
MMR Deficiency Defines Distinct Molecular Subtype of Breast Cancer with Histone Proteomic Networks

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
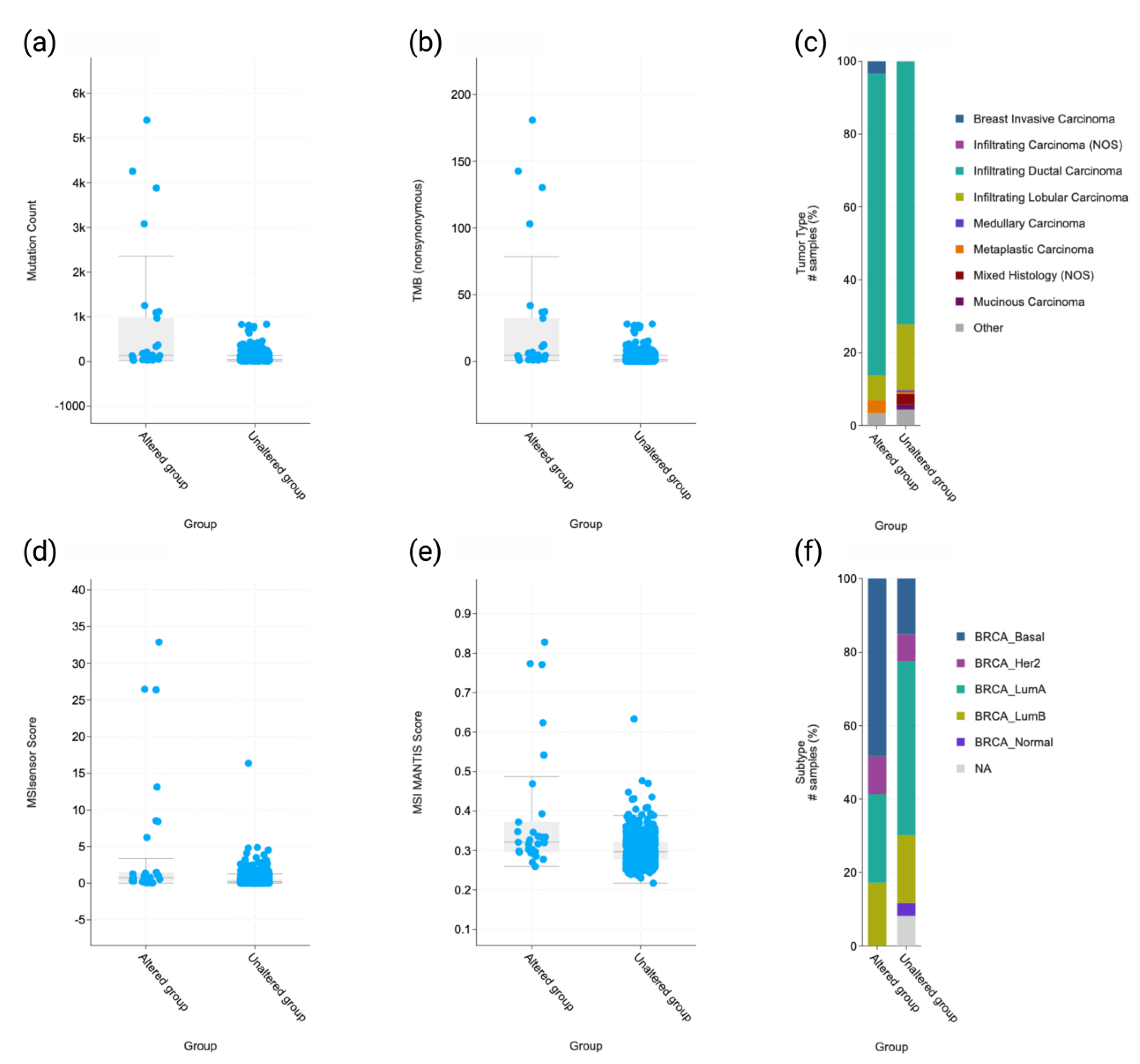
2.1. Mutational Portraits of MMR Genes Signatures in Human Breast Cancer
2.2. MMR-Deficient and MMR-Intact Patients Have Distinct PPI Networks
2.3. Molecular and Clinicopathologic Profile
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Proteinarium
4.3. Separation Test
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heer, E.; Harper, A.; Escandor, N.; Sung, H.; McCormack, V.; Fidler-Benaoudia, M.M. Global burden and trends in premenopausal and postmenopausal breast cancer: A population-based study. Lancet Glob. Health 2020, 8, e1027–e1037. [Google Scholar] [CrossRef] [PubMed]
- Buza, N.; Ziai, J.; Hui, P. Mismatch repair deficiency testing in clinical practice. Expert Rev. Mol. Diagn. 2016, 16, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Ozer, E.; Yuksel, E.; Kizildag, S.; Sercan, O.; Ozen, E.; Canda, T.; Sakizli, M. Microsatellite instability in early-onset breast cancer. Pathol. Res. Pract. 2002, 198, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Eliyatkın, N.; Yalçın, E.; Zengel, B.; Aktaş, S.; Vardar, E. Molecular Classification of Breast Carcinoma: From Traditional, Old-Fashioned Way to A New Age, and A New Way. J. Breast Health 2015, 11, 59–66. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Lipkin, S.M.; Wang, V.; Jacoby, R.; Banerjee-Basu, S.; Baxevanis, A.D.; Lynch, H.T.; Elliott, R.M.; Collins, F.S. MLH3: A DNA mismatch repair gene associated with mammalian microsatellite instability. Nat. Genet. 2000, 24, 27–35. [Google Scholar] [CrossRef]
- Clark, N.; Wu, X.; Her, C. MutS Homologues hMSH4 and hMSH5: Genetic Variations, Functions, and Implications in Human Diseases. Curr. Genom. 2013, 14, 81–90. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e2073. [Google Scholar] [CrossRef]
- Fusco, N.; Lopez, G.; Corti, C.; Pesenti, C.; Colapietro, P.; Ercoli, G.; Gaudioso, G.; Faversani, A.; Gambini, D.; Michelotti, A.; et al. Mismatch Repair Protein Loss as a Prognostic and Predictive Biomarker in Breast Cancers Regardless of Microsatellite Instability. JNCI Cancer Spectr. 2018, 2, pky056. [Google Scholar] [CrossRef]
- Paulson, T.G.; Wright, F.A.; Parker, B.A.; Russack, V.; Wahl, G.M. Microsatellite instability correlates with reduced survival and poor disease prognosis in breast cancer. Cancer Res. 1996, 56, 4021–4026. [Google Scholar] [PubMed]
