
SARS-CoV-2 S Mutations: A Lesson from the Viral World to Understand How Human Furin Works

 , ,
, ,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
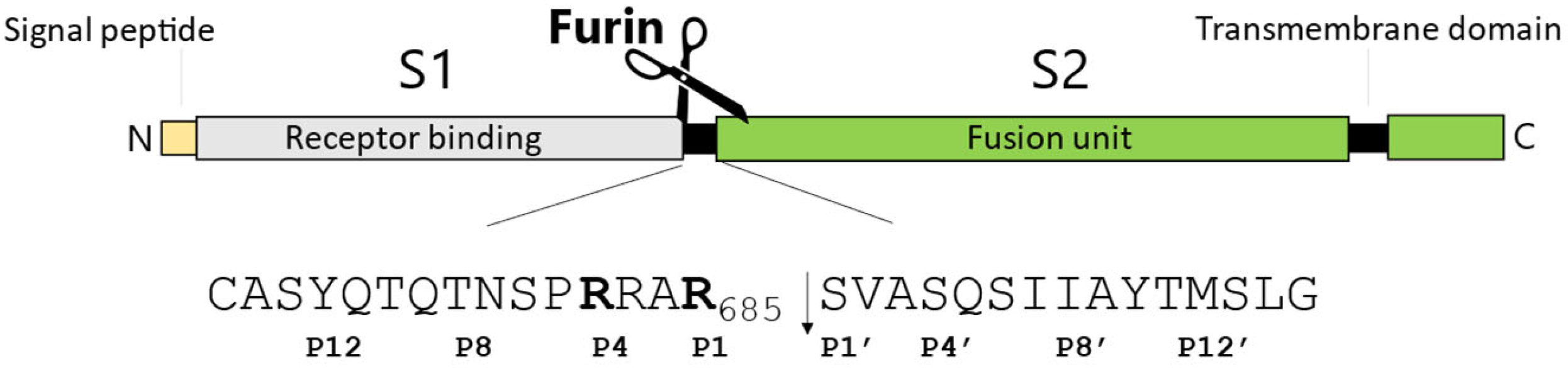
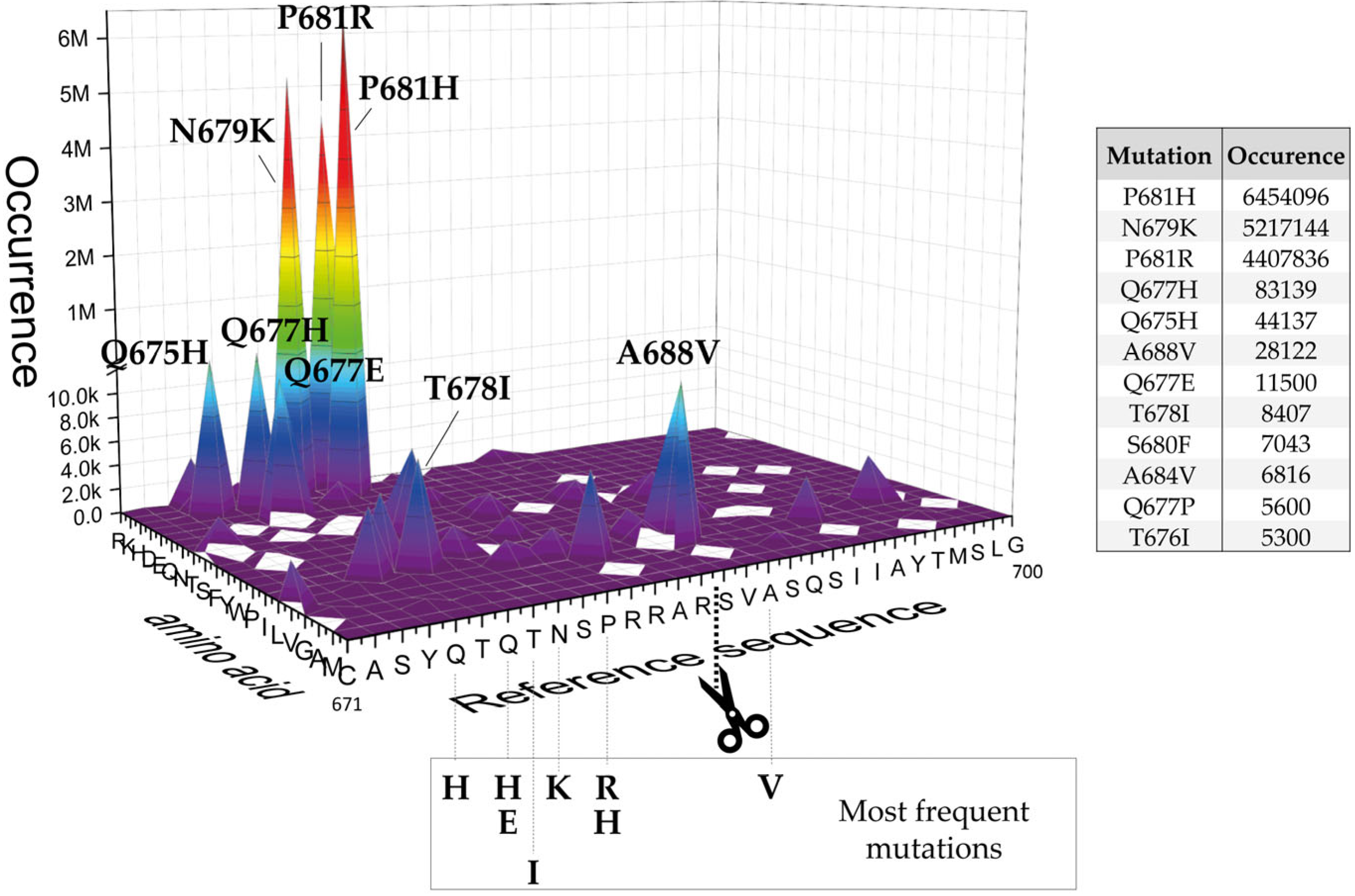
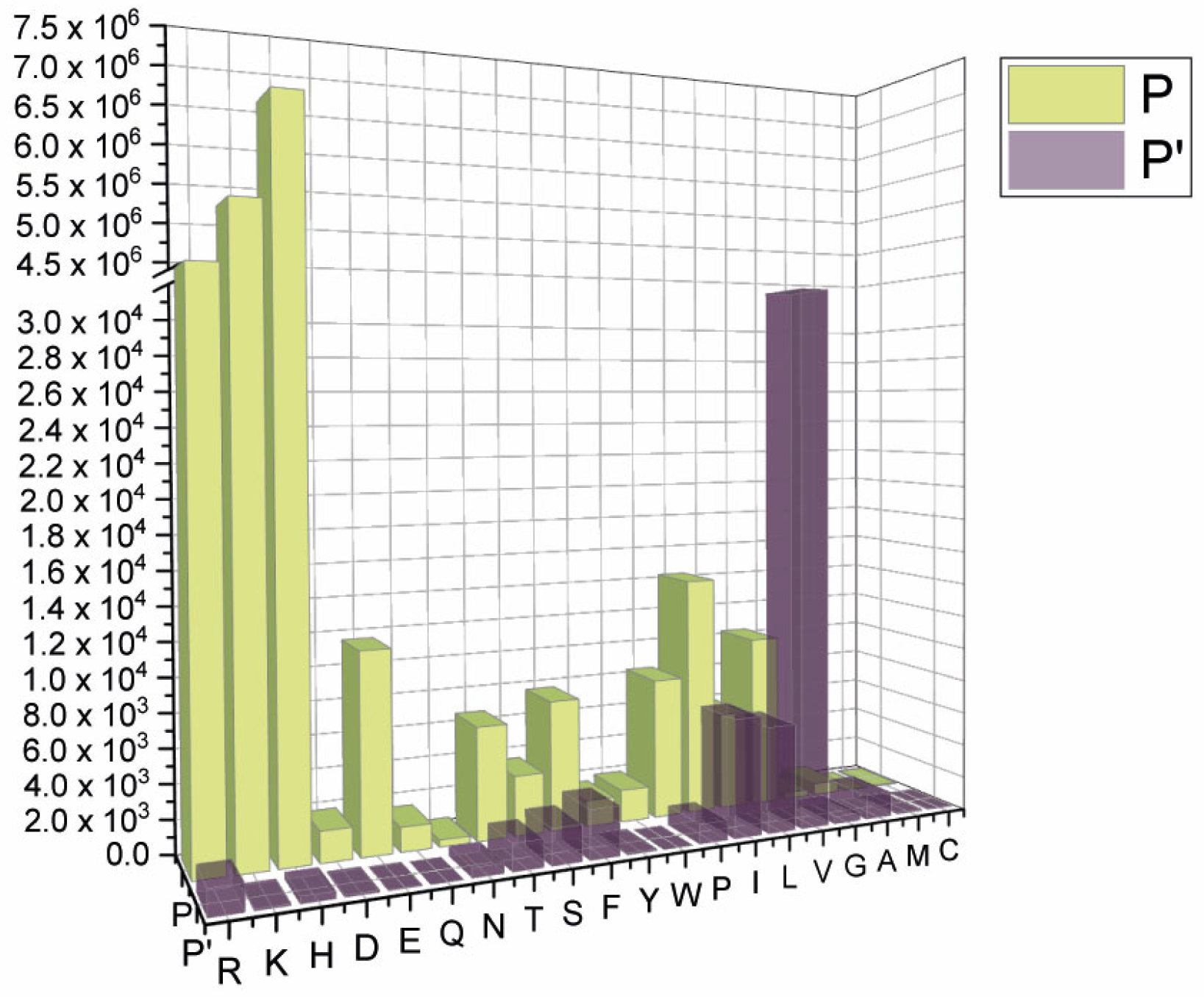
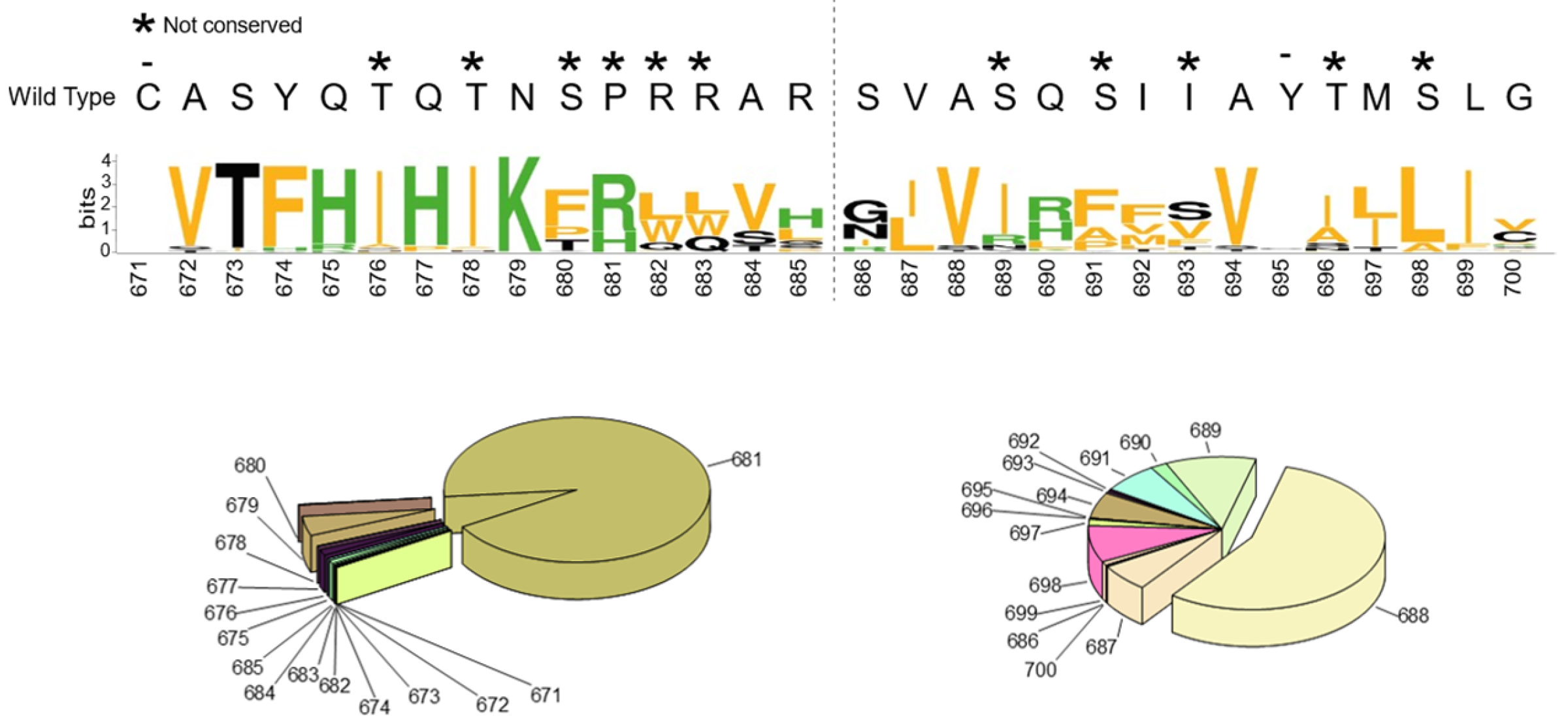
2.1. SARS-CoV-2 Spike S Varies Close to the Furin Cleavage Site Preferentially at P Positions
- The replacement of the key R685 and R682 at P4 and P1, as well as S686 at P1′, is a rare event (1108, 627, and 193 over 12.335.229 total sequences, respectively), as expected. Interestingly, we performed a similar database analysis on 10 January 2022, thus excluding the SARS-CoV-2 sequences uploaded in the GISAID platform from 11 January to 30 August 2022. Comparing the two analyses, we found that mutations at R682 are recent acquisitions. Indeed, more than 90% of the actual variants have been reported this year (52 vs. 575 from 11 January to 30 August 2022);
- The most popular variations occur at odd positions (P5, P7, P9, and P11) by replacing WT residues with basic amino acids. Of note, the closer the position is to the cleavage bond, the higher the number of recorded variants.Position P1, cleavage site, R685↓Position P5, P681H + P681R ---10.861.932 mutantsPosition P7, N679K -------------- 5.217.144 mutantsPosition P9, Q677H -------------- 83.139 mutantsPosition P11, Q675H ------------ 44.137 mutants
- The variation of residues at even-numbered positions (P6, P8, and P10) is much less frequent. Here, mutations involve the accommodation of amino acids with a hydrophobic character;
- Finally, there is nothing noteworthy within the P’ region, except the conservative A688V mutation at P3′.
