
VRK1 Kinase Activity Modulating Histone H4K16 Acetylation Inhibited by SIRT2 and VRK-IN-1

Abstract
1. Introduction
2. Results
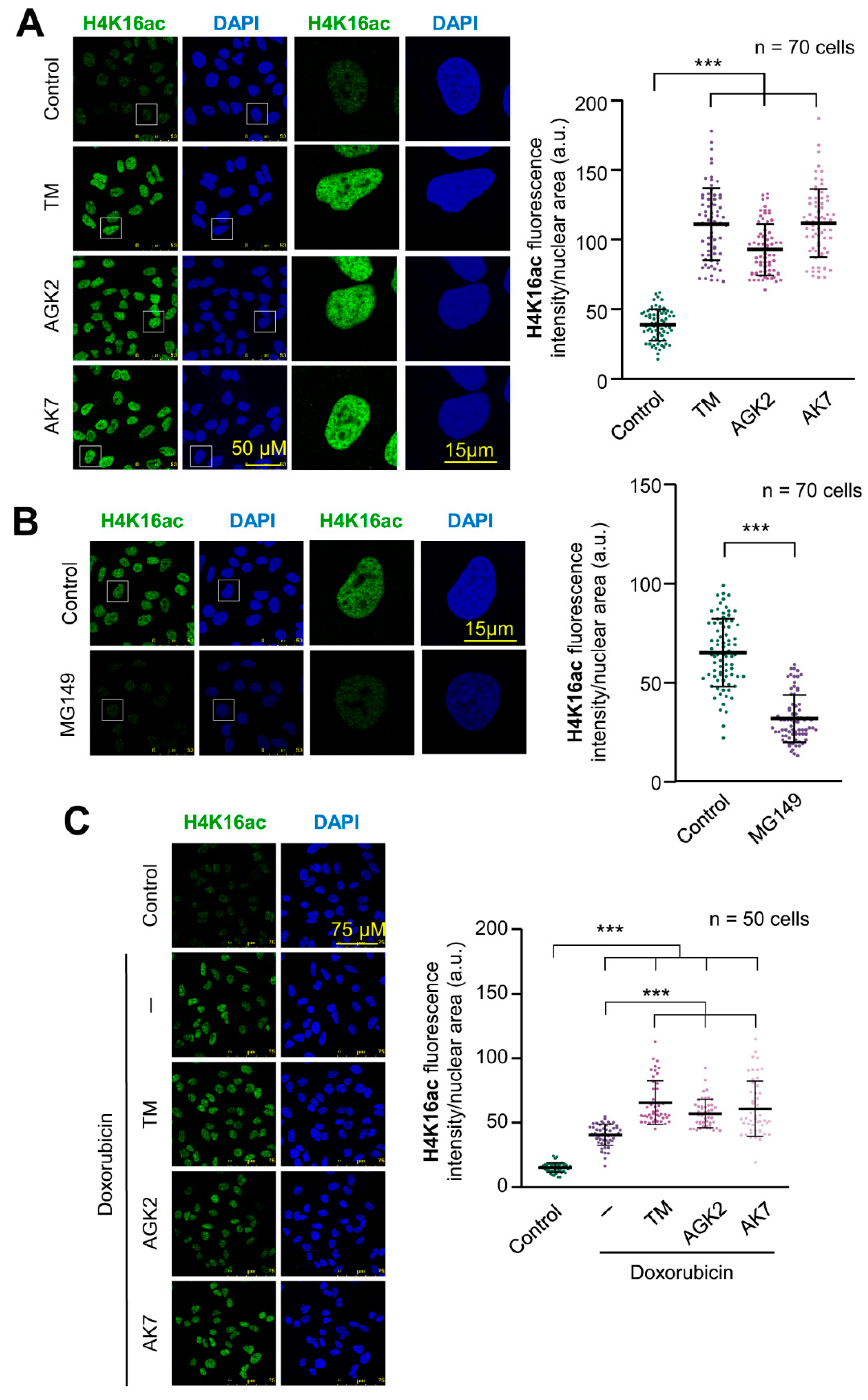
2.1. SIRT2 Inhibition and Its Combination with Doxorubicin Facilitate Histone H4K16 Acetylation
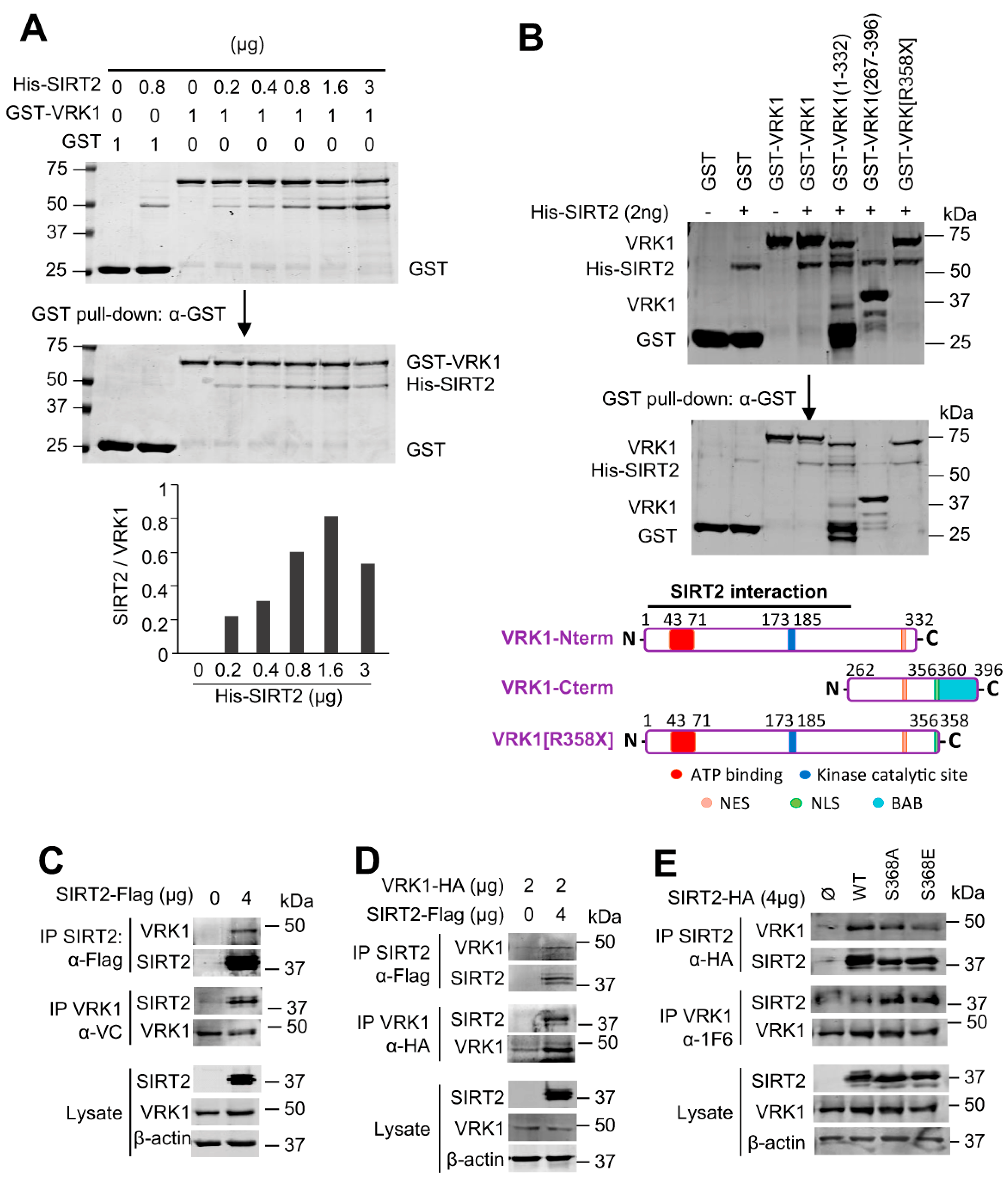
2.2. VRK1 Directly Interacts with SIRT2 In Vitro and In Vivo
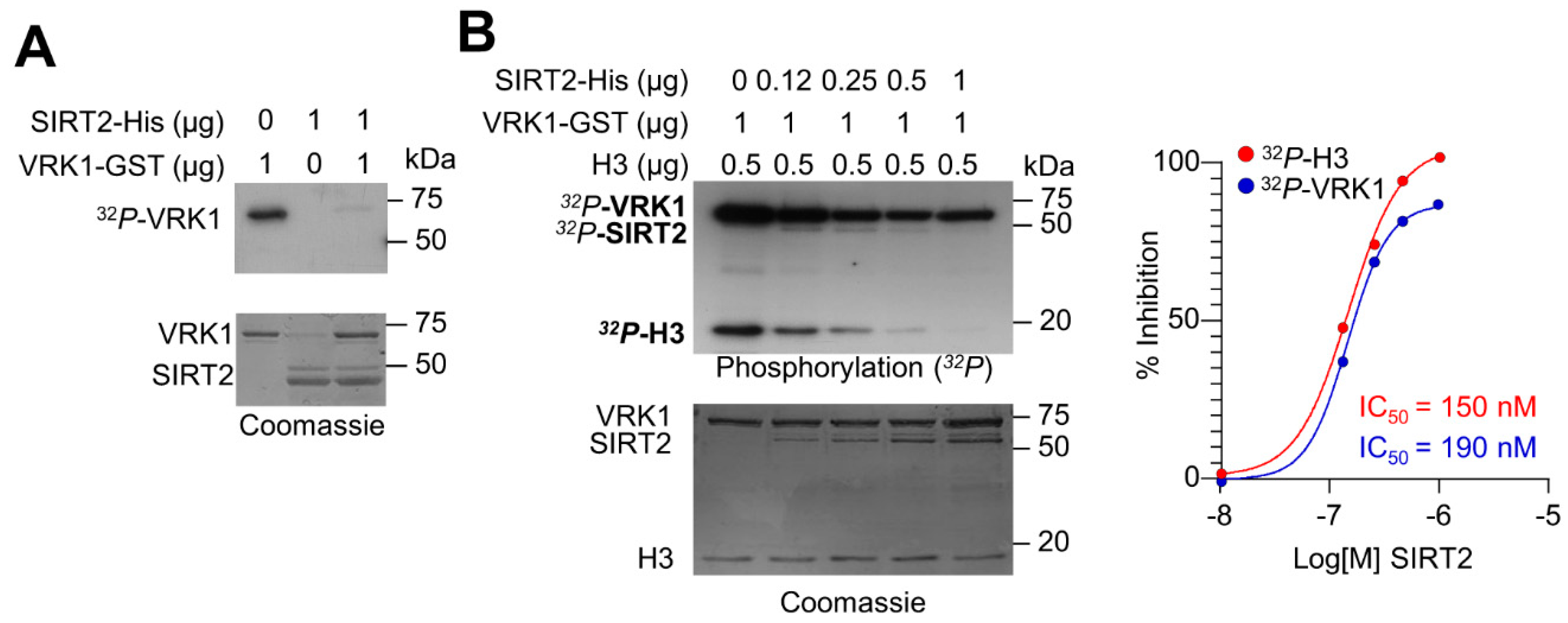
2.3. SIRT2 Inhibits the Kinase Activity of VRK1
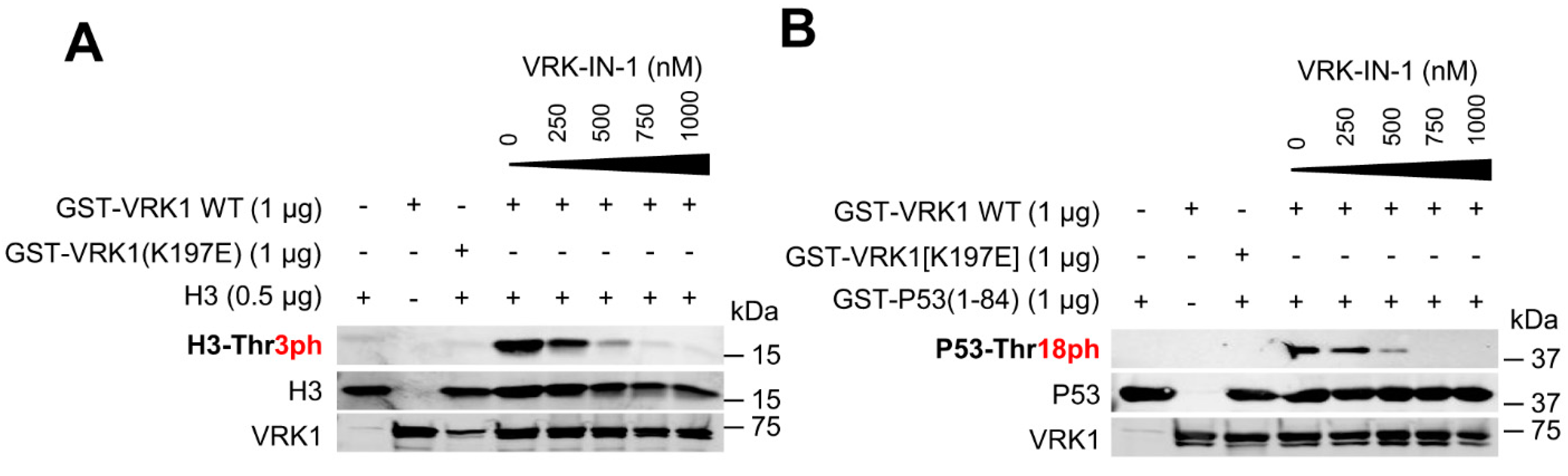
2.4. The VRK-IN-1 Inhibitor Impairs the Phosphorylation of Histone H3 and p53
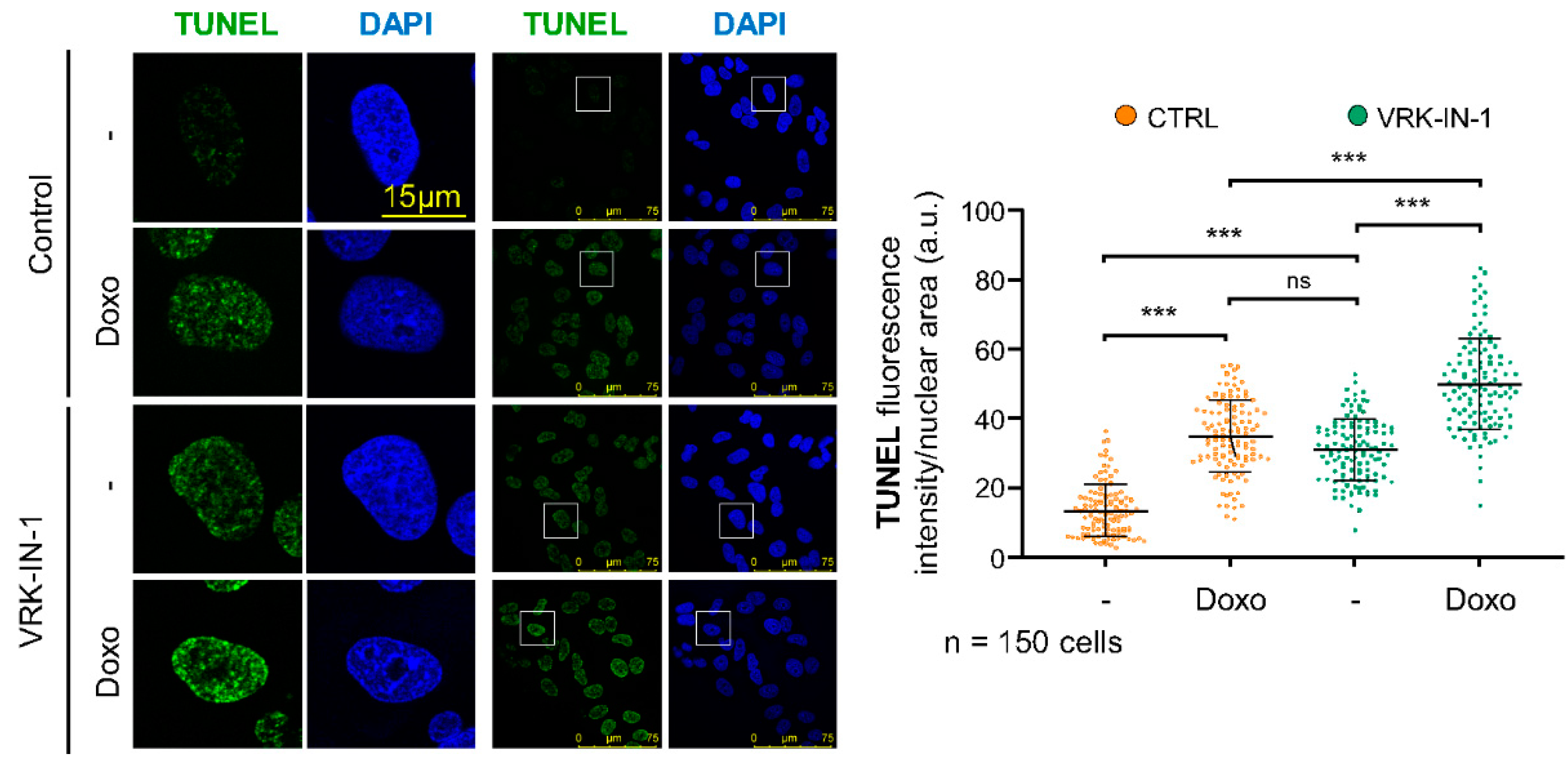
2.5. The VRK-IN-1 Inhibitor Facilitates the Accumulation of Endogenous DNA Strand Breaks
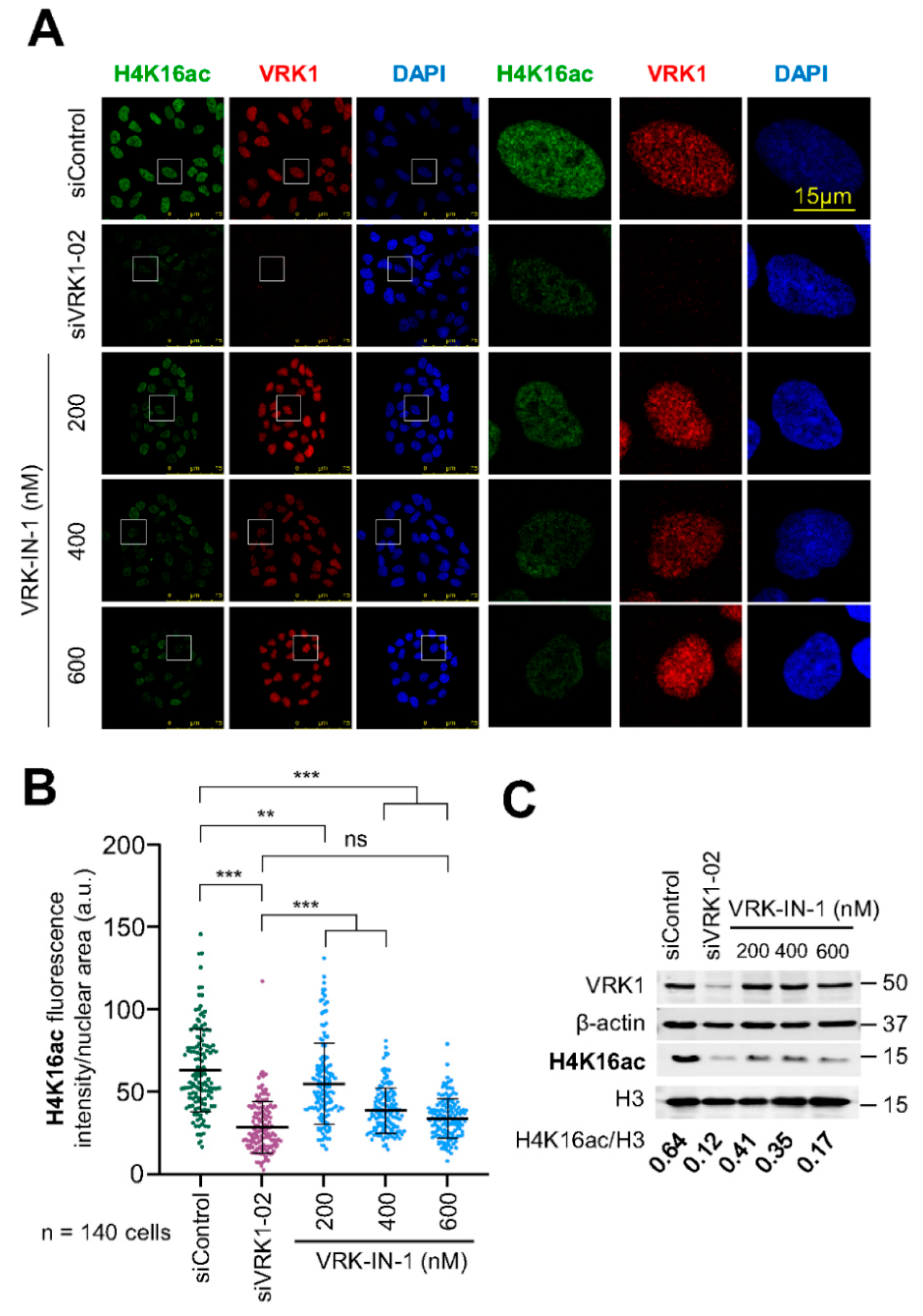
2.6. The VRK-IN-1 Inhibitor Reduces H4K16 Acetylation Levels
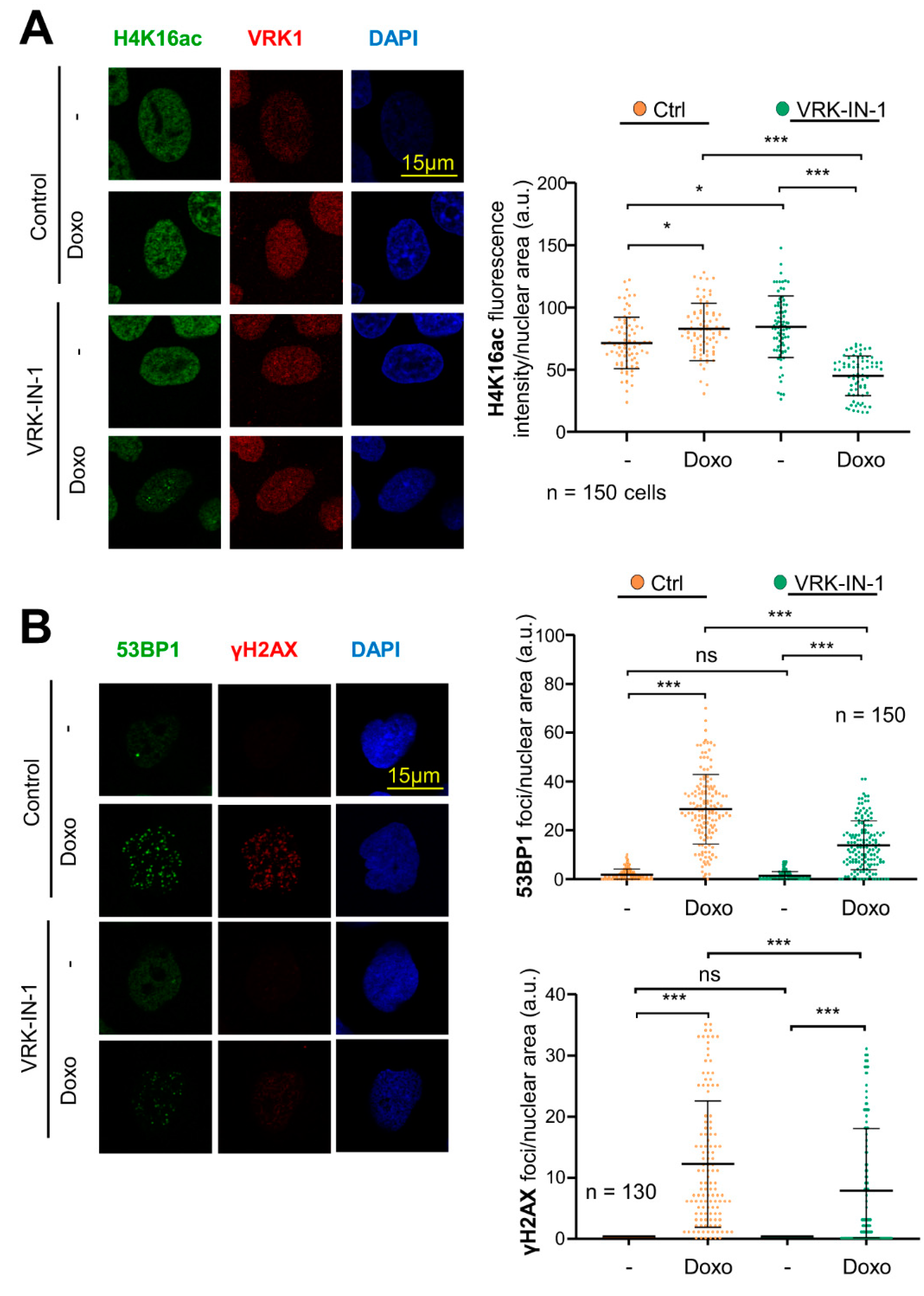
2.7. The VRK-IN-1 Inhibitor Impairs the DNA Damage Response Induced by Doxorubicin
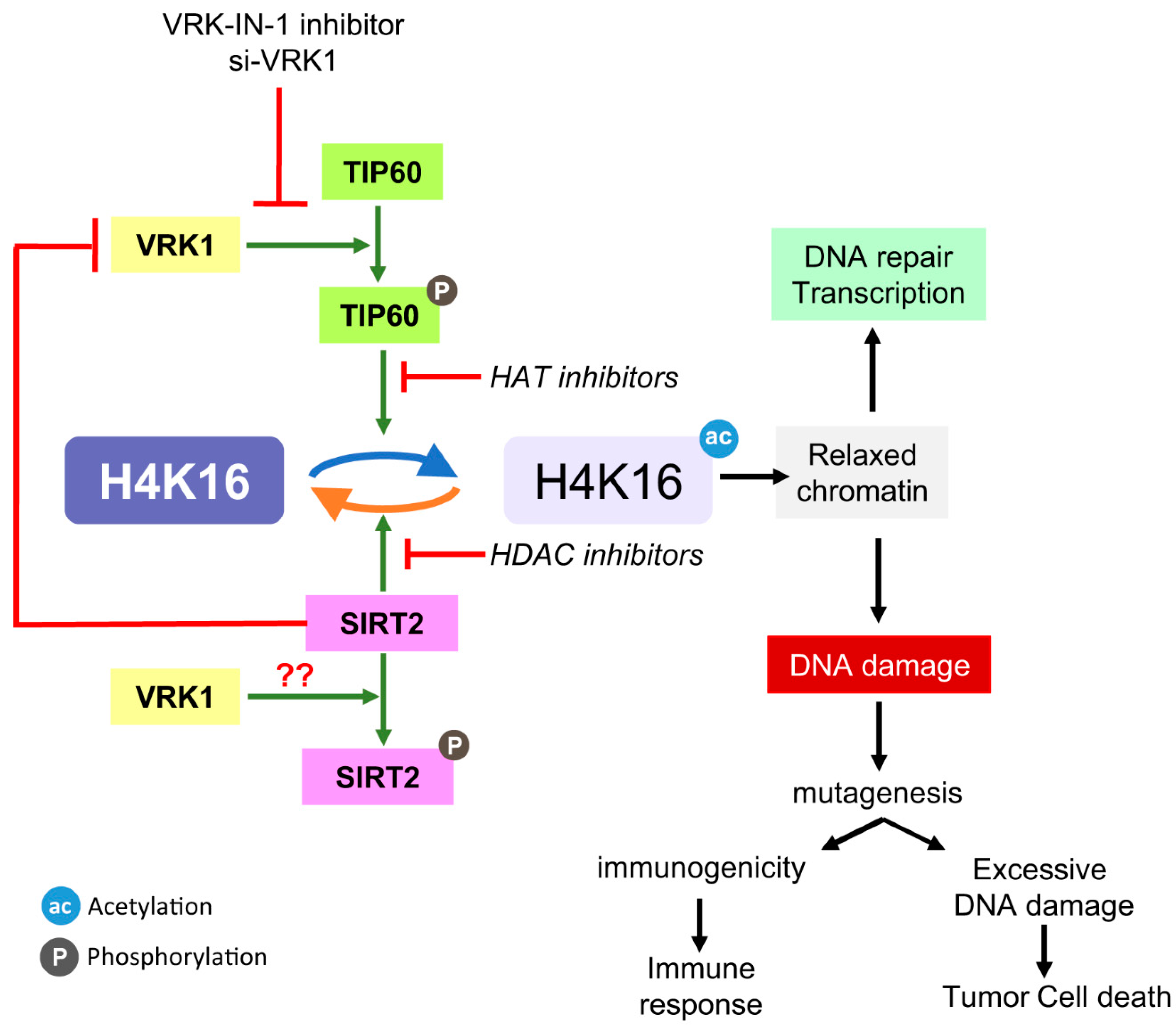
3. Discussion
4. Materials and Methods
4.1. Reagents and Inhibitors
4.2. Plasmids
4.3. Cell Lines, Culture and Transfections
4.4. VRK1 Depletion by siRNA
4.5. Cell Lysates and Acidic Histone Extraction
4.6. Antibodies
4.7. Immunoprecipitations
4.8. Immunoblots
4.9. Immunofluorescence and Confocal Microscopy
4.10. TUNEL Assay
4.11. In vitro Protein Interaction (Pull-Down Assays)
4.12. In vitro Kinase Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shogren-Knaak, M.; Ishii, H.; Sun, J.M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef]
- Yamagata, K.; Shino, M.; Aikawa, Y.; Fujita, S.; Kitabayashi, I. Tip60 activates Hoxa9 and Meis1 expression through acetylation of H2A.Z, promoting MLL-AF10 and MLL-ENL acute myeloid leukemia. Leukemia 2021, 35, 2840–2853. [Google Scholar] [CrossRef]
- Chakraborty, S.; Singh, M.; Pandita, R.K.; Singh, V.; Lo, C.S.C.; Leonard, F.; Horikoshi, N.; Moros, E.G.; Guha, D.; Hunt, C.R.; et al. Heat-induced SIRT1-mediated H4K16ac deacetylation impairs resection and SMARCAD1 recruitment to double strand breaks. iScience 2022, 25, 104142. [Google Scholar] [CrossRef]
