
Selectivity of Hydroxamate- and Difluoromethyloxadiazole-Based Inhibitors of Histone Deacetylase 6 In Vitro and in Cells

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. HDAC Inhibitors and Substrates
2.2. Chemicals
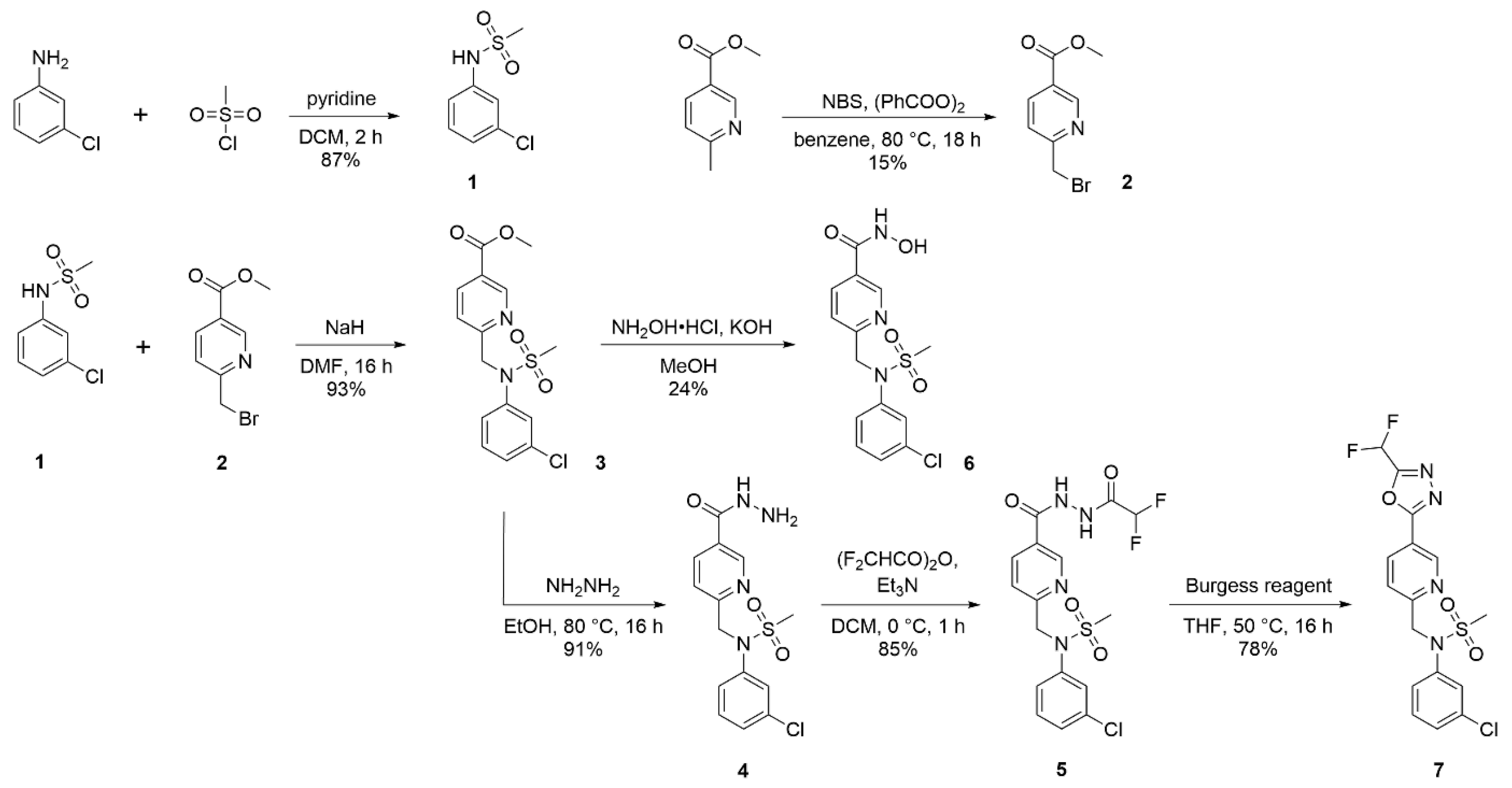
2.2.1. N-(3-chlorophenyl)methanesulfonamide (1)
2.2.2. Methyl 6-(bromomethyl)nicotinate (2)