2.2. In Vitro Digestion of Peptides Mimicking the Cleavage Site of SARS-CoV-2 Spike Variants
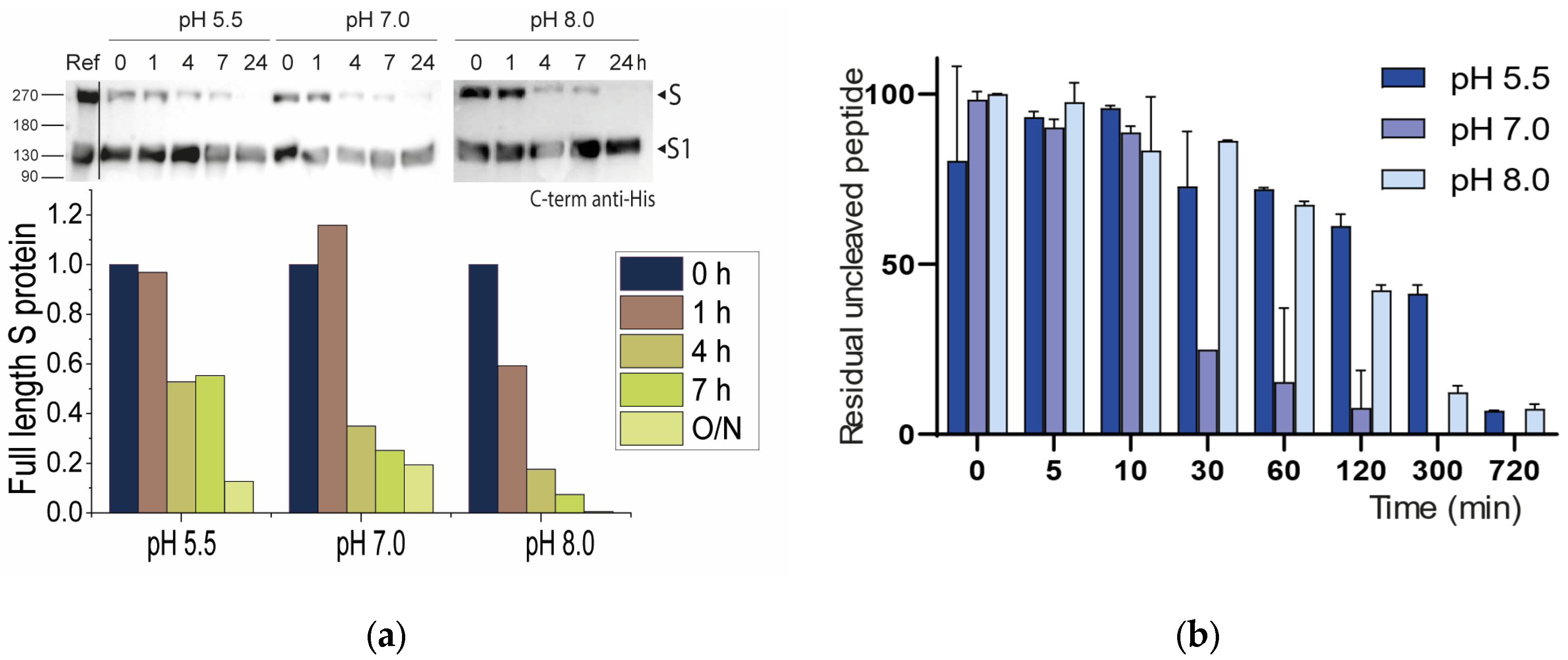
2.2.1. Neutral pH Privileges Cleavage of SARS-CoV-2 S WT
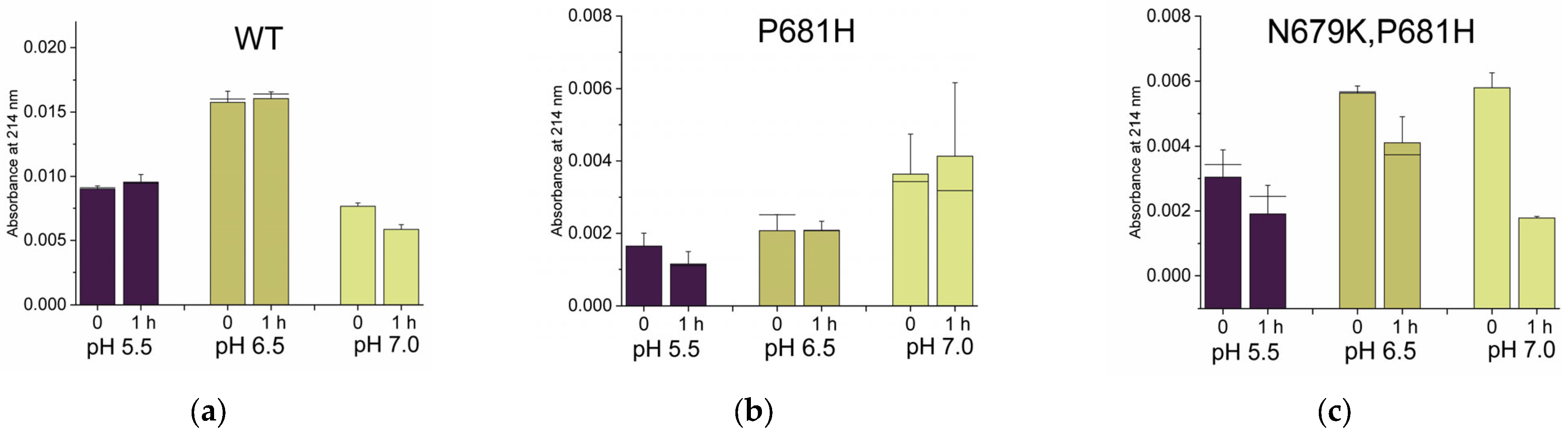
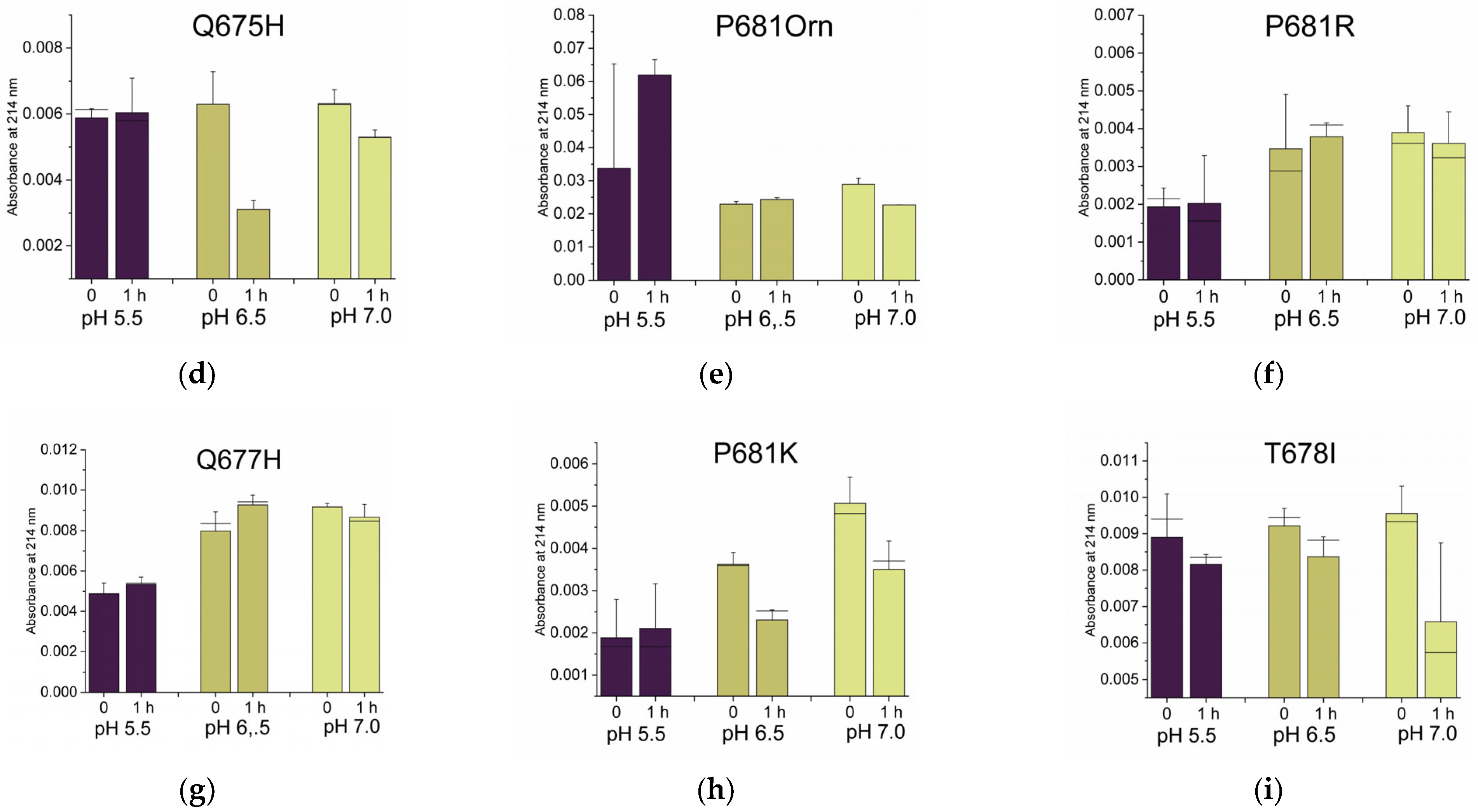
2.2.2. Histidines Upstream of the Cleavage Site Result in Gain-of-Function (GOF) Variants
2.2.3. Why Are There No P681K Variants of the SARS-CoV-2 Spike?
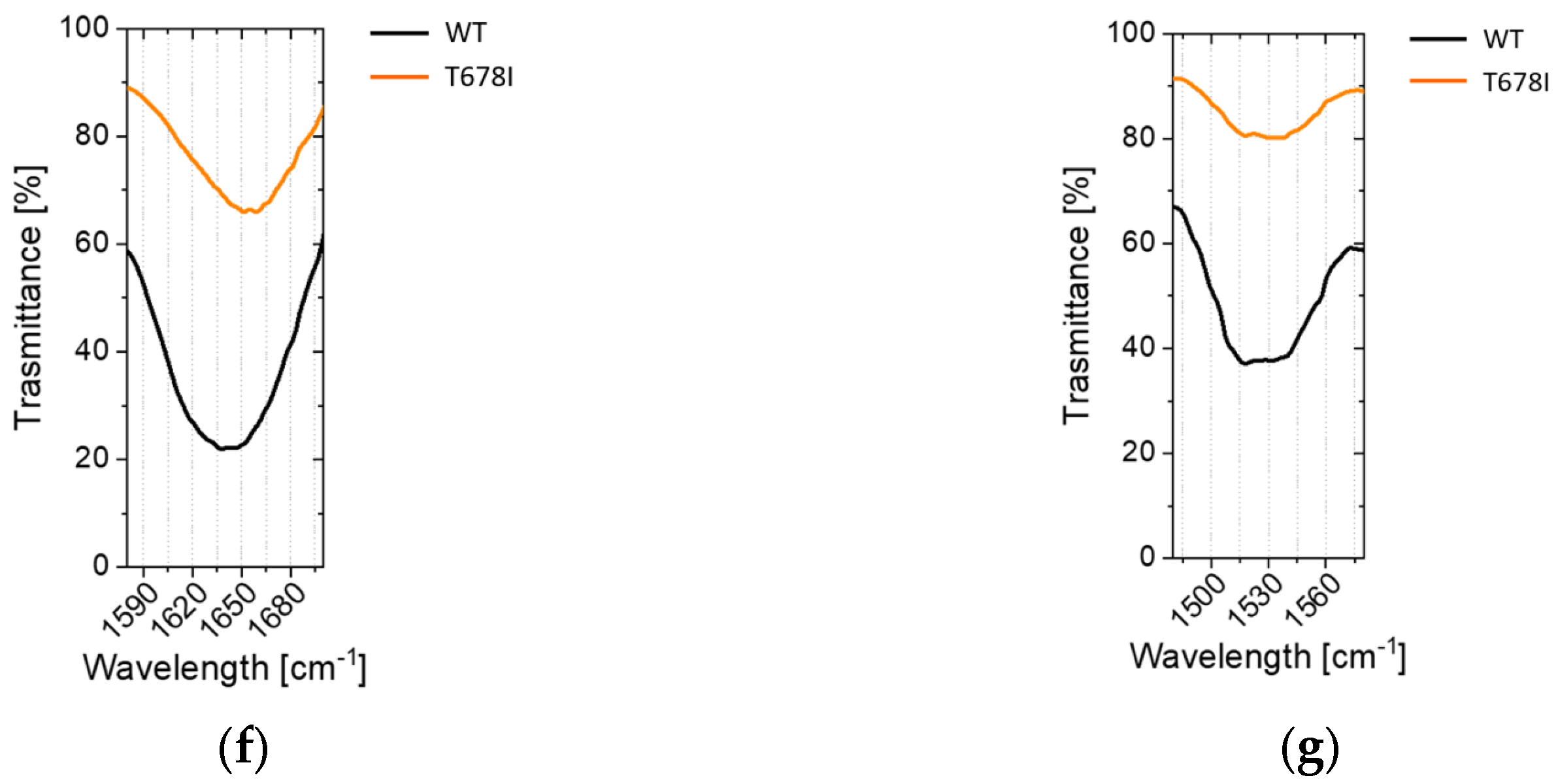
2.2.4. Non-Conservative T678I Replacement Enhances Processing
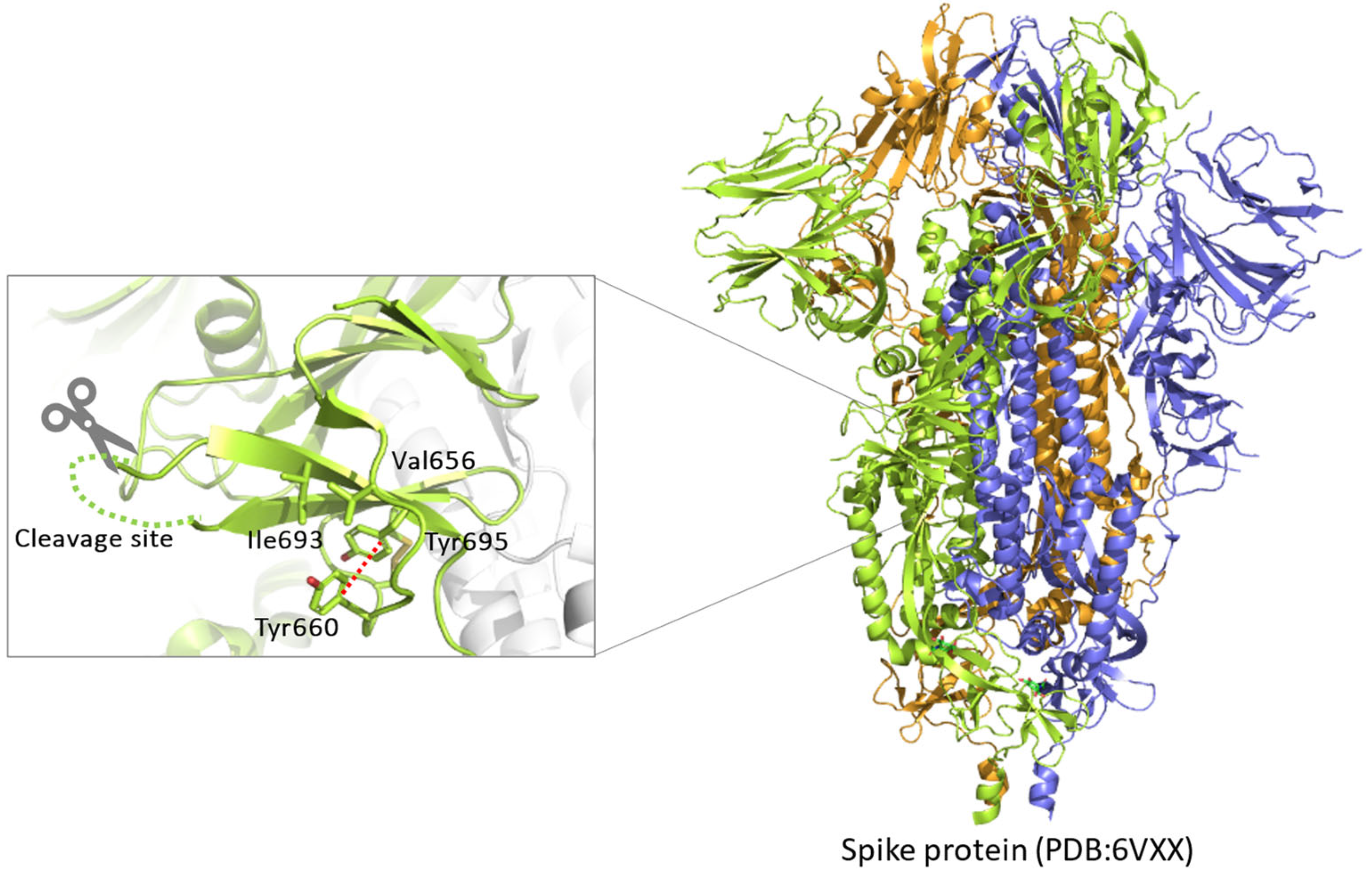
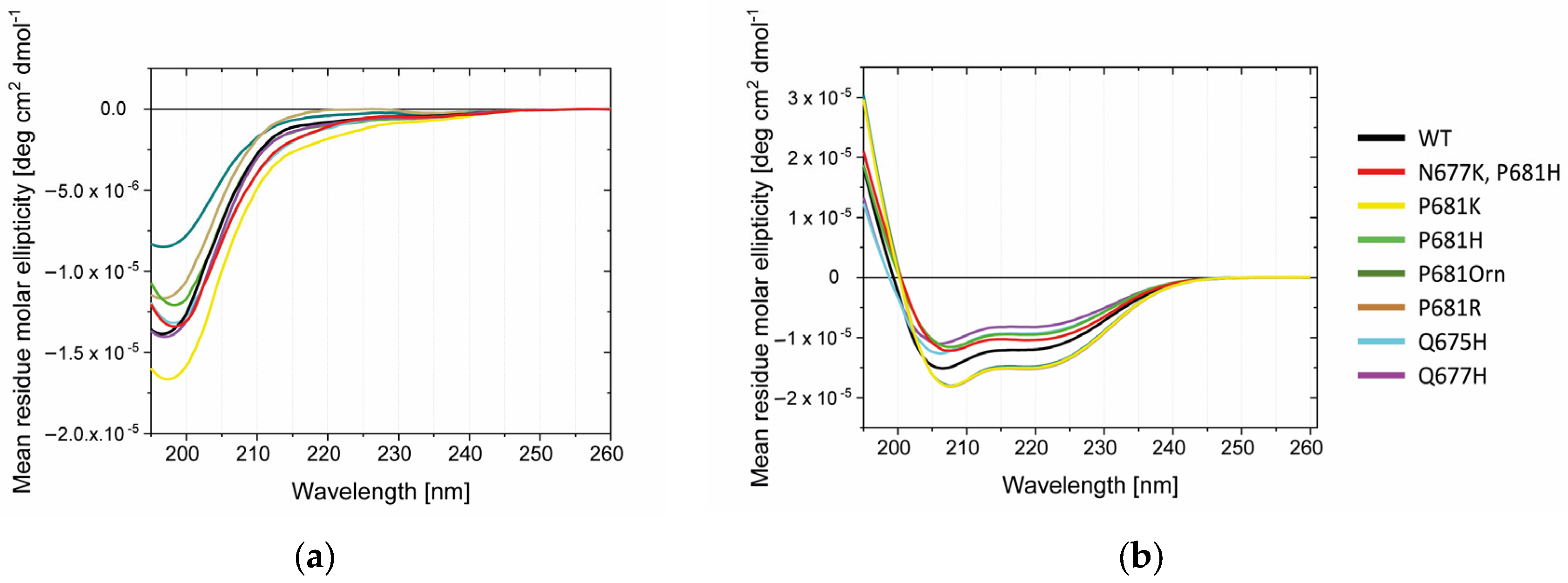
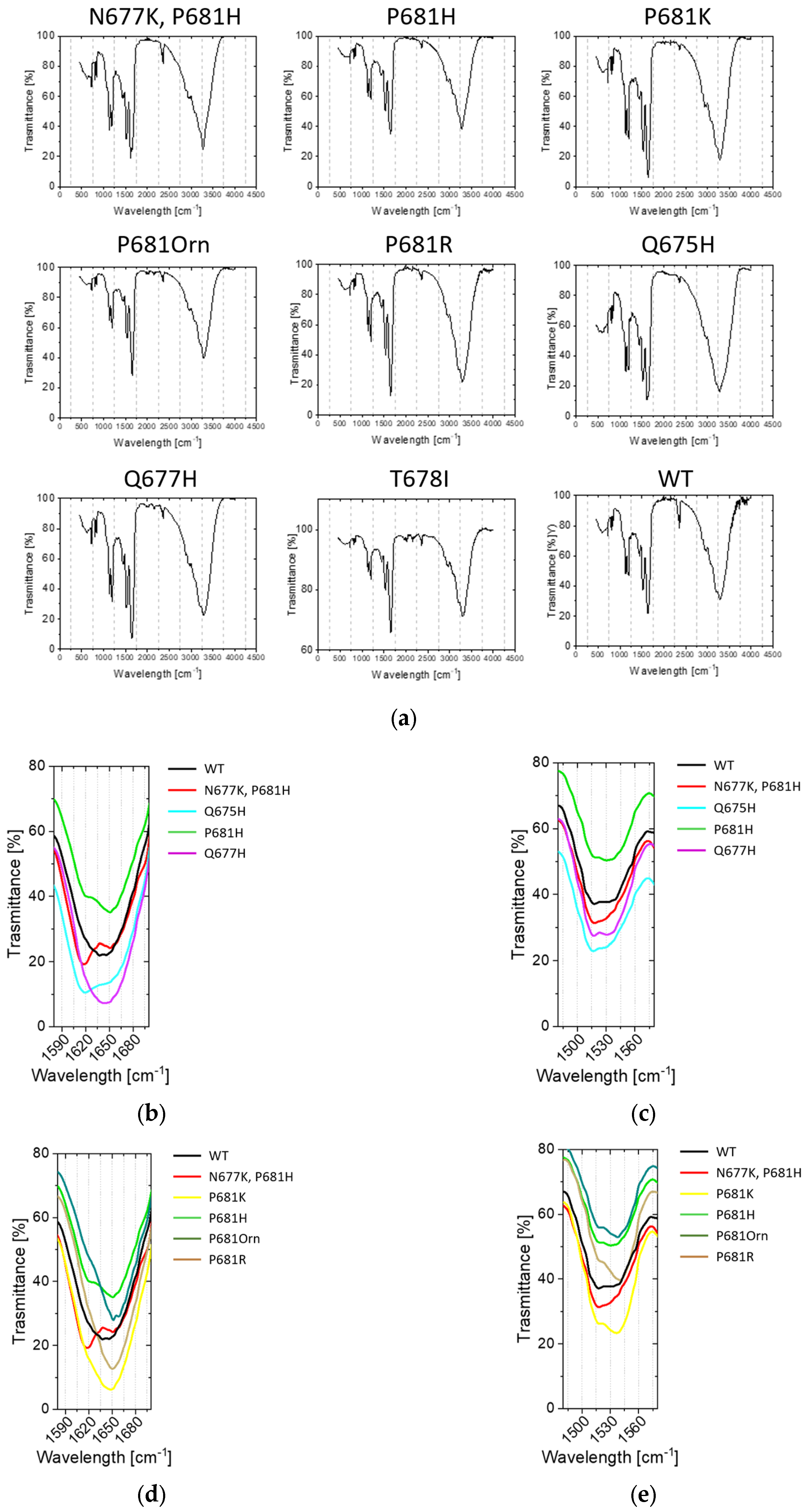
2.3. Conformational Investigations of the SARS-CoV-2 Wild-Type Cleavage Site and Its Mutants
2.4. Sustainability
3. Discussion
4. Materials and Methods
4.1. sFurin and SARS-CoV-2 S Production
4.2. In Vitro SARS-CoV-2 S Digestion
4.3. Western Blot
4.4. Peptide Synthesis
4.5. Peptide In Vitro Digestions
4.6. Circular Dichroism (CD) Analyses
4.7. Attenuated Total Reflectance Fourier-Transform Infrared Spectroscopy Investigation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127–e20. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and Bat RaTG13 Spike Glycoprotein Structures Inform on Virus Evolution and Furin-Cleavage Effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Herrera-Esposito, D.; de los Campos, G. Age-Specific Rate of Severe and Critical SARS-CoV-2 Infections Estimated with Multi-Country Seroprevalence Studies. BMC Infect. Dis. 2022, 22, 311. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Mohanty, A. Gut Microbiota and Covid-19- Possible Link and Implications. Virus Res. 2020, 285, 198018. [Google Scholar] [CrossRef]
- Rishi, P.; Thakur, K.; Vij, S.; Rishi, L.; Singh, A.; Kaur, I.P.; Patel, S.K.S.; Lee, J.-K.; Kalia, V.C. Diet, Gut Microbiota and COVID-19. Indian J. Microbiol. 2020, 60, 420–429. [Google Scholar] [CrossRef]
- Zahradník, J.; Nunvar, J.; Schreiber, G. Perspectives: SARS-CoV-2 Spike Convergent Evolution as a Guide to Explore Adaptive Advantage. Front. Cell. Infect. Microbiol. 2022, 12, 748948. [Google Scholar] [CrossRef]
- Thakur, V.; Bhola, S.; Thakur, P.; Patel, S.K.S.; Kulshrestha, S.; Ratho, R.K.; Kumar, P. Waves and Variants of SARS-CoV-2: Understanding the Causes and Effect of the COVID-19 Catastrophe. Infection 2022, 50, 309–325. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, W. Fast-Spreading SARS-CoV-2 Variants: Challenges to and New Design Strategies of COVID-19 Vaccines. Signal Transduct. Target. Ther. 2021, 6, 226. [Google Scholar] [CrossRef]
- Luo, W.-R.; Wu, X.-M.; Wang, W.; Yu, J.-L.; Chen, Q.-Q.; Zhou, X.; Huang, X.; Pan, H.-F.; Liu, Z.-R.; Gao, Y.; et al. Novel Coronavirus Mutations: Vaccine Development and Challenges. Microb. Pathog. 2022, 173, 105828. [Google Scholar] [CrossRef]
- Wang, R.; Simoneau, C.R.; Kulsuptrakul, J.; Bouhaddou, M.; Travisano, K.A.; Hayashi, J.M.; Carlson-Stevermer, J.; Zengel, J.R.; Richards, C.M.; Fozouni, P.; et al. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell 2021, 184, 106–119.e14. [Google Scholar] [CrossRef] [PubMed]
- Santopolo, S.; Riccio, A.; Santoro, M.G. The Biogenesis of SARS-CoV-2 Spike Glycoprotein: Multiple Targets for Host-Directed Antiviral Therapy. Biochem. Biophys. Res. Commun. 2021, 538, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The Furin Cleavage Site in the SARS-CoV-2 Spike Protein Is Required for Transmission in Ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and Furin Are Both Essential for Proteolytic Activation of SARS-CoV-2 in Human Airway Cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andréo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A.; et al. Distinctive Roles of Furin and TMPRSS2 in SARS-CoV-2 Infectivity. J. Virol. 2022, 96, e0012822. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-W.; Chao, T.-L.; Li, C.-L.; Chiu, M.-F.; Kao, H.-C.; Wang, S.-H.; Pang, Y.-H.; Lin, C.-H.; Tsai, Y.-M.; Lee, W.-H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020, 33, 108254. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Prat, A. The Biology and Therapeutic Targeting of the Proprotein Convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef]