- Yasuda, T.; Takizawa, K.; Ui, A.; Hama, M.; Kagawa, W.; Sugasawa, K.; Tajima, K. Human SIRT2 and SIRT3 deacetylases function in DNA homologous recombinational repair. Genes Cells 2021, 26, 328–335. [Google Scholar] [CrossRef]
- Mir, U.S.; Bhat, A.; Mushtaq, A.; Pandita, S.; Altaf, M.; Pandita, T.K. Role of histone acetyltransferases MOF and Tip60 in genome stability. DNA Repair 2021, 107, 103205. [Google Scholar] [CrossRef]
- Voss, A.K.; Thomas, T. MYST family histone acetyltransferases take center stage in stem cells and development. Bioessays 2009, 31, 1050–1061. [Google Scholar] [CrossRef]
- Urdinguio, R.G.; Lopez, V.; Bayon, G.F.; Diaz de la Guardia, R.; Sierra, M.I.; Garcia-Torano, E.; Perez, R.F.; Garcia, M.G.; Carella, A.; Pruneda, P.C.; et al. Chromatin regulation by Histone H4 acetylation at Lysine 16 during cell death and differentiation in the myeloid compartment. Nucleic. Acids Res. 2019, 47, 5016–5037. [Google Scholar] [CrossRef]
- Farria, A.; Li, W.; Dent, S.Y. KATs in cancer: Functions and therapies. Oncogene 2015, 34, 4901–4913. [Google Scholar] [CrossRef]
- Simon, R.P.; Robaa, D.; Alhalabi, Z.; Sippl, W.; Jung, M. KATching-Up on Small Molecule Modulators of Lysine Acetyltransferases. J. Med. Chem. 2016, 59, 1249–1270. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Lazo, P.A. Targeting Histone Epigenetic Modifications and DNA Damage Responses in Synthetic Lethality Strategies in Cancer? Cancers 2022, 14, 4050. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Wang, F.; Cai, Y.; Jin, J. The Functional Analysis of Histone Acetyltransferase MOF in Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef]
- Hoffmann, G.; Breitenbucher, F.; Schuler, M.; Ehrenhofer-Murray, A.E. A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J. Biol. Chem. 2014, 289, 5208–5216. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Sternglanz, R.; Reinberg, D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 2007, 26, 5505–5520. [Google Scholar] [CrossRef]