- Wild, P.J.; Reichle, A.; Andreesen, R.; Rockelein, G.; Dietmaier, W.; Ruschoff, J.; Blaszyk, H.; Hofstadter, F.; Hartmann, A. Microsatellite instability predicts poor short-term survival in patients with advanced breast cancer after high-dose chemotherapy and autologous stem-cell transplantation. Clin. Cancer Res. 2004, 10, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Kamat, N.; Khidhir, M.A.; Jaloudi, M.; Hussain, S.; Alashari, M.M.; Al Qawasmeh, K.H.; Rannug, U. High incidence of microsatellite instability and loss of heterozygosity in three loci in breast cancer patients receiving chemotherapy: A prospective study. BMC Cancer 2012, 12, 373. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.C.; Magalhaes, W.C.S.; Gonzalez-Bosquet, J.; Chanock, S.J. Genome-wide association studies in cancer—Current and future directions. Carcinogenesis 2010, 31, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Armanious, D.; Schuster, J.; Tollefson, G.A.; Agudelo, A.; DeWan, A.T.; Istrail, S.; Padbury, J.; Uzun, A. Proteinarium: Multi-sample protein-protein interaction analysis and visualization tool. Genomics 2020, 112, 4288–4296. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, E.W. A note on two problems in connexion with graphs. Numer. Math. 1959, 1, 269–271. [Google Scholar] [CrossRef]
- Ciccarelli, F.D.; Doerks, T.; von Mering, C.; Creevey, C.J.; Snel, B.; Bork, P. Toward automatic reconstruction of a highly resolved tree of life. Science 2006, 311, 1283–1287. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef]
- Okada, M.; Cheeseman, I.M.; Hori, T.; Okawa, K.; McLeod, I.X.; Yates, J.R., 3rd; Desai, A.; Fukagawa, T. The CENP-H-I complex is required for the efficient incorporation of newly synthesized CENP-A into centromeres. Nat. Cell. Biol. 2006, 8, 446–457. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Lin, C.; Jiang, M.; Tang, X.; Xie, D.; Chen, J.; Ke, R. CENPL, ISG20L2, LSM4, MRPL3 are four novel hub genes and may serve as diagnostic and prognostic markers in breast cancer. Sci. Rep. 2021, 11, 15610. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.L.; Xu, Y.H.; Wang, G. Identification of Potential Crucial Genes and Key Pathways in Breast Cancer Using Bioinformatic Analysis. Front. Genet. 2019, 10, 695. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Jin, L.T.; Wang, T.J.; Feng, Y.J.; Pan, C.P.; Zhao, D.M.; Shao, J. Identification of potential core genes in triple negative breast cancer using bioinformatics analysis. OncoTargets Ther. 2018, 11, 4105–4112. [Google Scholar] [CrossRef]
- Qiu, P.; Guo, Q.; Yao, Q.; Chen, J.; Lin, J. Hsa-mir-3163 and CCNB1 may be potential biomarkers and therapeutic targets for androgen receptor positive triple-negative breast cancer. PLoS ONE 2021, 16, e0254283. [Google Scholar] [CrossRef]
- Eshleman, J.R.; Markowitz, S.D. Mismatch repair defects in human carcinogenesis. Hum. Mol. Genet. 1996, 5 (Suppl. 1), 1489–1494. [Google Scholar] [CrossRef]
- Wheeler, J.M.; Bodmer, W.F.; Mortensen, N.M. DNA mismatch repair genes and colorectal cancer. Gut 2000, 47, 148–153. [Google Scholar] [CrossRef]