- Stout, A.E.; Guo, Q.; Millet, J.K.; Whittaker, G.R. Viral and Host Attributes Underlying the Origins of Zoonotic Coronaviruses in Bats. Comp. Med. 2021, 71, 442–450. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; Zhang, L.; et al. Furin Cleavage Site Is Key to SARS-CoV-2 Pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Böttcher-Friebertshäuser, E.; Klenk, H.-D.; Garten, W. Activation of Influenza Viruses by Proteases from Host Cells and Bacteria in the Human Airway Epithelium. Pathog. Dis. 2013, 69, 87–100. [Google Scholar] [CrossRef]
- Kido, H. Influenza Virus Pathogenicity Regulated by Host Cellular Proteases, Cytokines and Metabolites, and Its Therapeutic Options. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2015, 91, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The Spike Glycoprotein of the New Coronavirus 2019-NCoV Contains a Furin-like Cleavage Site Absent in CoV of the Same Clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 2020, 30, 1346–1351.e2. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, S. Furin Cleavage Sites Naturally Occur in Coronaviruses. Stem Cell Res. 2021, 50, 102115. [Google Scholar] [CrossRef]
- Chan, Y.A.; Zhan, S.H. The Emergence of the Spike Furin Cleavage Site in SARS-CoV-2. Mol. Biol. Evol. 2022, 39, msab327. [Google Scholar] [CrossRef]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic Cleavage of the SARS-CoV-2 Spike Protein and the Role of the Novel S1/S2 Site. iScience 2020, 23, 101212. [Google Scholar] [CrossRef]
- Mishra, S.; Mindermann, S.; Sharma, M.; Whittaker, C.; Mellan, T.A.; Wilton, T.; Klapsa, D.; Mate, R.; Fritzsche, M.; Zambon, M.; et al. Changing Composition of SARS-CoV-2 Lineages and Rise of Delta Variant in England. eClinicalMedicine 2021, 39, 101064. [Google Scholar] [CrossRef]
- Zhao, L.P.; Lybrand, T.; Gilbert, P.; Payne, T.H.; Pyo, C.-W.; Geraghty, D.; Jerome, K. Rapidly Identifying New Coronavirus Mutations of Potential Concern in the Omicron Variant Using an Unsupervised Learning Strategy. Res. Sq. 2022, 12, 1–16. [Google Scholar] [CrossRef]
- Winstone, H.; Lista, M.J.; Reid, A.C.; Bouton, C.; Pickering, S.; Galao, R.P.; Kerridge, C.; Doores, K.J.; Swanson, C.M.; Neil, S.J.D. The Polybasic Cleavage Site in SARS-CoV-2 Spike Modulates Viral Sensitivity to Type I Interferon and IFITM2. J. Virol. 2021, 95, e02422-20. [Google Scholar] [CrossRef] [PubMed]
- Michihito, S.; Shinsuke, T.; Yukari, I.; Herman, M.C.; Mai, K.; Koshiro, T.; Kittiya, I.; Kentaro, U.; Takao, S.; Akihiko, S.; et al. SARS-CoV-2 Bearing a Mutation at the S1/S2 Cleavage Site Exhibits Attenuated Virulence and Confers Protective Immunity. mBio 2021, 12, e0141521. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of Furin Cleavage Site Attenuates SARS-CoV-2 Pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; de la Cruz, M.O. Enhanced Binding of SARS-CoV-2 Spike Protein to Receptor by Distal Polybasic Cleavage Sites. ACS Nano 2020, 14, 10616–10623. [Google Scholar] [CrossRef]