- Campillo-Marcos, I.; García-González, R.; Navarro-Carrasco, E.; Lazo, P.A. The human VRK1 chromatin kinase in cancer biology. Cancer Lett. 2021, 503, 117–128. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Neth, B.J.; Balakrishnan, S.N.; Carabenciov, I.D.; Uhm, J.H.; Daniels, D.J.; Kizilbash, S.H.; Ruff, M.W. Panobinostat in adults with H3 K27M-mutant diffuse midline glioma: A single-center experience. J. Neurooncol. 2022, 157, 91–100. [Google Scholar] [CrossRef]
- Degorre, C.; Tofilon, P.; Camphausen, K.; Mathen, P. Bench to bedside radiosensitizer development strategy for newly diagnosed glioblastoma. Radiat. Oncol. 2021, 16, 191. [Google Scholar] [CrossRef]
- Serrano, L.; Martinez-Redondo, P.; Marazuela-Duque, A.; Vazquez, B.N.; Dooley, S.J.; Voigt, P.; Beck, D.B.; Kane-Goldsmith, N.; Tong, Q.; Rabanal, R.M.; et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013, 27, 639–653. [Google Scholar] [CrossRef]
- Kang, T.H.; Park, D.Y.; Kim, W.; Kim, K.T. VRK1 phosphorylates CREB and mediates CCND1 expression. J. Cell Sci. 2008, 121, 3035–3041. [Google Scholar] [CrossRef]
- Valbuena, A.; Lopez-Sanchez, I.; Lazo, P.A. Human VRK1 is an early response gene and its loss causes a block in cell cycle progression. PLoS ONE 2008, 3, e1642. [Google Scholar] [CrossRef]
- Salzano, M.; Sanz-Garcia, M.; Monsalve, D.M.; Moura, D.S.; Lazo, P.A. VRK1 chromatin kinase phosphorylates H2AX and is required for foci formation induced by DNA damage. Epigenetics 2015, 10, 373–383. [Google Scholar] [CrossRef]
- Sanz-Garcia, M.; Monsalve, D.M.; Sevilla, A.; Lazo, P.A. Vaccinia-related Kinase 1 (VRK1) is an upstream nucleosomal kinase required for the assembly of 53BP1 foci in response to ionizing radiation-induced DNA damage. J. Biol. Chem. 2012, 287, 23757–23768. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalez, R.; Morejon-Garcia, P.; Campillo-Marcos, I.; Salzano, M.; Lazo, P.A. VRK1 Phosphorylates Tip60/KAT5 and Is Required for H4K16 Acetylation in Response to DNA Damage. Cancers 2020, 12, 2986. [Google Scholar] [CrossRef] [PubMed]
- García-González, R.; Monte-Serrano, E.; Morejón-García, P.; Navarro-Carrasco, E.; Lazo, P.A. The VRK1 chromatin kinase regulates the acetyltransferase activity of Tip60/KAT5 by sequential phosphorylations in response to DNA damage. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194887. [Google Scholar] [CrossRef]