- Stelloo, E.; Jansen, A.M.L.; Osse, E.M.; Nout, R.A.; Creutzberg, C.L.; Ruano, D.; Church, D.N.; Morreau, H.; Smit, V.T.H.B.M.; van Wezel, T.; et al. Practical guidance for mismatch repair-deficiency testing in endometrial cancer. Ann. Oncol. 2017, 28, 96–102. [Google Scholar] [CrossRef]
- Prasad, V.; Kaestner, V.; Mailankody, S. Cancer Drugs Approved Based on Biomarkers and Not Tumor Type-FDA Approval of Pembrolizumab for Mismatch Repair-Deficient Solid Cancers. JAMA Oncol. 2018, 4, 157–158. [Google Scholar] [CrossRef]
- Polk, A.; Svane, I.M.; Andersson, M.; Nielsen, D. Checkpoint inhibitors in breast cancer—Current status. Cancer Treat. Rev. 2018, 63, 122–134. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.S.; Leung, S.C.Y.; Gao, D.; Burugu, S.; Anurag, M.; Ellis, M.J.; Nielsen, T.O. Mismatch repair protein loss in breast cancer: Clinicopathological associations in a large British Columbia cohort. Breast Cancer Res. Treat. 2020, 179, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Goellner, E.M. Chromatin remodeling and mismatch repair: Access and excision. DNA Repair 2020, 85, 102733. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-M. Decoding the histone code: Role of H3K36me3 in mismatch repair and implications for cancer susceptibility and therapy. Cancer Res. 2013, 73, 6379–6383. [Google Scholar] [CrossRef]
- Li, F.; Tian, L.; Gu, L.; Li, G.-M. Evidence that nucleosomes inhibit mismatch repair in eukaryotic cells. J. Biol. Chem. 2009, 284, 33056–33061. [Google Scholar] [CrossRef]
- Li, F.; Ortega, J.; Gu, L.; Li, G.-M. Regulation of mismatch repair by histone code and posttranslational modifications in eukaryotic cells. DNA Repair 2016, 38, 68–74. [Google Scholar] [CrossRef]
- Huang, Y.; Gu, L.; Li, G.-M. H3K36me3-mediated mismatch repair preferentially protects actively transcribed genes from mutation. J. Biol. Chem. 2018, 293, 7811–7823. [Google Scholar] [CrossRef]
- Zhao, W.; Neyt, P.; Van Lijsebettens, M.; Shen, W.H.; Berr, A. Interactive and noninteractive roles of histone H2B monoubiquitination and H3K36 methylation in the regulation of active gene transcription and control of plant growth and development. New Phytol. 2019, 221, 1101–1116. [Google Scholar] [CrossRef]
- Chen, H.; Feng, H.; Zhang, X.; Zhang, C.; Wang, T.; Dong, J. An Arabidopsis E3 ligase HUB2 increases histone H2B monoubiquitination and enhances drought tolerance in transgenic cotton. Plant Biotechnol. J. 2019, 17, 556–568. [Google Scholar] [CrossRef]
- Haque, W.; Kee Yuan, D.M.; Verma, V.; Butler, E.B.; Teh, B.S.; Wiederhold, L.; Hatch, S. Radiation therapy utilization and outcomes for older women with breast cancer: Impact of molecular subtype and tumor grade. Breast 2017, 35, 34–41. [Google Scholar] [CrossRef]
- Li, W.; Wu, H.; Sui, S.; Wang, Q.; Xu, S.; Pang, D. Targeting Histone Modifications in Breast Cancer: A Precise Weapon on the Way. Front. Cell Dev. Biol. 2021, 9, 736935. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Zou, J.X.; Wang, H.; Duan, Z.J.; Wang, H.B.; Chen, P.; Liu, P.Q.; Xu, J.Z.; Chen, H.W. Histone methyltransferase NSD2 mediates the survival and invasion of triple-negative breast cancer cells via stimulating ADAM9-EGFR-AKT signaling. Acta Pharmacol. Sin. 2019, 40, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—Expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2022, 19, 91–113. [Google Scholar] [CrossRef]
- Cosgrove, N.; Varešlija, D.; Keelan, S.; Elangovan, A.; Atkinson, J.M.; Cocchiglia, S.; Bane, F.T.; Singh, V.; Furney, S.; Hu, C.; et al. Mapping molecular subtype specific alterations in breast cancer brain metastases identifies clinically relevant vulnerabilities. Nat. Commun. 2022, 13, 514. [Google Scholar] [CrossRef]
- Willis, J.A.; Reyes-Uribe, L.; Chang, K.; Lipkin, S.M.; Vilar, E. Immune Activation in Mismatch Repair-Deficient Carcinogenesis: More Than Just Mutational Rate. Clin. Cancer Res. 2020, 26, 11–17. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018, 6, 271–281.e277. [Google Scholar] [CrossRef]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e673. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell. Rep. 2018, 23, 227–238.e223. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Bailey, M.H.; Porta-Pardo, E.; Thorsson, V.; Colaprico, A.; Bertrand, D.; Gibbs, D.L.; Weerasinghe, A.; Huang, K.L.; Tokheim, C.; et al. Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 2018, 173, 305–320.e310. [Google Scholar] [CrossRef]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 2017, 1–15. [Google Scholar] [CrossRef]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabási, A.L. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hacking, S.; Chou, C.; Baykara, Y.; Wang, Y.; Uzun, A.; Gamsiz Uzun, E.D. MMR Deficiency Defines Distinct Molecular Subtype of Breast Cancer with Histone Proteomic Networks. Int. J. Mol. Sci. 2023, 24, 5327. https://doi.org/10.3390/ijms24065327
Hacking S, Chou C, Baykara Y, Wang Y, Uzun A, Gamsiz Uzun ED. MMR Deficiency Defines Distinct Molecular Subtype of Breast Cancer with Histone Proteomic Networks. International Journal of Molecular Sciences. 2023; 24(6):5327. https://doi.org/10.3390/ijms24065327
Chicago/Turabian StyleHacking, Sean, Charissa Chou, Yigit Baykara, Yihong Wang, Alper Uzun, and Ece D. Gamsiz Uzun. 2023. "MMR Deficiency Defines Distinct Molecular Subtype of Breast Cancer with Histone Proteomic Networks" International Journal of Molecular Sciences 24, no. 6: 5327. https://doi.org/10.3390/ijms24065327
APA StyleHacking, S., Chou, C., Baykara, Y., Wang, Y., Uzun, A., & Gamsiz Uzun, E. D. (2023). MMR Deficiency Defines Distinct Molecular Subtype of Breast Cancer with Histone Proteomic Networks. International Journal of Molecular Sciences, 24(6), 5327. https://doi.org/10.3390/ijms24065327