- Ludovico, C.-C.; Ravi, O.; Liliana, P.; Minou, D.; Jonas, F.; Suvi, K.; van der Meer, F.; Katri, K.; Tuğberk, K.; Maria, A.; et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jin, W.; Sood, A.; Montgomery, D.W.; Grant, O.C.; Fuster, M.M.; Fu, L.; Dordick, J.S.; Woods, R.J.; Zhang, F.; et al. Characterization of Heparin and Severe Acute Respiratory Syndrome-Related Coronavirus 2 (SARS-CoV-2) Spike Glycoprotein Binding Interactions. Antivir. Res. 2020, 181, 104873. [Google Scholar] [CrossRef]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia Formation by SARS-CoV-2-Infected Cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef]
- Chaudhry, M.Z.; Eschke, K.; Hoffmann, M.; Grashoff, M.; Abassi, L.; Kim, Y.; Brunotte, L.; Ludwig, S.; Kröger, A.; Klawonn, F.; et al. Rapid SARS-CoV-2 Adaptation to Available Cellular Proteases. J. Virol. 2022, 96, e0218621. [Google Scholar] [CrossRef]
- Nagy, A.; Basiouni, S.; Parvin, R.; Hafez, H.M.; Shehata, A.A. Evolutionary Insights into the Furin Cleavage Sites of SARS-CoV-2 Variants from Humans and Animals. Arch. Virol. 2021, 166, 2541–2549. [Google Scholar] [CrossRef]
- Liu, Z.; Zheng, H.; Lin, H.; Li, M.; Yuan, R.; Peng, J.; Xiong, Q.; Sun, J.; Li, B.; Wu, J.; et al. Identification of Common Deletions in the Spike Protein of Severe Acute Respiratory Syndrome Coronavirus 2. J. Virol. 2020, 94, e00790-20. [Google Scholar] [CrossRef]
- Anand, P.; Puranik, A.; Aravamudan, M.; Venkatakrishnan, A.J.; Soundararajan, V. SARS-CoV-2 Strategically Mimics Proteolytic Activation of Human ENaC. eLife 2020, 9, e58603. [Google Scholar] [CrossRef]
- Dahms, S.O.; Hardes, K.; Steinmetzer, T.; Than, M.E. X-Ray Structures of the Proprotein Convertase Furin Bound with Substrate Analogue Inhibitors Reveal Substrate Specificity Determinants beyond the S4 Pocket. Biochemistry 2018, 57, 925–934. [Google Scholar] [CrossRef]
- Tian, S.; Huajun, W.; Wu, J. Computational Prediction of Furin Cleavage Sites by a Hybrid Method and Understanding Mechanism Underlying Diseases. Sci. Rep. 2012, 2, 261. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Chernov, A.V.; Golubkov, V.S.; Thomsen, E.R.; Chudin, E.; Chee, M.S.; Kozlov, I.A.; Strongin, A.Y.; Cieplak, P. High-Resolution Analysis and Functional Mapping of Cleavage Sites and Substrate Proteins of Furin in the Human Proteome. PLoS ONE 2013, 8, e54290. [Google Scholar] [CrossRef]
- Örd, M.; Faustova, I.; Loog, M. The Sequence at Spike S1/S2 Site Enables Cleavage by Furin and Phospho-Regulation in SARS-CoV2 but Not in SARS-CoV1 or MERS-CoV. Sci. Rep. 2020, 10, 16944. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; de Silva, T.I.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 Variant Biology: Immune Escape, Transmission and Fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Gangavarapu, K.; Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.M.; Zaiets, K.; et al. Outbreak.Info Genomic Reports: Scalable and Dynamic Surveillance of SARS-CoV-2 Variants and Mutations. medRxiv 2022. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Molloy, S.S.; Bresnahan, P.A.; Leppla, S.H.; Klimpel, K.R.; Thomas, G. Human Furin Is a Calcium-Dependent Serine Endoprotease That Recognizes the Sequence Arg-X-X-Arg and Efficiently Cleaves Anthrax Toxin Protective Antigen. J. Biol. Chem. 1992, 267, 16396–16402. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G. Furin at the Cutting Edge: From Protein Traffic to Embryogenesis and Disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef]
- Ginefra, P.; Filippi, B.G.H.; Donovan, P.; Bessonnard, S.; Constam, D.B. Compartment-Specific Biosensors Reveal a Complementary Subcellular Distribution of Bioactive Furin and PC7. Cell Rep. 2018, 22, 2176–2189. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and Regulators of Intracellular PH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Stieneke-Gröber, A.; Vey, M.; Angliker, H.; Shaw, E.; Thomas, G.; Roberts, C.; Klenk, H.D.; Garten, W. Influenza Virus Hemagglutinin with Multibasic Cleavage Site Is Activated by Furin, a Subtilisin-like Endoprotease. EMBO J. 1992, 11, 2407–2414. [Google Scholar] [CrossRef]