- Sevilla, A.; Santos, C.R.; Barcia, R.; Vega, F.M.; Lazo, P.A. c-Jun phosphorylation by the human vaccinia-related kinase 1 (VRK1) and its cooperation with the N-terminal kinase of c-Jun (JNK). Oncogene 2004, 23, 8950–8958. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, A.; Santos, C.R.; Vega, F.M.; Lazo, P.A. Human vaccinia-related kinase 1 (VRK1) activates the ATF2 transcriptional activity by novel phosphorylation on Thr-73 and Ser-62 and cooperates with JNK. J. Biol. Chem. 2004, 279, 27458–27465. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Marcos, I.; Lazo, P.A. Implication of the VRK1 chromatin kinase in the signaling responses to DNA damage: A therapeutic target? Cell. Mol. Life Sci. 2018, 75, 2375–2388. [Google Scholar] [CrossRef]
- Salzano, M.; Vazquez-Cedeira, M.; Sanz-Garcia, M.; Valbuena, A.; Blanco, S.; Fernandez, I.F.; Lazo, P.A. Vaccinia-related kinase 1 (VRK1) confers resistance to DNA-damaging agents in human breast cancer by affecting DNA damage response. Oncotarget 2014, 5, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, D.M.; Campillo-Marcos, I.; Salzano, M.; Sanz-Garcia, M.; Cantarero, L.; Lazo, P.A. VRK1 phosphorylates and protects NBS1 from ubiquitination and proteasomal degradation in response to DNA damage. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Valbuena, A.; Lopez-Sanchez, I.; Vega, F.M.; Sevilla, A.; Sanz-Garcia, M.; Blanco, S.; Lazo, P.A. Identification of a dominant epitope in human vaccinia-related kinase 1 (VRK1) and detection of different intracellular subpopulations. Arch. Biochem. Biophys. 2007, 465, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Nahhas, F.; Dryden, S.C.; Abrams, J.; Tainsky, M.A. Mutations in SIRT2 deacetylase which regulate enzymatic activity but not its interaction with HDAC6 and tubulin. Mol. Cell. Biochem. 2007, 303, 221–230. [Google Scholar] [CrossRef]
- North, B.J.; Verdin, E. Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J. Biol. Chem. 2007, 282, 19546–19555. [Google Scholar] [CrossRef]
