on Bleomycin-Enhanced Collagen Fiber Production and Fibrogenic Markers through AS605240
2.3. Suppression of the Effects of MV on Bleomycin-Stimulated PI3K-γ Protein Expression through AS605240
2.4. Inhibition of Bleomycin-Stimulated MV-Induced Lung Inflammation and EMT in PI3K-γ-Deficient Mice
2.5. Reduction of the Effects of MV on Bleomycin-Enhanced Expression of Caspase-3 and Epithelial Apoptosis in PI3K-γ-Deficient Mice
3. Discussion
4. Materials and Methods
4.1. Ethics of Experimental Animals
4.2. Bleomycin Administration
4.3. Pharmacological Inhibitors
4.4. Experimental Groups
4.5. Measurement of Inflammatory Cytokines
4.6. Measurement of Oxidative Stress and Antioxidant Enzyme Expression
4.7. Immunoblot Analysis
4.8. Immunofluorescence Labeling
4.9. Immunohistochemistry
4.10. Masson’s Trichrome Stain and Fibrosis Scoring
4.11. Micro-Computer Tomography
4.12. Analysis of Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | serine/threonine kinase-protein kinase B |
| ALI | acute lung injury |
| ARDS | acute respiratory distress syndrome |
| BAL | bronchoalveolar lavage |
| EBD | Evans blue dye |
| ECM | extracellular matrix |
| EMT | epithelial–mesenchymal transition |
| FiO2 | fraction of inspired oxygen |
| HU | Hounsfield Unit |
| IL | interleukin |
| MDA | malondialdehyde |
| MV | mechanical ventilation |
| ROS | reactive oxygen species |
| PAI-1 | plasminogen activator inhibitor-1 |
| Penh | enhanced pause |
| PI3K-γ-/- | PI3K-γ-deficient mice |
| α-SMA | α-smooth muscle actin |
| TAC | total antioxidant capacity |
| VILI | ventilator-induced lung injury |
| ZO-1 | Zonula occludens |
References
- Li, L.F.; Liu, Y.Y.; Kao, K.C.; Wu, C.T.; Chang, C.H.; Hung, C.Y.; Yang, C.T. Mechanical ventilation augments bleomycin-induced epithelial-mesenchymal transition through the Src pathway. Lab. Investig. 2014, 94, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Li, L.F.; Kao, K.C.; Liu, Y.Y.; Lin, C.W.; Chen, N.H.; Lee, C.S.; Wang, C.W.; Yang, C.T. Nintedanib reduces ventilation-augmented bleomycin-induced epithelial-mesenchymal transition and lung fibrosis through suppression of the Src pathway. J. Cell. Mol. Med. 2017, 21, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Savin, I.A.; Zenkova, M.A.; Sen’kova, A.V. Pulmonary Fibrosis as a Result of Acute Lung Inflammation: Molecular Mechanisms, Relevant In Vivo Models, Prognostic and Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 14959. [Google Scholar] [CrossRef]
- Lin, C.; Zheng, X.; Lin, S.; Zhang, Y.; Wu, J.; Li, Y. Mechanotransduction Regulates the Interplays Between Alveolar Epithelial and Vascular Endothelial Cells in Lung. Front. Physiol. 2022, 13, 818394. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Pan, X.; Wang, L.; Yu, G. Alveolar cells under mechanical stressed niche: Critical contributors to pulmonary fibrosis. Mol. Med. 2020, 26, 95. [Google Scholar] [CrossRef]
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L740–L752. [Google Scholar] [CrossRef]
- Marudamuthu, A.S.; Shetty, S.K.; Bhandary, Y.P.; Karandashova, S.; Thompson, M.; Sathish, V.; Florova, G.; Hogan, T.B.; Pabelick, C.M.; Prakash, Y.S.; et al. Plasminogen activator inhibitor-1 suppresses profibrotic responses in fibroblasts from fibrotic lungs. J. Biol. Chem. 2015, 290, 9428–9441. [Google Scholar] [CrossRef]
- Salton, F.; Ruaro, B.; Confalonieri, P.; Confalonieri, M. Epithelial-Mesenchymal Transition: A Major Pathogenic Driver in Idiopathic Pulmonary Fibrosis? Medicina 2020, 56, 608. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef]
- Delbrel, E.; Uzunhan, Y.; Soumare, A.; Gille, T.; Marchant, D.; Planes, C.; Boncoeur, E. ER Stress is Involved in Epithelial-To-Mesenchymal Transition of Alveolar Epithelial Cells Exposed to a Hypoxic Microenvironment. Int. J. Mol. Sci. 2019, 20, 1299. [Google Scholar] [CrossRef] [PubMed]
- Baratella, E.; Ruaro, B.; Marrocchio, C.; Starvaggi, N.; Salton, F.; Giudici, F.; Quaia, E.; Confalonieri, M.; Cova, M.A. Interstitial Lung Disease at High Resolution CT after SARS-CoV-2-Related Acute Respiratory Distress Syndrome According to Pulmonary Segmental Anatomy. J. Clin. Med. 2021, 10, 3985. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Liu, S.; Li, S.; Xia, Y. Targeting Growth Factor and Cytokine Pathways to Treat Idiopathic Pulmonary Fibrosis. Front. Pharmacol. 2022, 13, 918771. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, K.; Cai, X.; Yang, B.; He, Q.; Wang, J.; Weng, Q. Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharm. Sinica B 2022, 12, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Azad, N.; Wang, L.; Iyer, A.K.; Castranova, V.; Jiang, B.H.; Rojanasakul, Y. Phosphatidylinositol-3-kinase/akt regulates bleomycin-induced fibroblast proliferation and collagen production. Am. J. Respir. Cell Mol. Biol. 2010, 42, 432–441. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef]
- Barratt, S.L.; Blythe, T.; Ourradi, K.; Jarrett, C.; Welsh, G.I.; Bates, D.O.; Millar, A.B. Effects of hypoxia and hyperoxia on the differential expression of VEGF-A isoforms and receptors in Idiopathic Pulmonary Fibrosis (IPF). Respir. Res. 2018, 19, 9. [Google Scholar] [CrossRef]
- Wei, X.; Han, J.; Chen, Z.Z.; Qi, B.W.; Wang, G.C.; Ma, Y.H.; Zheng, H.; Luo, Y.F.; Wei, Y.Q.; Chen, L.J. A phosphoinositide 3-kinase-gamma inhibitor, AS605240 prevents bleomycin-induced pulmonary fibrosis in rats. Biochem. Biophys. Res. Commun. 2010, 397, 311–317. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Li, L.F.; Liu, Y.Y.; Lin, S.W.; Chang, C.H.; Chen, N.H.; Hung, C.Y.; Lee, C.S. Low-Molecular-Weight Heparin Reduces Ventilation-Induced Lung Injury through Hypoxia Inducible Factor-1alpha in a Murine Endotoxemia Model. Int. J. Mol. Sci. 2020, 21, 3097. [Google Scholar] [CrossRef]
- Cabrera-Benitez, N.E.; Laffey, J.G.; Parotto, M.; Spieth, P.M.; Villar, J.; Zhang, H.; Slutsky, A.S. Mechanical ventilation-associated lung fibrosis in acute respiratory distress syndrome: A significant contributor to poor outcome. Anesthesiology 2014, 121, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Ichikado, K.; Muranaka, H.; Gushima, Y.; Kotani, T.; Nader, H.M.; Fujimoto, K.; Johkoh, T.; Iwamoto, N.; Kawamura, K.; Nagano, J.; et al. Fibroproliferative changes on high-resolution CT in the acute respiratory distress syndrome predict mortality and ventilator dependency: A prospective observational cohort study. BMJ Open 2012, 2, e000545. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Wurfel, M.M.; Lee, J.S.; Park, D.R.; Frevert, C.W.; Madtes, D.K.; Shapiro, S.D.; Martin, T.R. Essential role of MMP-12 in Fas-induced lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 37, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Tager, A.M.; Kradin, R.L.; LaCamera, P.; Bercury, S.D.; Campanella, G.S.; Leary, C.P.; Polosukhin, V.; Zhao, L.H.; Sakamoto, H.; Blackwell, T.S.; et al. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am. J. Respir. Cell Mol. Biol. 2004, 31, 395–404. [Google Scholar] [CrossRef]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell Mol. Life Sci. 2021, 78, 2031–2057. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zimmerman, G.A.; Esmon, C.; Bhattacharya, J.; Coller, B.; Doerschuk, C.M.; Floros, J.; Gimbrone, M.A., Jr.; Hoffman, E.; Hubmayr, R.D.; et al. Future research directions in acute lung injury: Summary of a National Heart, Lung, and Blood Institute working group. Am. J. Respir. Crit. Care Med. 2003, 167, 1027–1035. [Google Scholar] [CrossRef]
- ARDSNet, The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. [comment]. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial(-)Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina 2019, 55, 83. [Google Scholar] [CrossRef]
- Sakashita, A.; Nishimura, Y.; Nishiuma, T.; Takenaka, K.; Kobayashi, K.; Kotani, Y.; Yokoyama, M. Neutrophil elastase inhibitor (sivelestat) attenuates subsequent ventilator-induced lung injury in mice. Eur. J. Pharmacol. 2007, 571, 62–71. [Google Scholar] [CrossRef]
- Ma, H.; Wu, X.; Li, Y.; Xia, Y. Research Progress in the Molecular Mechanisms, Therapeutic Targets, and Drug Development of Idiopathic Pulmonary Fibrosis. Front. Pharmacol. 2022, 13, 963054. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, L.; Xia, H.; Chen, L.; Cui, S.; Wang, Y.; Zhou, T.; Xiong, W.; Song, L.; Li, S.; et al. Resolvin D1 attenuates mechanical stretch-induced pulmonary fibrosis via epithelial-mesenchymal transition. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L1013–L1024. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.Z.; Li, M.; Wang, Y.X.; Zhang, P.; Sun, M.M.; Xu, J.X.; Yang, Y.Y.; He, Y.J.; Yu, Y.; Li, R.T.; et al. Mechanosensitive ion channel Piezo1 mediates mechanical ventilation-exacerbated ARDS-associated pulmonary fibrosis. J. Adv. Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Zhang, N.; Xia, J.; Zhan, Q.; Wang, C. Potential role of M2 macrophage polarization in ventilator-induced lung fibrosis. Int. Immunopharmacol. 2019, 75, 105795. [Google Scholar] [CrossRef] [PubMed]
- Laddha, A.P.; Kulkarni, Y.A. VEGF and FGF-2: Promising targets for the treatment of respiratory disorders. Respir. Med. 2019, 156, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Sala, V.; Della Sala, A.; Ghigo, A.; Hirsch, E. Roles of phosphatidyl inositol 3 kinase gamma (PI3Kgamma) in respiratory diseases. Cell Stress 2021, 5, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.M.; Li, Q.; Chen, K.J.; Xu, C.X.; Deng, M.S.; Li, T.; Zhang, D.D.; Duan, Z.X.; Chen, Z.Q.; Li, G.H.; et al. Bleomycin induces epithelial-to-mesenchymal transition via bFGF/PI3K/ESRP1 signaling in pulmonary fibrosis. Biosci. Rep. 2020, 40, BSR20190756. [Google Scholar] [CrossRef]
- Cheng, S.E.; Lee, I.T.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. Thrombin induces ICAM-1 expression in human lung epithelial cells via c-Src/PDGFR/PI3K/Akt-dependent NF-κB/p300 activation. Clin. Sci. 2014, 127, 171–183. [Google Scholar] [CrossRef]