- Bolt, G.; Pedersen, L.Ø.; Birkeslund, H.H. Cleavage of the Respiratory Syncytial Virus Fusion Protein Is Required for Its Surface Expression: Role of Furin. Virus Res. 2000, 68, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Hallenberger, S.; Bosch, V.; Angliker, H.; Shaw, E.; Klenk, H.-D.; Garten, W. Inhibition of Furin-Mediated Cleavage Activation of HIV-1 Glycoprotein Gpl60. Nature 1992, 360, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.-D. Processing of the Ebola Virus Glycoprotein by the Proprotein Convertase Furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762. [Google Scholar] [CrossRef]
- Pasquato, A.; Dettin, M.; Basak, A.; Gambaretto, R.; Tonin, L.; Seidah, N.G.; Di Bello, C. Heparin Enhances the Furin Cleavage of HIV-1 Gp160 Peptides. FEBS Lett. 2007, 581, 5807–5813. [Google Scholar] [CrossRef] [PubMed]
- Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations-NCoV-2019 Genomic Epidemiology-Virological. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 14 September 2022).
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á. Assessing Transmissibility of SARS-CoV-2 Lineage B. 1.1. 7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.; Abubaker, J.; Al-Mulla, F. Structural Modelling of SARS-CoV-2 Alpha Variant (B.1.1.7) Suggests Enhanced Furin Binding and Infectivity. Virus Res. 2021, 303, 198522. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta Spike P681R Mutation Enhances SARS-CoV-2 Fitness over Alpha Variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef]
- Arora, P.; Sidarovich, A.; Graichen, L.; Hörnich, B.; Hahn, A.; Hoffmann, M.; Pöhlmann, S. Functional Analysis of Polymorphisms at the S1/S2 Site of SARS-CoV-2 Spike Protein. PLoS ONE 2022, 17, e0265453. [Google Scholar] [CrossRef]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional Evaluation of the P681H Mutation on the Proteolytic Activation of the SARS-CoV-2 Variant B.1.1.7 (Alpha) Spike. iScience 2022, 25, 103589. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, A.; D’Ursi, P.; Campisi, G.; Messali, S.; Milanesi, M.; Giovanetti, M.; Ciccozzi, M.; Caccuri, F.; Caruso, A. Role of Q675H Mutation in Improving SARS-CoV-2 Spike Interaction with the Furin Binding Pocket. Viruses 2021, 13, 2511. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Domman, D.B.; Snyder, D.J.; Oguntuyo, K.Y.; Van Diest, M.; Densmore, K.H.; Schwalm, K.C.; Femling, J.; Carroll, J.L.; Scott, R.S.; et al. Emergence in Late 2020 of Multiple Lineages of SARS-CoV-2 Spike Protein Variants Affecting Amino Acid Position 677. medRxiv 2021. [Google Scholar] [CrossRef]
- Grabowski, F.; Preibisch, G.; Giziński, S.; Kochańczyk, M.; Lipniacki, T. SARS-CoV-2 Variant of Concern 202012/01 Has about Twofold Replicative Advantage and Acquires Concerning Mutations. Viruses 2021, 13, 392. [Google Scholar] [CrossRef]
- Jung, C.; Kmiec, D.; Koepke, L.; Zech, F.; Jacob, T.; Sparrer, K.M.J.; Kirchhoff, F. Omicron: What Makes the Latest SARS-CoV-2 Variant of Concern So Concerning? J. Virol. 2022, 96, e02077-21. [Google Scholar] [CrossRef] [PubMed]
- Yamasoba, D.; Kimura, I.; Nasser, H.; Morioka, Y.; Nao, N.; Ito, J.; Uriu, K.; Tsuda, M.; Zahradnik, J.; Shirakawa, K.; et al. Virological Characteristics of SARS-CoV-2 BA.2 Variant. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lubinski, B.; Jaimes, J.A.; Whittaker, G.R. Intrinsic Furin-Mediated Cleavability of the Spike S1/S2 Site from SARS-CoV-2 Variant B.1.529 (Omicron). bioRxiv 2022. [Google Scholar] [CrossRef]
- Singh, J.; Rahman, S.A.; Ehtesham, N.Z.; Hira, S.; Hasnain, S.E. SARS-CoV-2 Variants of Concern Are Emerging in India. Nat. Med. 2021, 27, 1131–1133. [Google Scholar] [CrossRef]