- Kang, T.H.; Park, D.Y.; Choi, Y.H.; Kim, K.J.; Yoon, H.S.; Kim, K.T. Mitotic histone H3 phosphorylation by vaccinia-related kinase 1 in mammalian cells. Mol. Cell. Biol. 2007, 27, 8533–8546. [Google Scholar] [CrossRef] [PubMed]
- Budziszewski, G.R.; Zhao, Y.; Spangler, C.J.; Kedziora, K.M.; Williams, M.R.; Azzam, D.N.; Skrajna, A.; Koyama, Y.; Cesmat, A.P.; Simmons, H.C.; et al. Multivalent DNA and nucleosome acidic patch interactions specify VRK1 mitotic localization and activity. Nucleic. Acids Res. 2022, 50, 4355–4371. [Google Scholar] [CrossRef]
- Moura, D.S.; Campillo-Marcos, I.; Vazquez-Cedeira, M.; Lazo, P.A. VRK1 and AURKB form a complex that cross inhibit their kinase activity and the phosphorylation of histone H3 in the progression of mitosis. Cell. Mol. Life Sci. 2018, 76, 2591–2611. [Google Scholar] [CrossRef]
- Vazquez-Cedeira, M.; Barcia-Sanjurjo, I.; Sanz-Garcia, M.; Barcia, R.; Lazo, P.A. Differential Inhibitor Sensitivity between Human Kinases VRK1 and VRK2. PLoS ONE 2011, 6, e23235. [Google Scholar] [CrossRef]
- Fedorov, O.; Marsden, B.; Pogacic, V.; Rellos, P.; Muller, S.; Bullock, A.N.; Schwaller, J.; Sundstrom, M.; Knapp, S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 20523–20528. [Google Scholar] [CrossRef]
- Counago, R.M.; Allerston, C.K.; Savitsky, P.; Azevedo, H.; Godoi, P.H.; Wells, C.I.; Mascarello, A.; de Souza Gama, F.H.; Massirer, K.B.; Zuercher, W.J.; et al. Structural characterization of human Vaccinia-Related Kinases (VRK) bound to small-molecule inhibitors identifies different P-loop conformations. Sci. Rep. 2017, 7, 7501. [Google Scholar] [CrossRef] [PubMed]
- Serafim, R.A.M.; de Souza Gama, F.H.; Dutra, L.A.; Dos Reis, C.V.; Vasconcelos, S.N.S.; da Silva Santiago, A.; Takarada, J.E.; Di Pillo, F.; Azevedo, H.; Mascarello, A.; et al. Development of Pyridine-based Inhibitors for the Human Vaccinia-related Kinases 1 and 2. ACS Med. Chem. Lett. 2019, 10, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Borges, S.; Lazo, P.A. The human vaccinia-related kinase 1 (VRK1) phosphorylates threonine-18 within the mdm-2 binding site of the p53 tumour suppressor protein. Oncogene 2000, 19, 3656–3664. [Google Scholar] [CrossRef]