2.2.3. Methyl 6-((N-(3-chlorophenyl)methylsulfonamido)methyl)nicotinate (3)
2.2.4. N-(3-chlorophenyl)-N-((5-(hydrazinecarbonyl)pyridin-2-yl)methyl)methanesulfonamide (4)
2.2.5. N-(3-Chlorophenyl)-N-((5-(2-(2,2-difluoroacetyl)hydrazine-1-carbonyl)pyridin-2-yl)methyl)methanesulfonamide (5)
2.2.6. 6-((N-(3-Chlorophenyl)methylsulfonamido)methyl)-N-hydroxynicotinamide (6)
2.2.7. N-(3-chlorophenyl)-N-((5-(5-(difluoromethyl)-1,3,4-oxadiazol-2-yl)pyridin-2-yl)methyl)methanesulfonamide (7)
3. Cloning of Expression Plasmids
3.1. Heterologous Expression and Purification of Recombinant HDACs
3.2. Deacetylation Assays In Vitro
3.3. MTT Viability Assay
3.4. HDAC1/HDAC6 Engagement Assay
3.5. IC50 Determination in Cells
3.6. Quantitative Western Blotting
3.7. Correlation Analysis
4. Results and Discussion
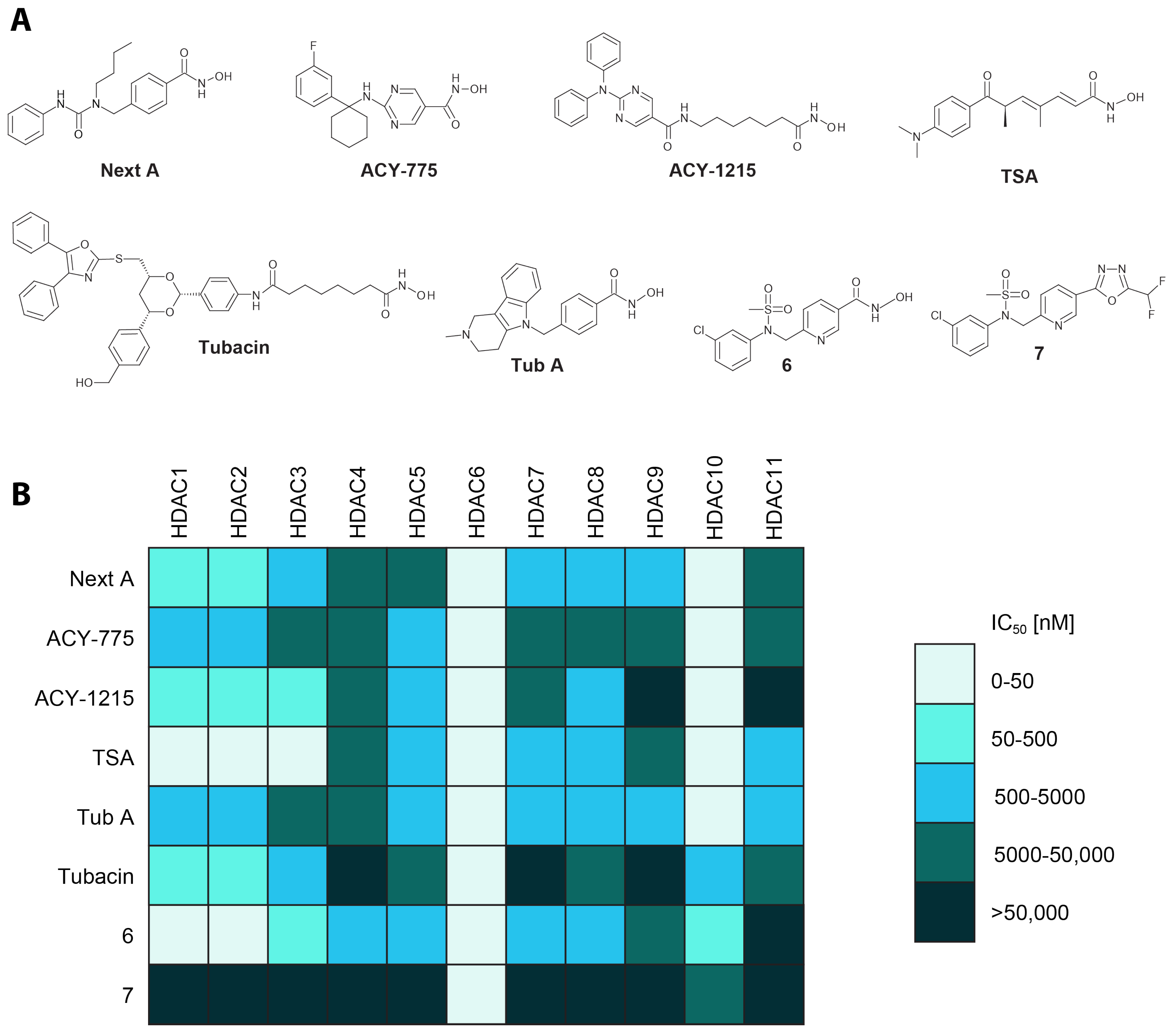
4.1. Selectivity of Hydroxamate-Based HDAC6-Specific Inhibitors (HDAC6is) Is Limited In Vitro
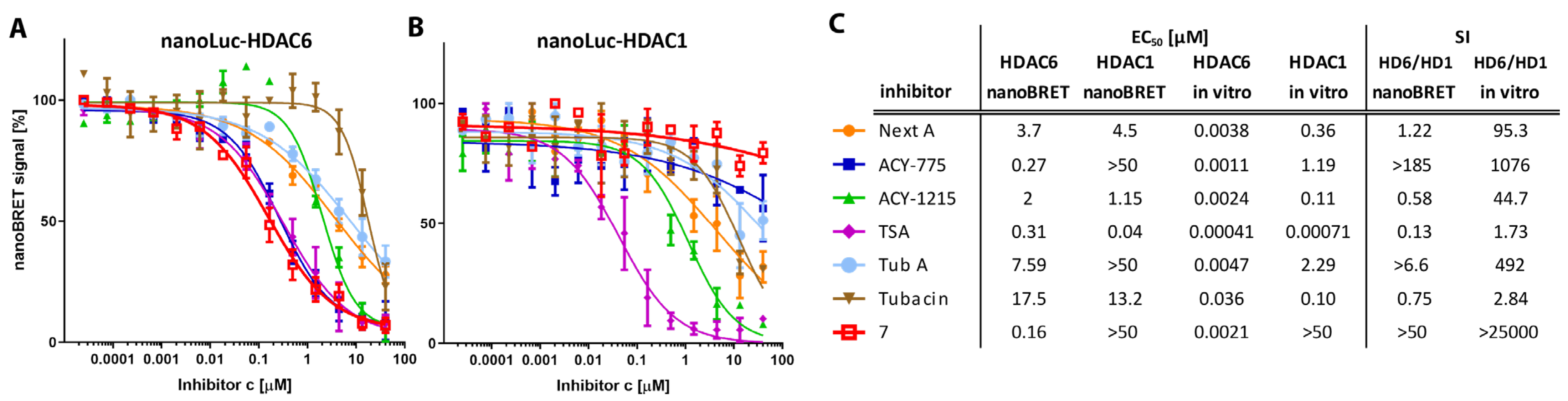
4.2. The Difluoromethyl-1,3,4-Oxadiazole Zinc-Binding Group Imparts High HDAC6 Selectivity
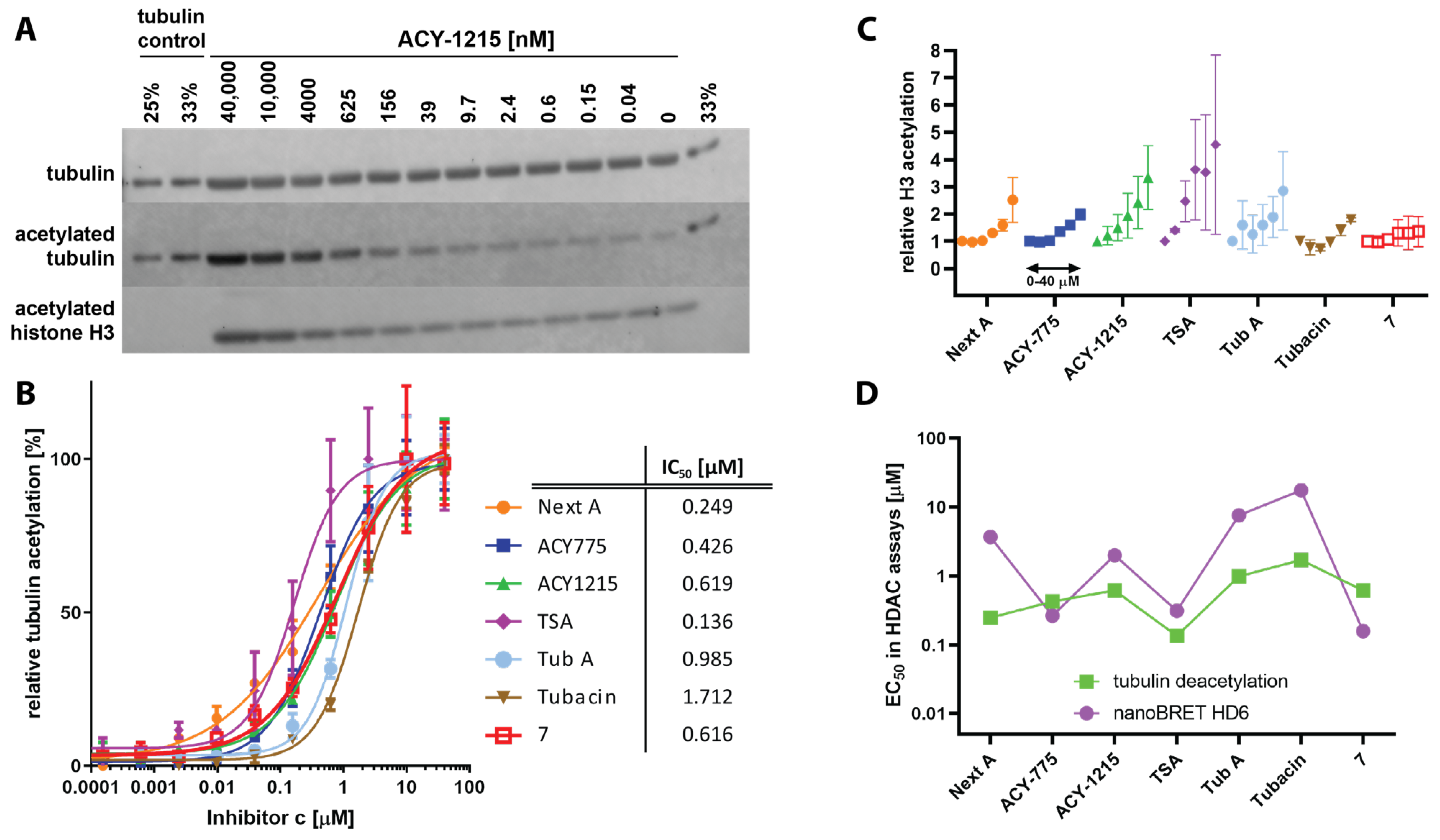
4.3. