
Hepatocyte-Specific Triggering of Hepatic Stellate Cell Profibrotic Activation by Apoptotic Bodies: The Role of Hepatoma-Derived Growth Factor, HIV, and Ethanol

 ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. HSC Profibrotic Activation after Internalization of Acetaldehyde- and HIV-Derived Hepatocyte ABSS
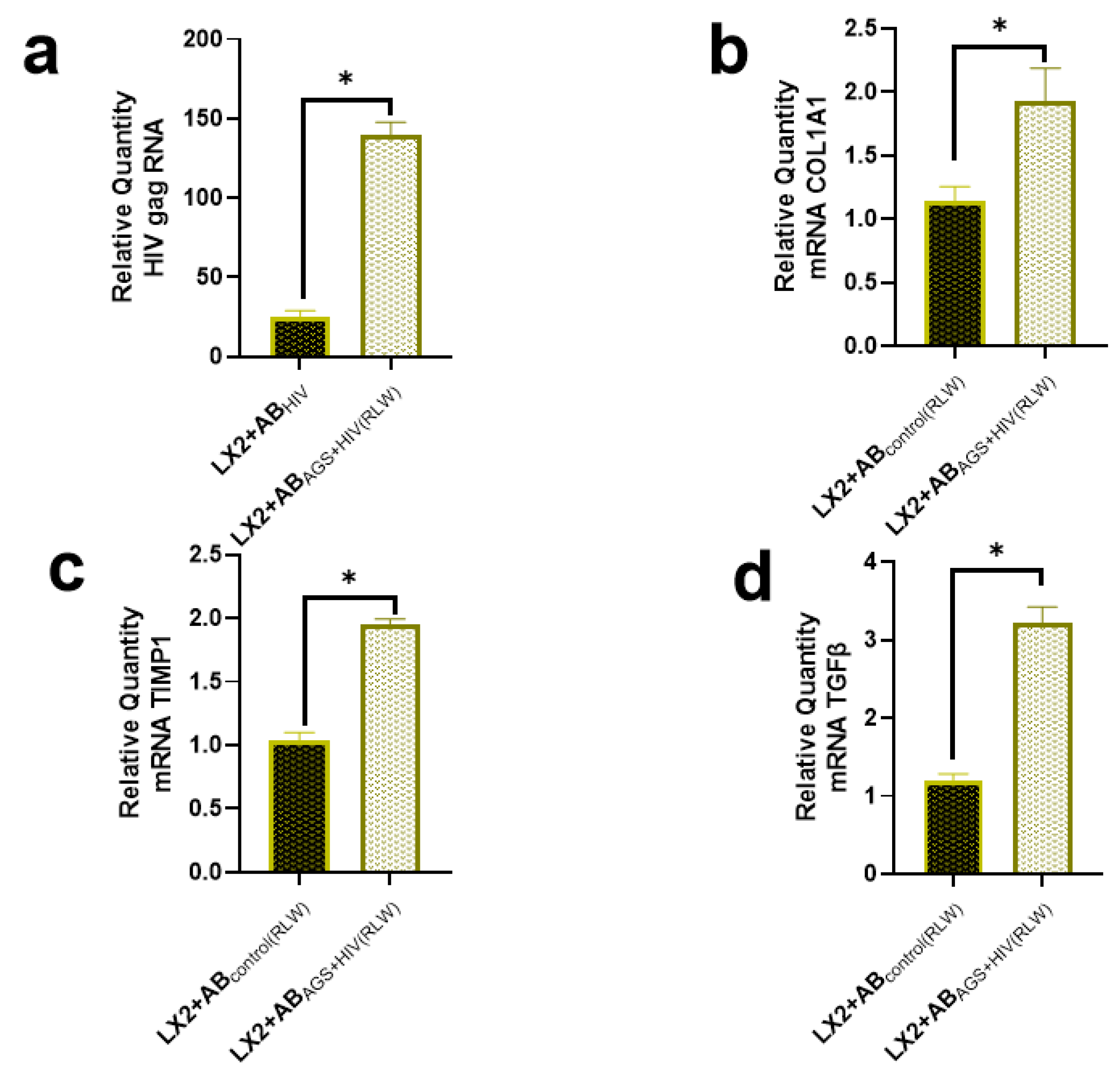
2.2. Lack of Profibrotic Changes in HSC after Internalization of ABs Derived from HIV- and AGS-Exposed Lymphocytes
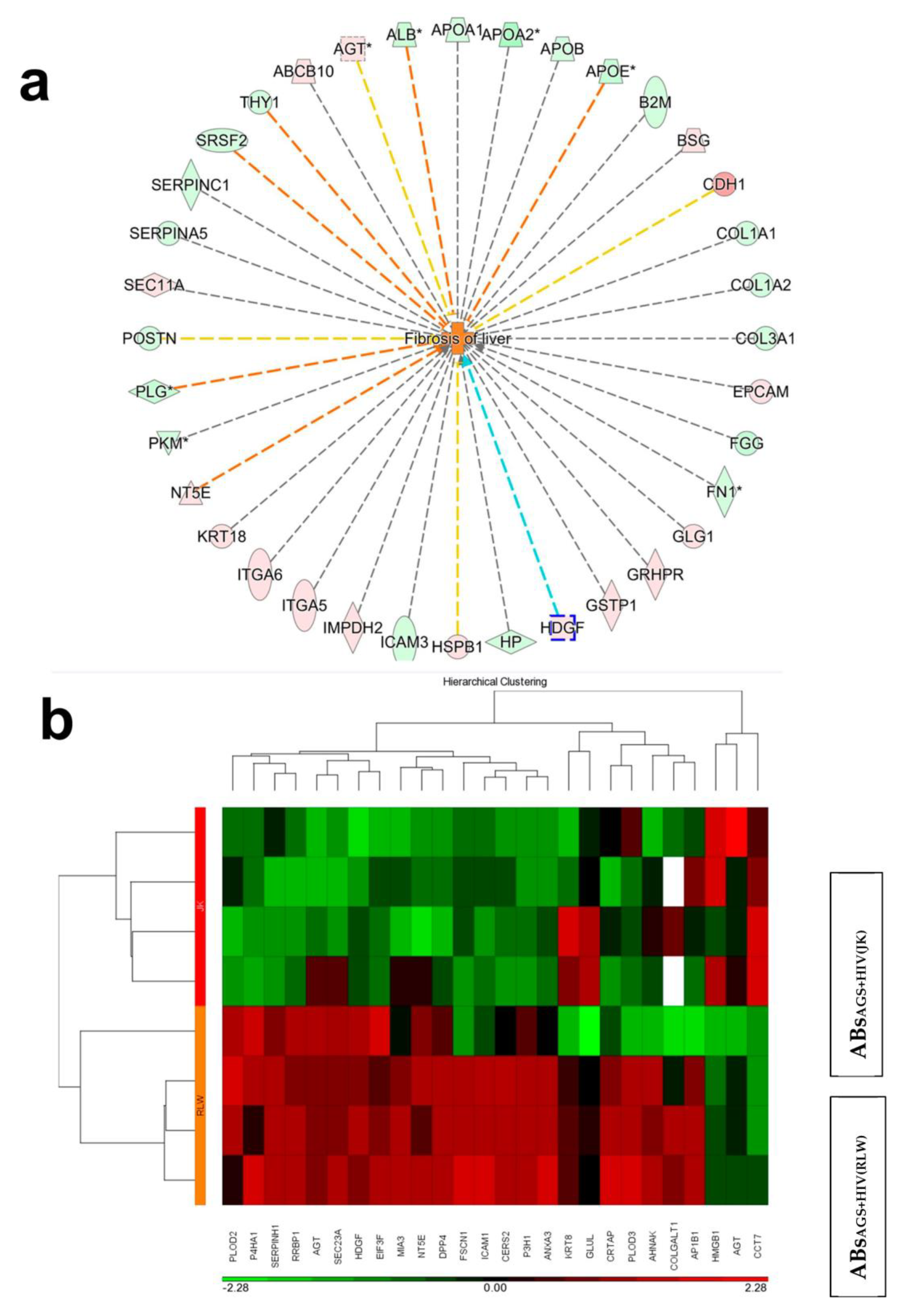
2.3. Proteins Expressed in ABsAGS+HIV(RLW) and ABsAGS+HIV(JK) Associated with Hepatotoxicity
2.4. Proteins in Hepatocyte ABS vs. Lymphocyte ABs Associated with Liver Fibrosis
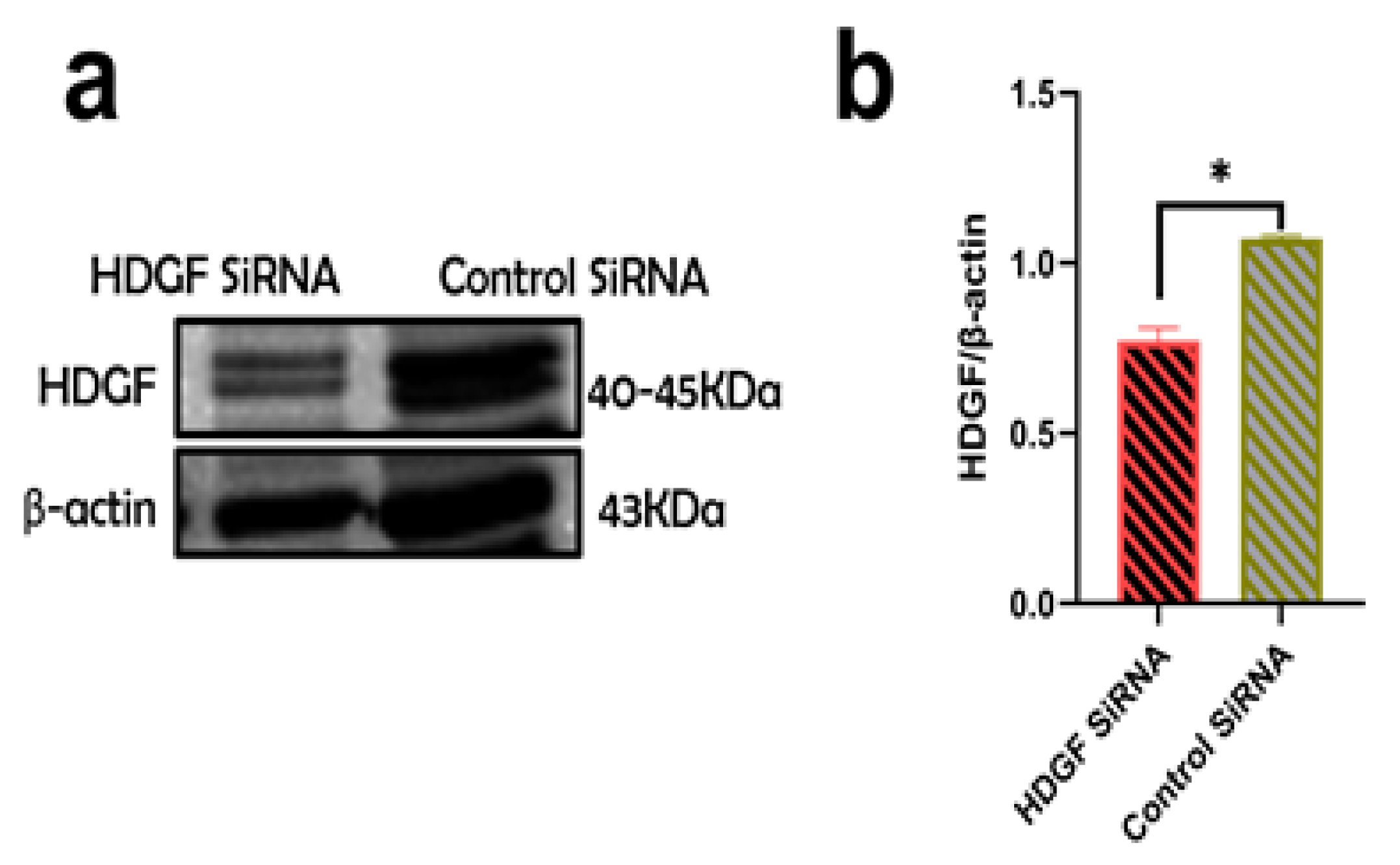
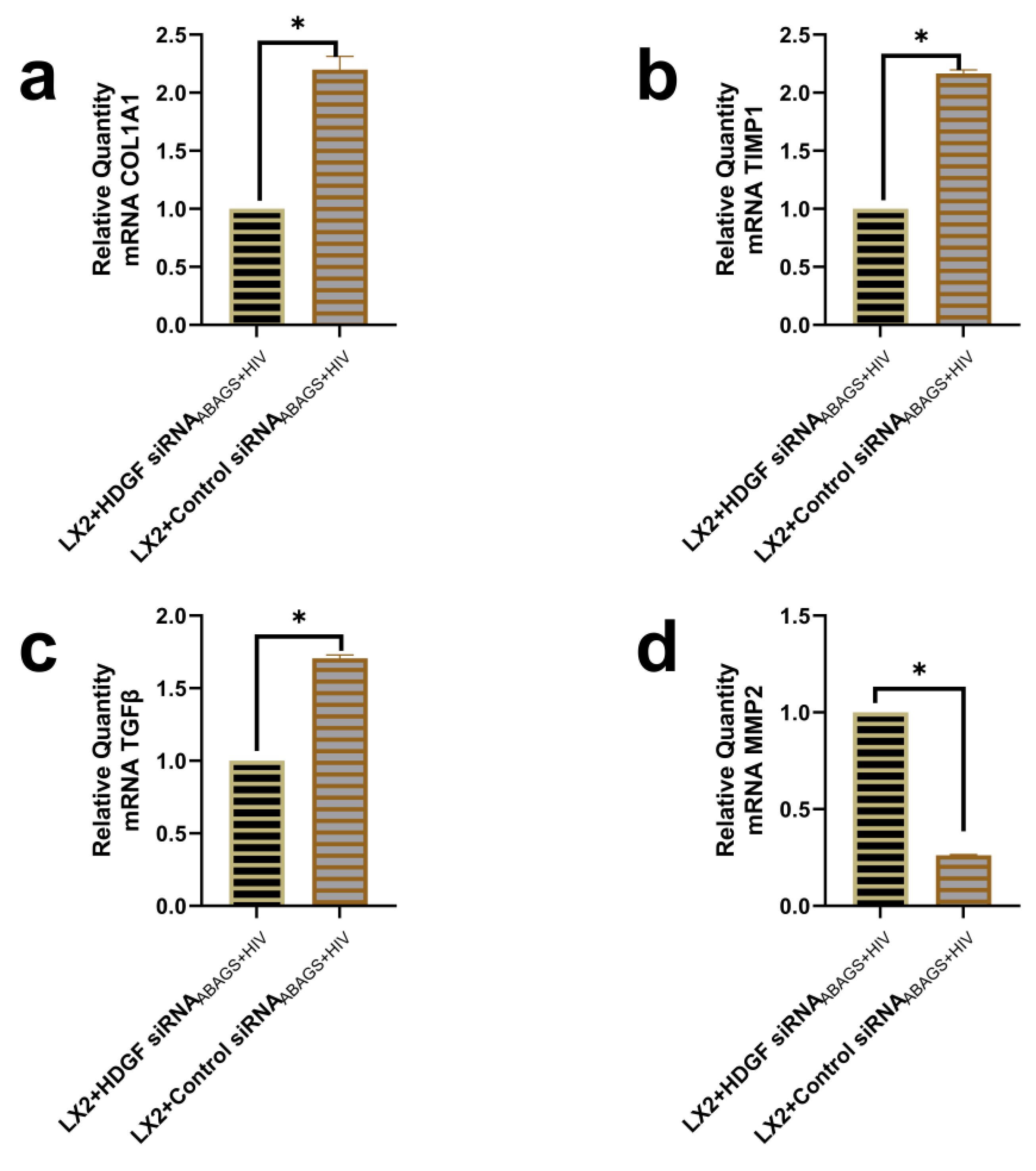
2.5. Suppression of Profibrotic Genes in LX2 Cells after Internalization of ABsAGS+HIV Derived from HDGF-Knockdown RLW Cells
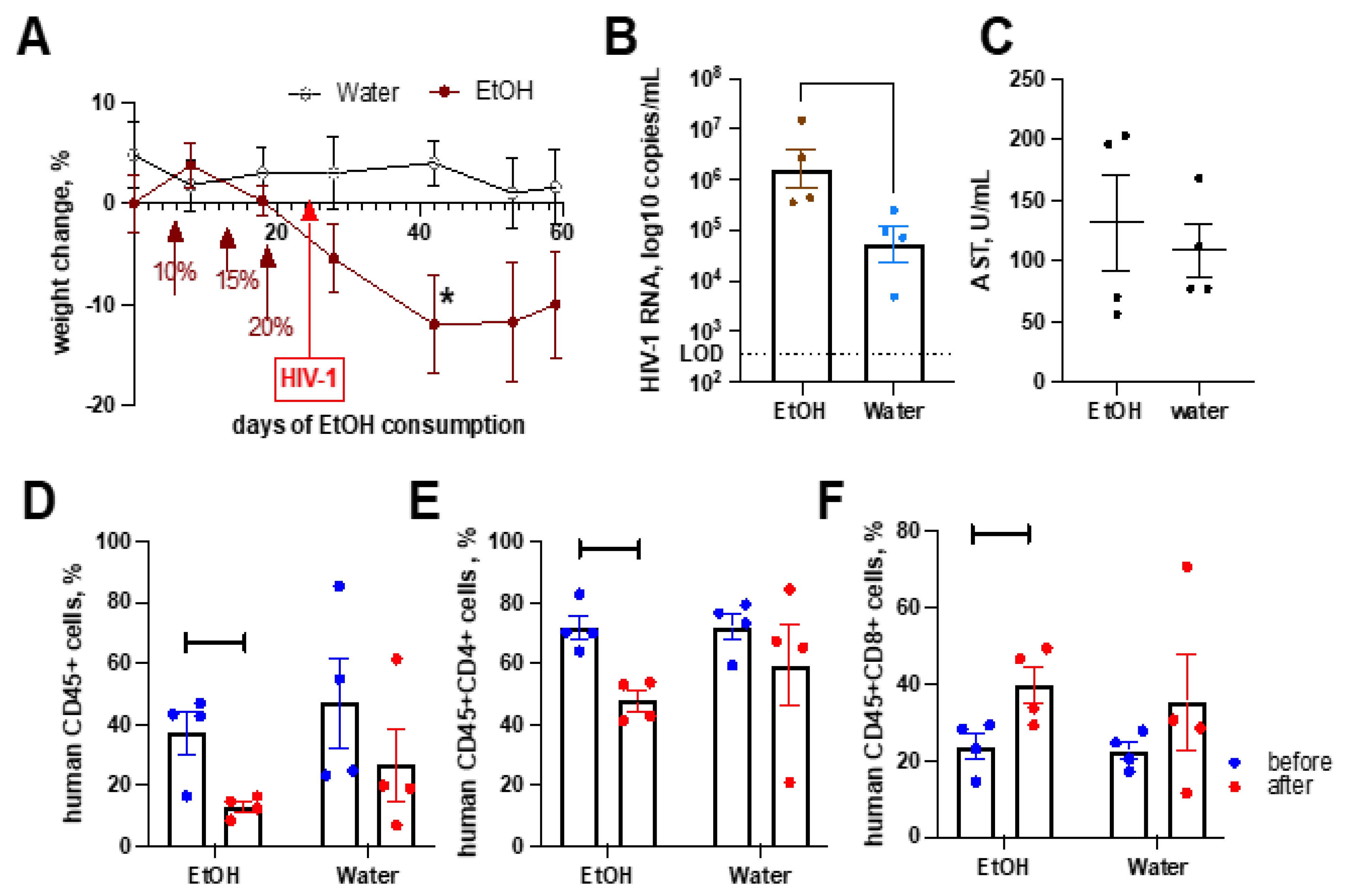
2.6. Alcohol Consumption Exacerbated HIV-1-Induced Immunopathology but Did Not Affect Liver Fibrosis in the Absence of Transplanted Human Hepatocytes
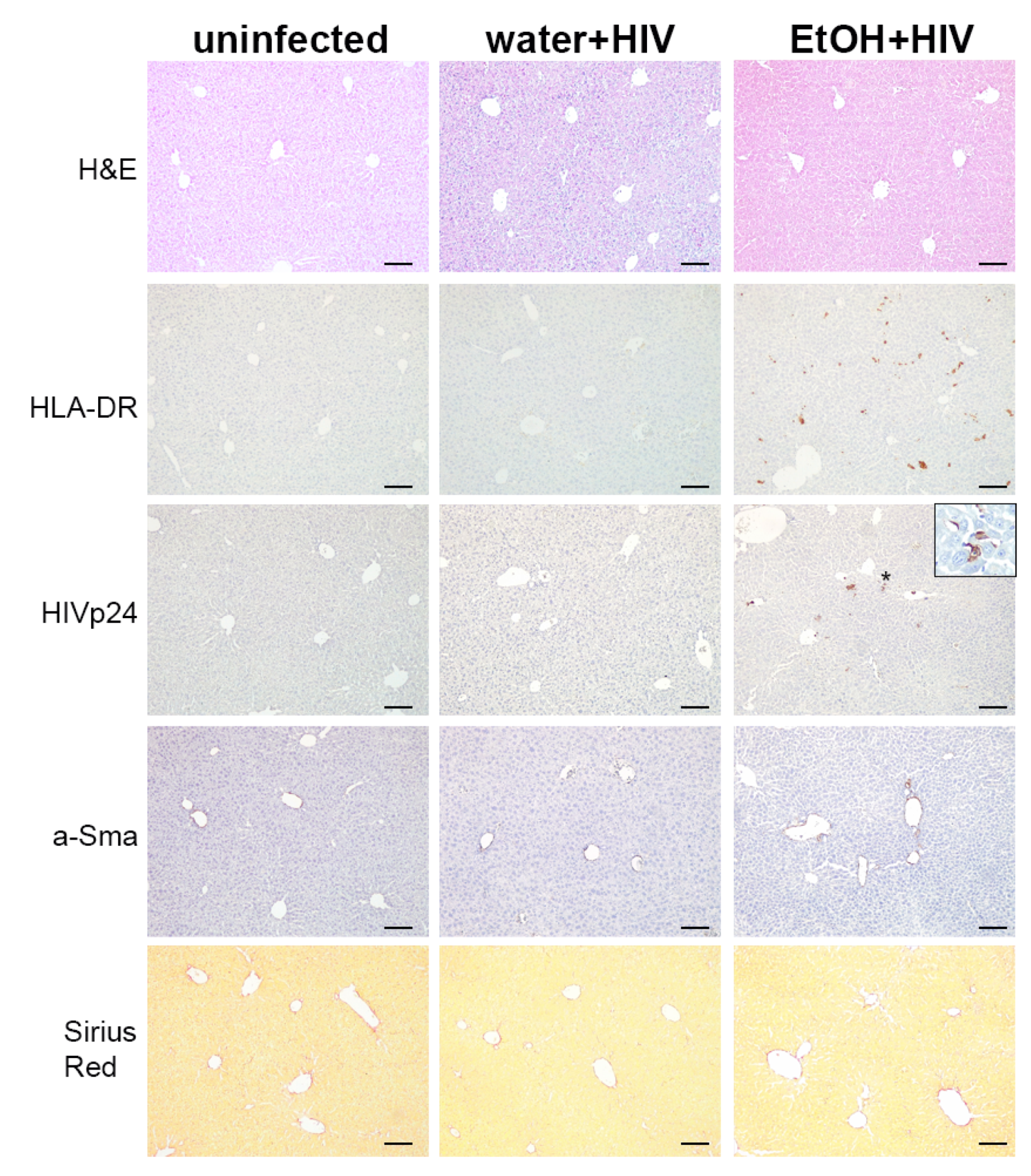
2.7. Alcohol Consumption Increased Human Immune Cells Infiltration but Did Not Induce Fibrotic Changes in the Liver of HIV-1-Infected Humanized Mice
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. In Vitro Studies
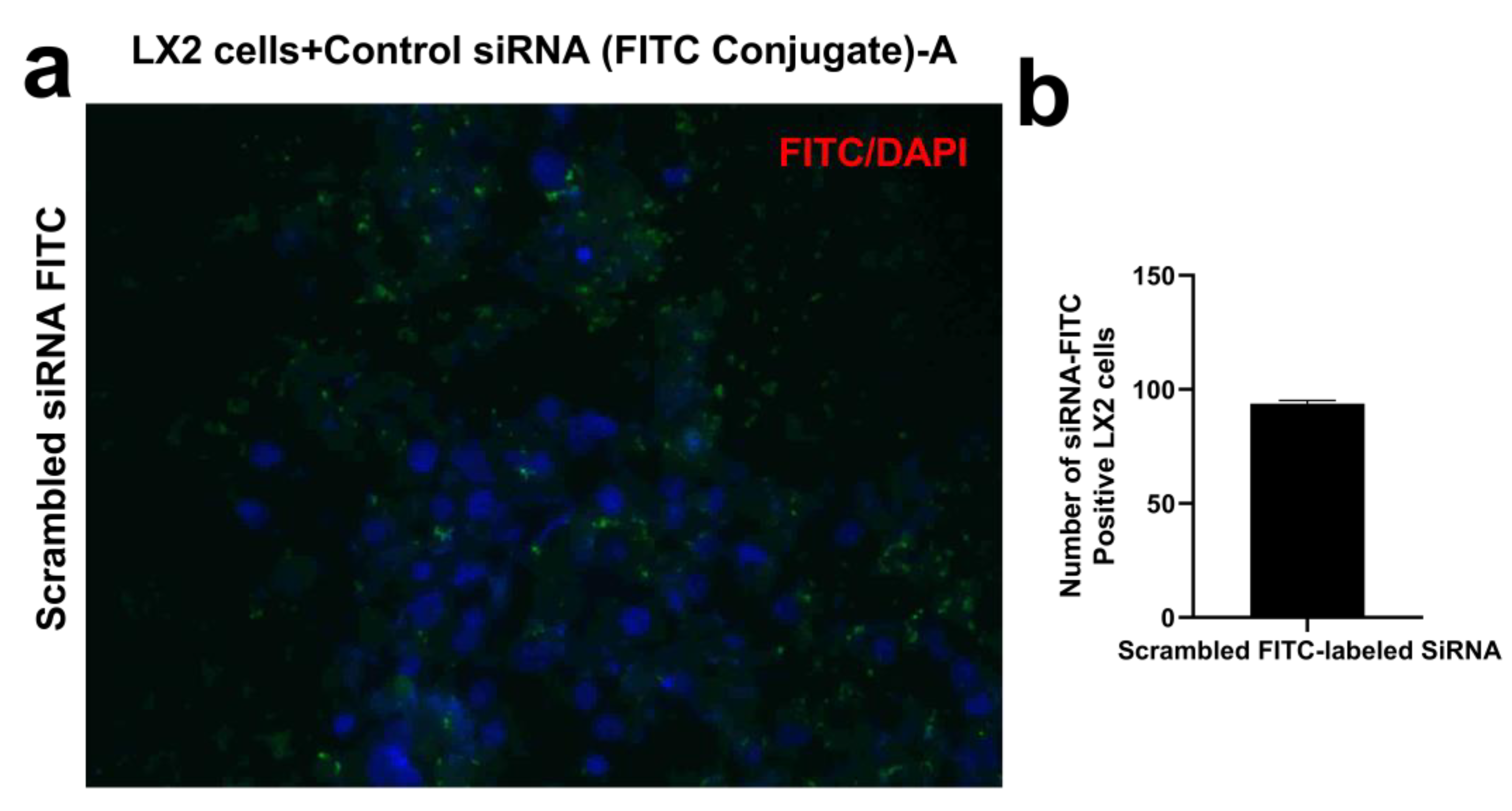
4.3. siRNA Transfection of LX2 Cells
4.4. Sample Preparation for Mass Spectrometry
4.5. LC-MS/MS
4.6. Data Analysis and Bioinformatics Analysis
4.7. RNA Isolation and RT-PCR
4.8. Immunoblotting/Western Blotting
4.9. In Vivo Studies
4.9.1. Humanized Mice
4.9.2. Immunohistochemistry
4.9.3. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tattevin, P.; Camus, C.; Arvieux, C.; Ruffault, A.; Michelet, C. Multiple Organ Failure during Primary HIV Infection. Clin. Infect. Dis. 2007, 44, e28–e29. [Google Scholar] [CrossRef] [PubMed]
- Lorenc, A.; Ananthavarathan, P.; Lorigan, J.; Jowata, M.; Brook, G.; Banarsee, R. The prevalence of comorbidities among people living with HIV in Brent: A diverse London Borough. Lond. J. Prim. Care 2014, 6, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.; Hsue, P.; Budd, D.; Meyer, N. Healthcare utilization and direct costs of non-infectious comorbidities in HIV-infected patients in the USA. Curr. Med. Res. Opin. 2018, 34, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, F.; Miners, A.; Smith, C.J.; Simmons, R.; Lodwick, R.K.; Cambiano, V.; Lundgren, J.D.; Delpech, V.; Phillips, A.N. Projected Lifetime Healthcare Costs Associated with HIV Infection. PLoS ONE 2015, 10, e0125018. [Google Scholar] [CrossRef]
- Bonnet, F.; Le Marec, F.; Leleux, O.; Gerard, Y.; Neau, D.; Lazaro, E.; Duffau, P.; Caubet, O.; Vandenhende, M.A.; Mercie, P.; et al. Evolution of comorbidities in people living with HIV between 2004 and 2014: Cross-sectional analyses from ANRS CO3 Aquitaine cohort. BMC Infect. Dis. 2020, 20, 850. [Google Scholar] [CrossRef]
- Triant, V.A. Cardiovascular disease and HIV infection. Curr. HIV/AIDS Rep. 2013, 10, 199–206. [Google Scholar] [CrossRef]
- Sherman, K.E.; Rockstroh, J.; Thomas, D. Human immunodeficiency virus and liver disease: An update. Hepatology 2015, 62, 1871–1882. [Google Scholar] [CrossRef]
- Duko, B.; Ayalew, M.; Ayano, G. The prevalence of alcohol use disorders among people living with HIV/AIDS: A systematic review and meta-analysis. Subst. Abus. Treat. Prev. Policy 2019, 14, 52. [Google Scholar] [CrossRef]
- Ganesan, M.; New-Aaron, M.; Dagur, R.S.; Makarov, E.; Wang, W.; Kharbanda, K.K.; Kidambi, S.; Poluektova, L.Y.; Osna, N.A. Alcohol Metabolism Potentiates HIV-Induced Hepatotoxicity: Contribution to End-Stage Liver Disease. Biomolecules 2019, 9, 851. [Google Scholar] [CrossRef]
- New-Aaron, M.; Thomes, P.G.; Ganesan, M.; Dagur, R.S.; Donohue, T.M., Jr.; Kusum, K.K.; Poluektova, L.Y.; Osna, N.A. Alcohol-induced lysosomal damage and suppression of lysosome biogenesis contribute to hepatotoxicity in HIV-exposed liver cells. Biomolecules 2021, 11, 1497. [Google Scholar] [CrossRef]
- New-Aaron, M.; Dagur, R.S.; Koganti, S.S.; Ganesan, M.; Wang, W.; Makarov, E.; Ogunnaike, M.; Kharbanda, K.K.; Poluektova, L.Y.; Osna, N.A. Alcohol and HIV-Derived Hepatocyte Apoptotic Bodies Induce Hepatic Stellate Cell Activation. Biology 2022, 11, 1059. [Google Scholar] [CrossRef] [PubMed]
- Kitto, L.J.; Henderson, N.C. Hepatic Stellate Cell Regulation of Liver Regeneration and Repair. Hepatol. Commun. 2021, 5, 358–370. [Google Scholar] [CrossRef]
- Jiao, J.; Friedman, S.L.; Aloman, C. Hepatic fibrosis. Curr. Opin. Gastroenterol. 2009, 25, 223–229. [Google Scholar] [CrossRef] [PubMed]
- New-Aaron, M.O. Hepatocyte-Hepatic Stellate Cell Axis in Potentiation of Alcohol and HIV-Induced Liver Injury. Ph.D. Dissertation, University of Nebraska Medical Center, Omaha, NE, USA, 2022. [Google Scholar]
- Lee, C.H.; Wu, S.B.; Hong, C.H.; Yu, H.S.; Wei, Y.H. Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy. Int. J. Mol. Sci. 2013, 14, 6414–6435. [Google Scholar] [CrossRef]
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic Body Engulfment by a Human Stellate Cell Line Is Profibrogenic. Lab. Investig. 2003, 83, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Koma, T.; Suzuki, A.; Nagamatsu, K.; Yasui, T.; Yasutomo, K.; Adachi, A.; Minamikawa, T.; Nomaguchi, M. Major target for UV-induced complete loss of HIV-1 infectivity: A model study of single-stranded RNA enveloped viruses. Front. Virol. 2022, 86. [Google Scholar] [CrossRef]
- Pallet, N.; Hebert, M.-J. The apoptotic program promotes tissue remodeling and fibrosis. Kidney Int. 2011, 80, 1108. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.; DiPietro, L.A. Apoptosis and angiogenesis: An evolving mechanism for fibrosis. FASEB J. 2013, 27, 3893–3901. [Google Scholar] [CrossRef] [PubMed]
- Janssen, W.J.; Morimoto, K. Apoptotic cell clearance and fibrotic lung disease. Eur. Respir. J. 2012, 40, 289–290. [Google Scholar] [CrossRef]
- Crispe, I.N. Death and destruction of activated T lymphocytes. Immunol. Res. 1999, 19, 143–157. [Google Scholar] [CrossRef]
- Bertolino, P.; Bowen, D.G.; Benseler, V.; Kowdley, K.; McCaughan, G.; Trautwein, C. T cells in the liver: There is life beyond the graveyard. Hepatology 2007, 45, 1580–1582. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, H.; Nakamura, H.; Liu, W.; Nishiguchi, S. Hepatoma-Derived Growth Factor: Its Possible Involvement in the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2015, 16, 14086–14097. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Tang, H.; Liang, Y.; Huang, S.; Wang, Y.; Huang, W.; Zhou, Y. Alcohol Consumption and Risk of Liver Fibrosis in People Living With HIV: A Systematic Review and Meta-Analysis. Front. Immunol. 2022, 13, 841314. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Davies, S.P.; Reynolds, G.M.; Stamataki, Z. Clearance of Apoptotic Cells by Tissue Epithelia: A Putative Role for Hepatocytes in Liver Efferocytosis. Front. Immunol. 2018, 9, 44. [Google Scholar] [CrossRef]
- Nelson, S.; Bagby, G.J. Alcohol and HIV Infection. Trans. Am. Clin. Climatol. Assoc. 2011, 122, 244–253. [Google Scholar]
- Enomoto, H.; Nakamura, H.; Nishikawa, H.; Nishiguchi, S.; Iijima, H. Hepatoma-Derived Growth Factor: An Overview and Its Role as a Potential Therapeutic Target Molecule for Digestive Malignancies. Int. J. Mol. Sci. 2020, 21, 4216. [Google Scholar] [CrossRef]
- Kao, Y.H.; Chen, C.L.; Jawan, B.; Chung, Y.H.; Sun, C.K.; Kuo, S.M.; Hu, T.H.; Lin, Y.C.; Chan, H.H.; Cheng, K.H.; et al. Upregulation of hepatoma-derived growth factor is involved in murine hepatic fibrogenesis. J. Hepatol. 2010, 52, 96–105. [Google Scholar] [CrossRef]
- Luo, Y.; Huo, Y.; Song, P.; Zhang, X.; Liao, M. Validation and functional analysis of the critical proteins in combination with taurine, epigallocatechin gallate and genistein against liver fibrosis in rats. Biomed. Pharmacother. 2019, 115, 108975. [Google Scholar] [CrossRef]
- Tsang, T.Y.; Tang, W.Y.; Tsang, W.P.; Co, N.N.; Kong, S.K.; Kwok, T.T. Downregulation of hepatoma-derived growth factor activates the Bad-mediated apoptotic pathway in human cancer cells. Apoptosis 2008, 13, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Ma, Y.; Chen, X.; Zhou, L.; Zhang, H.; Zhong, G.; Zhang, L.; Tang, J. Heparin-binding growth factor (HDGF) drives radioresistance in breast cancer by activating the STAT3 signaling pathway. J. Transl. Med. 2021, 19, 344. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.; McVicker, B.; Poluektova, L.; Ganesan, M.; Kharbanda, K. Mode of Oral Ethanol Feeding Affects Liver Oxidative Stress Levels And Methylation Status. Int. J. Biochem. Res. Rev. 2014, 4, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, M.; O’Neil, M.; Villar, M.T.; Artigues, A.; Averilla, J.; Gunewardena, S.; Weinman, S.A.; Tikhanovich, I. A Western diet with alcohol in drinking water recapitulates features of alcohol-associated liver disease in mice. Alcohol. Clin. Exp. Res. 2021, 45, 1980–1993. [Google Scholar] [CrossRef] [PubMed]
- Akil, A.; Endsley, M.; Shanmugam, S.; Saldarriaga, O.; Somasunderam, A.; Spratt, H.; Stevenson, H.L.; Utay, N.S.; Ferguson, M.; Yi, M. Fibrogenic Gene Expression in Hepatic Stellate Cells Induced by HCV and HIV Replication in a Three Cell Co-Culture Model System. Sci. Rep. 2019, 9, 568. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Osna, N.A.; Eguchi, A.; Feldstein, A.E.; Tsukamoto, H.; Dagur, R.S.; Ganesan, M.; New-Aaron, M.; Arumugam, M.K.; Chava, S.; Ribeiro, M.; et al. Cell-to-Cell Communications in Alcohol-Associated Liver Disease. Front. Physiol. 2022, 13, 831004. [Google Scholar] [CrossRef]
- Gorantla, S.; Makarov, E.; Finke-Dwyer, J.; Castanedo, A.; Holguin, A.; Gebhart, C.L.; Gendelman, H.E.; Poluektova, L. Links between progressive HIV-1 infection of humanized mice and viral neuropathogenesis. Am. J. Pathol. 2010, 177, 2938–2949. [Google Scholar] [CrossRef]
- Powers, J.; Zhang, H.; Battrell, L.; Meadows, G.G.; Trobridge, G.D. Establishment of an immunodeficient alcohol mouse model to study the effects of alcohol on human cells in vivo. J. Stud. Alcohol Drugs 2012, 73, 933–937. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
New-Aaron, M.; Koganti, S.S.; Ganesan, M.; Kanika, S.; Kumar, V.; Wang, W.; Makarov, E.; Kharbanda, K.K.; Poluektova, L.Y.; Osna, N.A. Hepatocyte-Specific Triggering of Hepatic Stellate Cell Profibrotic Activation by Apoptotic Bodies: The Role of Hepatoma-Derived Growth Factor, HIV, and Ethanol. Int. J. Mol. Sci. 2023, 24, 5346. https://doi.org/10.3390/ijms24065346
New-Aaron M, Koganti SS, Ganesan M, Kanika S, Kumar V, Wang W, Makarov E, Kharbanda KK, Poluektova LY, Osna NA. Hepatocyte-Specific Triggering of Hepatic Stellate Cell Profibrotic Activation by Apoptotic Bodies: The Role of Hepatoma-Derived Growth Factor, HIV, and Ethanol. International Journal of Molecular Sciences. 2023; 24(6):5346. https://doi.org/10.3390/ijms24065346
Chicago/Turabian StyleNew-Aaron, Moses, Siva Sankar Koganti, Murali Ganesan, Sharma Kanika, Vikas Kumar, Weimin Wang, Edward Makarov, Kusum K. Kharbanda, Larisa Y. Poluektova, and Natalia A. Osna. 2023. "Hepatocyte-Specific Triggering of Hepatic Stellate Cell Profibrotic Activation by Apoptotic Bodies: The Role of Hepatoma-Derived Growth Factor, HIV, and Ethanol" International Journal of Molecular Sciences 24, no. 6: 5346. https://doi.org/10.3390/ijms24065346
APA StyleNew-Aaron, M., Koganti, S. S., Ganesan, M., Kanika, S., Kumar, V., Wang, W., Makarov, E., Kharbanda, K. K., Poluektova, L. Y., & Osna, N. A. (2023). Hepatocyte-Specific Triggering of Hepatic Stellate Cell Profibrotic Activation by Apoptotic Bodies: The Role of Hepatoma-Derived Growth Factor, HIV, and Ethanol. International Journal of Molecular Sciences, 24(6), 5346. https://doi.org/10.3390/ijms24065346