- Barcia, R.; Lopez-Borges, S.; Vega, F.M.; Lazo, P.A. Kinetic properties of p53 phosphorylation by the human vaccinia-related kinase 1. Arch. Biochem. Biophys. 2002, 399, 1–5. [Google Scholar] [CrossRef]
- Faucher, F.; Doublie, S.; Jia, Z. 8-oxoguanine DNA glycosylases: One lesion, three subfamilies. Int. J. Mol. Sci. 2012, 13, 6711–6729. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Sevilla, A.; Lazo, P.A. p53 Stabilization and accumulation induced by human vaccinia-related kinase 1. Mol. Cell. Biol. 2004, 24, 10366–10380. [Google Scholar] [CrossRef]
- Aihara, H.; Nakagawa, T.; Yasui, K.; Ohta, T.; Hirose, S.; Dhomae, N.; Takio, K.; Kaneko, M.; Takeshima, Y.; Muramatsu, M.; et al. Nucleosomal histone kinase-1 phosphorylates H2A Thr 119 during mitosis in the early Drosophila embryo. Genes Dev. 2004, 18, 877–888. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef]
- Luo, H.; Shan, J.; Zhang, H.; Song, G.; Li, Q.; Xu, C.X. Targeting the epigenetic processes to enhance antitumor immunity in small cell lung cancer. Semin. Cancer Biol. 2022, 86, 960–970. [Google Scholar] [CrossRef]
- Lopez-Sanchez, I.; Sanz-Garcia, M.; Lazo, P.A. Plk3 interacts with and specifically phosphorylates VRK1 in Ser342, a downstream target in a pathway that induces Golgi fragmentation. Mol. Cell. Biol. 2009, 29, 1189–1201. [Google Scholar] [CrossRef]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Carrasco, E.; Lazo, P.A. VRK1 Depletion Facilitates the Synthetic Lethality of Temozolomide and Olaparib in Glioblastoma Cells. Front. Cell Dev. Biol. 2021, 9, 683038. [Google Scholar] [CrossRef]
- Shechter, D.; Dormann, H.L.; Allis, C.D.; Hake, S.B. Extraction, purification and analysis of histones. Nat. Protoc. 2007, 2, 1445–1457. [Google Scholar] [CrossRef]
- Dubey, S.; Jaiswal, B.; Gupta, A. TIP60 acts as a regulator of genes involved in filopodia formation and cell migration during wound healing. J. Biol. Chem. 2022, 298, 102015. [Google Scholar] [CrossRef]
- Campillo-Marcos, I.; Lazo, P.A. Olaparib and ionizing radiation trigger a cooperative DNA-damage repair response that is impaired by depletion of the VRK1 chromatin kinase. J. Exp. Clin. Cancer Res. 2019, 38, 203. [Google Scholar] [CrossRef] [PubMed]
- Cantarero, L.; Sanz-Garcia, M.; Vinograd-Byk, H.; Renbaum, P.; Levy-Lahad, E.; Lazo, P.A. VRK1 regulates Cajal body dynamics and protects coilin from proteasomal degradation in cell cycle. Sci. Rep. 2015, 5, 10543. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, M.; Lopez-Sanchez, I.; Lazo, P.A. Proteomics identification of nuclear Ran GTPase as an inhibitor of human VRK1 and VRK2 (vaccinia-related kinase) activities. Mol. Cell. Proteomics 2008, 7, 2199–2214. [Google Scholar] [CrossRef]
- Martin-Doncel, E.; Rojas, A.M.; Cantarero, L.; Lazo, P.A. VRK1 functional insufficiency due to alterations in protein stability or kinase activity of human VRK1 pathogenic variants implicated in neuromotor syndromes. Sci. Rep. 2019, 9, 13381. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Target | Concentration and Time | Reference | Supplier |
|---|---|---|---|---|
| AGK2 | SIRT2 | 5 µM/24 h | S7577 | SelleckChem |
| AK7 | SIRT2 | 8 µM/24 h | S5914 | SelleckChem |
| Thiomyristoyl | SIRT2 | 5 µM/24 h | S8245 | SelleckChem |
| MG149 | KAT5 (TIP60) | 1 µM/24 h | 1785 | Axon MedChem |
| VRK-IN-1 | VRK1 | 0–600 nM/24 h | HY-126542 | MedChemExpress |
| Doxorubicin | Intercalates DNA Top2A inhibitor | 3 µM/2 h | 25316-40-9 | Sigma-Aldrich |
| Antibody | Type | Dilution (WB/IF) | Clone, Reference | Supplier |
|---|---|---|---|---|
| 53BP1 | Rabbit polyclonal | 1:1000/1:400 | NB100-304 | Novus Biologicals |
| β-actin | Murine mAb | 1:2000/- | AC15, A5441 | Sigma-Aldrich |
| Flag Tag | Murine mAb | 1:1000/- | M5, F4042 | Sigma-Aldrich |
| Flag Tag | Rabbit polyclonal | 1:1000/- | F7425/ab1162 | Sigma-Aldrich/ Abcam |
| GST Tag | Murine mAb | 1:1000/- | B-14, Sc-138 | Santa Cruz Biotech. |
| HA.11 Tag | Murine mAb | 1:1000/1:1000 | 901514,16B12 | BioLegend |
| HA Tag | Rabbit polyclonal | 1:1000/1:1000 | H6908 | Sigma-Aldrich |
| His Tag | Murine mAb | 1:1000/- | HIS-1, H1029 | Sigma-Aldrich |
| H3-T3ph | Rabbit polyclonal | 1:1000/- | 07-424 | Millipore |