- Russo, R.C.; Garcia, C.C.; Barcelos, L.S.; Rachid, M.A.; Guabiraba, R.; Roffe, E.; Souza, A.L.; Sousa, L.P.; Mirolo, M.; Doni, A.; et al. Phosphoinositide 3-kinase gamma plays a critical role in bleomycin-induced pulmonary inflammation and fibrosis in mice. J. Leukoc. Biol. 2011, 89, 269–282. [Google Scholar] [CrossRef]
- Camara, J.; Jarai, G. Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis Tissue Repair 2010, 3, 2. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, H.; Jiang, K.; Wang, Y.; Zhang, W.; Chu, Q.; Li, J.; Huang, H.; Cai, T.; Ji, H.; et al. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016, 16, 1810–1819. [Google Scholar] [CrossRef]
- Fullgrabe, J.; Hajji, N.; Joseph, B. Cracking the death code: Apoptosis-related histone modifications. Cell Death Differ. 2010, 17, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Criscione, S.W.; Teo, Y.V.; Neretti, N. The Chromatin Landscape of Cellular Senescence. Trends Genet. 2016, 32, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ma, H.; Jin, J.; Uttam, S.; Fu, R.; Huang, Y.; Liu, Y. Super-Resolution Imaging of Higher-Order Chromatin Structures at Different Epigenomic States in Single Mammalian Cells. Cell Rep. 2018, 24, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Pan, Y.; Fanelli, V.; Wu, S.; Luo, A.A.; Islam, D.; Han, B.; Mao, P.; Ghazarian, M.; Zeng, W.; et al. Mechanical Stress and the Induction of Lung Fibrosis via the Midkine Signaling Pathway. Am. J. Respir. Crit. Care Med. 2015, 192, 315–323. [Google Scholar] [CrossRef]
- Lionetti, V.; Lisi, A.; Patrucco, E.; De Giuli, P.; Milazzo, M.G.; Ceci, S.; Wymann, M.; Lena, A.; Gremigni, V.; Fanelli, V.; et al. Lack of phosphoinositide 3-kinase-gamma attenuates ventilator-induced lung injury. Crit. Care Med. 2006, 34, 134–141. [Google Scholar] [CrossRef]
- Jin, G.Y.; Bok, S.M.; Han, Y.M.; Chung, M.J.; Yoon, K.H.; Kim, S.R.; Lee, Y.C. Effectiveness of rosiglitazone on bleomycin-induced lung fibrosis: Assessed by micro-computed tomography and pathologic scores. Eur. J. Radiol. 2012, 81, 1901–1906. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.-F.; Yu, C.-C.; Huang, C.-Y.; Wu, H.-P.; Chu, C.-M.; Liu, P.-C.; Liu, Y.-Y. Attenuation of Ventilation-Enhanced Epithelial–Mesenchymal Transition through the Phosphoinositide 3-Kinase-γ in a Murine Bleomycin-Induced Acute Lung Injury Model. Int. J. Mol. Sci. 2023, 24, 5538. https://doi.org/10.3390/ijms24065538
Li L-F, Yu C-C, Huang C-Y, Wu H-P, Chu C-M, Liu P-C, Liu Y-Y. Attenuation of Ventilation-Enhanced Epithelial–Mesenchymal Transition through the Phosphoinositide 3-Kinase-γ in a Murine Bleomycin-Induced Acute Lung Injury Model. International Journal of Molecular Sciences. 2023; 24(6):5538. https://doi.org/10.3390/ijms24065538
Chicago/Turabian StyleLi, Li-Fu, Chung-Chieh Yu, Chih-Yu Huang, Huang-Pin Wu, Chien-Ming Chu, Ping-Chi Liu, and Yung-Yang Liu. 2023. "Attenuation of Ventilation-Enhanced Epithelial–Mesenchymal Transition through the Phosphoinositide 3-Kinase-γ in a Murine Bleomycin-Induced Acute Lung Injury Model" International Journal of Molecular Sciences 24, no. 6: 5538. https://doi.org/10.3390/ijms24065538
APA StyleLi, L.-F., Yu, C.-C., Huang, C.-Y., Wu, H.-P., Chu, C.-M., Liu, P.-C., & Liu, Y.-Y. (2023). Attenuation of Ventilation-Enhanced Epithelial–Mesenchymal Transition through the Phosphoinositide 3-Kinase-γ in a Murine Bleomycin-Induced Acute Lung Injury Model. International Journal of Molecular Sciences, 24(6), 5538. https://doi.org/10.3390/ijms24065538