- Lubinski, B.; Frazier, L.E.; Phan, M.V.T.; Bugembe, D.L.; Cunningham, J.L.; Tang, T.; Daniel, S.; Cotten, M.; Jaimes, J.A.; Whittaker, G.R. Spike Protein Cleavage-Activation in the Context of the SARS-CoV-2 P681R Mutation: An Analysis from Its First Appearance in Lineage A.23.1 Identified in Uganda. Microbiol. Spectr. 2022, 10, e01514-22. [Google Scholar] [CrossRef]
- Zhang, L.; Mann, M.; Syed, Z.; Reynolds, H.M.; Tian, E.; Samara, N.L.; Zeldin, D.C.; Tabak, L.A.; Ten Hagen, K.G. Furin Cleavage of the SARS-CoV-2 Spike Is Modulated by O-Glycosylation. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Dahms, S.O.; Creemers, J.W.M.; Schaub, Y.; Bourenkov, G.P.; Zögg, T.; Brandstetter, H.; Than, M.E. The Structure of a Furin-Antibody Complex Explains Non-Competitive Inhibition by Steric Exclusion of Substrate Conformers. Sci. Rep. 2016, 6, 34303. [Google Scholar] [CrossRef] [PubMed]
- Henrich, S.; Cameron, A.; Bourenkov, G.P.; Kiefersauer, R.; Huber, R.; Lindberg, I.; Bode, W.; Than, M.E. The Crystal Structure of the Proprotein Processing Proteinase Furin Explains Its Stringent Specificity. Nat. Struct. Mol. Biol. 2003, 10, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Long, S.W.; Olsen, R.J.; Christensen, P.A.; Bernard, D.W.; Davis, J.J.; Shukla, M.; Nguyen, M.; Saavedra, M.O.; Yerramilli, P.; Pruitt, L.; et al. Molecular Architecture of Early Dissemination and Massive Second Wave of the SARS-CoV-2 Virus in a Major Metropolitan Area. mBio 2020, 11, e02707-20. [Google Scholar] [CrossRef] [PubMed]
- Venyaminov, S.Y.; Baikalov, I.A.; Shen, Z.M.; Wu, C.S.C.; Yang, J.T. Circular Dichroic Analysis of Denatured Proteins: Inclusion of Denatured Proteins in the Reference Set. Anal. Biochem. 1993, 214, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.J. Using Circular Dichroism Spectra to Estimate Protein Secondary Structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Mantsch, H.H. The Use and Misuse of FTIR Spectroscopy in the Determination of Protein Structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Rabaan, A.; Al-Ahmed, S.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, I.; Bonilla-Aldana, K.; Rodriguez-Morales, A. SARS-CoV-2, SARS-CoV, and MERS-COV: A Comparative Overview. Infez. Med. 2020, 28, 174–184. [Google Scholar]
- Neumann, G.; Geisbert, T.W.; Ebihara, H.; Geisbert, J.B.; Daddario-DiCaprio, K.M.; Feldmann, H.; Kawaoka, Y. Proteolytic Processing of the Ebola Virus Glycoprotein Is Not Critical for Ebola Virus Replication in Nonhuman Primates. J. Virol. 2007, 81, 2995–2998. [Google Scholar] [CrossRef] [PubMed]
- Dahms, S.O.; Hardes, K.; Becker, G.L.; Steinmetzer, T.; Brandstetter, H.; Than, M.E. X-Ray Structures of Human Furin in Complex with Competitive Inhibitors. ACS Chem. Biol. 2014, 9, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Kadam, S.B.; Sukhramani, G.S.; Bishnoi, P.; Pable, A.A.; Barvkar, V.T. SARS-CoV-2, the Pandemic Coronavirus: Molecular and Structural Insights. J. Basic Microbiol. 2021, 61, 180–202. [Google Scholar] [CrossRef] [PubMed]
- Decroly, E.; Wouters, S.; Di Bello, C.; Lazure, C.; Ruysschaert, J.-M.; Seidah, N.G. Identification of the Paired Basic Convertases Implicated in HIV Gp160 Processing Based on in Vitro Assays and Expression in CD4+ Cell Lines*. J. Biol. Chem. 1996, 271, 30442–30450. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | P13 | P12 | P11 | P10 | P9 | P8 | P7 | P6 | P5 | P4 | P3 | P2 | P1 | P1’ | P2’ | P3’ | P4’ | Theoretical Mass [Da] | Experimental Mass [Da] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Naturally occurring | Wild Type (WT) | S | Y | Q | T | Q | T | N | S | P | R | R | A | R | S | V | A | S | 1908.05 | 1907.61 |
| Q675H | S | Y | H | T | Q | T | N | S | P | R | R | A | R | S | V | A | S | 1917.06 | 1917.69 | |
| Q677H | S | Y | Q | T | H | T | N | S | P | R | R | A | R | S | V | A | S | 1917.06 | 1917.52 | |
| T678I | S | Y | Q | T | Q | I | N | S | P | R | R | A | R | S | V | A | S | 1920.11 | 1920.63 | |
| N679K, P681H | S | Y | Q | T | Q | T | K | S | H | R | R | A | R | S | V | A | S | 1962.15 | 1962.46 | |
| P681H | S | Y | Q | T | Q | T | N | S | H | R | R | A | R | S | V | A | S | 1948.08 | 1948.55 | |
| P681R | S | Y | Q | T | Q | T | N | S | R | R | R | A | R | S | V | A | S | 1967.12 | 1968.61 | |
| Synthetic | P681K | S | Y | Q | T | Q | T | N | S | K | R | R | A | R | S | V | A | S | 1939.11 | 1939.87 |
| P681Orn | S | Y | Q | T | Q | T | N | S | Orn | R | R | A | R | S | V | A | S | 1925.07 | 1925.65 | |
| amino acid position | 673 | 674 | 675 | 676 | 677 | 678 | 679 | 680 | 681 | 682 | 683 | 684 | 685 | 686 | 687 | 688 | 689 |
| Use | Object | Quantity | Unit Weight [g] | TOT Weight [g] |
|---|---|---|---|---|
| Peptide synthesis | PTFE filter 0.45 μm (diameter 30 mm) | 9 | 2.30 | 20.70 |
| PVDF filter 0.45 μm (diameter 13 mm) | 9 | 12.00 | 108.00 | |
| Reactor | 9 | 4.00 | 36.00 | |
| Syringe (1 mL) | 9 | 2.20 | 19.80 | |
| Syringe (20 mL) | 12 | 10.00 | 120.00 | |
| Tips (200 μL) | 100 | 0.27 | 27.00 | |
| Tips (20 μL) | 100 | 0.12 | 12.00 | |
| Tube (1.5 mL) | 40 | 1.00 | 40.00 | |
| Tube (2 mL) | 10 | 1.15 | 11.50 | |
| Tube (50 mL) | 40 | 12.70 | 508.00 | |
| Tube (6 mL) | 30 | 3.50 | 105.00 | |
| Glove pairs | 9 | 2.30 | 20.70 | |
| In vitro—conformation studies | Flask (125 cm2) | 1 | 100.00 | 100.00 |
| Flask (250 cm2) | 1 | 200.00 | 200.00 | |
| Flask (75 cm2) | 6 | 60.00 | 360.00 | |
| Plate (24 well) | 16 | 65.40 | 1046.40 | |
| Plate (48 well) | 3 | 56.00 | 168.00 | |
| Plate (96 well) | 3 | 64.60 | 193.80 | |
| Sterile pipette (10 mL) | 80 | 14.00 | 1120.00 | |
| Sterile pipette (25 mL) | 15 | 15.60 | 234.00 | |
| Sterile pipette (2 mL) | 2 | 4.40 | 8.80 | |
| Sterile pipette (5 mL) | 60 | 7.90 | 474.00 | |
| Tips (10 μL) | 100 | 0.12 | 12.00 | |
| Tips (1000 μL) | 200 | 0.76 | 152.00 | |
| Tips (200 μL) | 1500 | 0.27 | 405.00 | |
| Tube (1.5 mL) | 1000 | 1.00 | 1000.00 | |
| Tube (15 mL) | 15 | 6.40 | 96.00 | |
| Tube (50 mL) | 30 | 12.70 | 381.00 | |
| Glove pairs | 200 | 6.20 | 1240.00 | |
| Other | 1000.00 | |||
| TOT | 9819.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cassari, L.; Pavan, A.; Zoia, G.; Chinellato, M.; Zeni, E.; Grinzato, A.; Rothenberger, S.; Cendron, L.; Dettin, M.; Pasquato, A. SARS-CoV-2 S Mutations: A Lesson from the Viral World to Understand How Human Furin Works. Int. J. Mol. Sci. 2023, 24, 4791. https://doi.org/10.3390/ijms24054791
Cassari L, Pavan A, Zoia G, Chinellato M, Zeni E, Grinzato A, Rothenberger S, Cendron L, Dettin M, Pasquato A. SARS-CoV-2 S Mutations: A Lesson from the Viral World to Understand How Human Furin Works. International Journal of Molecular Sciences. 2023; 24(5):4791. https://doi.org/10.3390/ijms24054791
Chicago/Turabian StyleCassari, Leonardo, Angela Pavan, Giulia Zoia, Monica Chinellato, Elena Zeni, Alessandro Grinzato, Sylvia Rothenberger, Laura Cendron, Monica Dettin, and Antonella Pasquato. 2023. "SARS-CoV-2 S Mutations: A Lesson from the Viral World to Understand How Human Furin Works" International Journal of Molecular Sciences 24, no. 5: 4791. https://doi.org/10.3390/ijms24054791
APA StyleCassari, L., Pavan, A., Zoia, G., Chinellato, M., Zeni, E., Grinzato, A., Rothenberger, S., Cendron, L., Dettin, M., & Pasquato, A. (2023). SARS-CoV-2 S Mutations: A Lesson from the Viral World to Understand How Human Furin Works. International Journal of Molecular Sciences, 24(5), 4791. https://doi.org/10.3390/ijms24054791