| H3 | Rabbit polyclonal | 1:1000/- | 9715 | Cell signaling |
| γH2AX | Murine mAb | -/1:500 | JBW301, 05-636 | Millipore |
| H2AX | Rabbit polyclonal | 1:1000/- | 2595S | Cell Signaling |
| H4K16ac | Rabbit monoclonal | 1:500/1:1.000 | Ab109463 | Abcam |
| P53-T18ph | Rabbit polyclonal | 1:1000/- | 2529 | Cell signaling |
| P53 | Murine mAb | 1:1000/- | DO-1, Sc-126 | Santa Cruz Biotech |
| VRK1 | Murine mAb | -/1:1000 | 1B5 | [54] |
| VRK1 | Murine mAb | 1:1000/- | 1F6 | [54] |
| VRK1 | Rabbit polyclonal | 1:1000/- | VC | [54] |
| Antibody | Application/ Dilution | Reference | Supplier |
|---|---|---|---|
| CyTM5-Goat Anti-Mouse | IF 1:1000 | 115-175-146 | Jackson ImmunoResearch |
| CyTM3-Goat Anti-Mouse | IF 1:1000 | 15-165-146 | Jackson ImmunoResearch |
| CyTM2-Goat Anti-Rabbit | IF 1:1000 | 111-225-144 | Jackson ImmunoResearch |
| Sheep ECL Anti-Mouse IgG, Peroxidase Conjugated | WB 1:10,000 | NA931 | Cytiva-Amersham |
| Goat ECL Anti-Rabbit IgG, Peroxidase Conjugated | WB 1:10,000 | A0545 | Sigma-Aldrich |
| Goat anti-Mouse IgG DyLight 680 | WB 1:10,000 | 35518 | Thermo-Fisher |
| Goat anti-Rabbit IgG DyLight 800 | WB 1:10,000 | 35571 | Thermo-Fisher |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monte-Serrano, E.; Lazo, P.A. VRK1 Kinase Activity Modulating Histone H4K16 Acetylation Inhibited by SIRT2 and VRK-IN-1. Int. J. Mol. Sci. 2023, 24, 4912. https://doi.org/10.3390/ijms24054912
Monte-Serrano E, Lazo PA. VRK1 Kinase Activity Modulating Histone H4K16 Acetylation Inhibited by SIRT2 and VRK-IN-1. International Journal of Molecular Sciences. 2023; 24(5):4912. https://doi.org/10.3390/ijms24054912
Chicago/Turabian StyleMonte-Serrano, Eva, and Pedro A. Lazo. 2023. "VRK1 Kinase Activity Modulating Histone H4K16 Acetylation Inhibited by SIRT2 and VRK-IN-1" International Journal of Molecular Sciences 24, no. 5: 4912. https://doi.org/10.3390/ijms24054912
APA StyleMonte-Serrano, E., & Lazo, P. A. (2023). VRK1 Kinase Activity Modulating Histone H4K16 Acetylation Inhibited by SIRT2 and VRK-IN-1. International Journal of Molecular Sciences, 24(5), 4912. https://doi.org/10.3390/ijms24054912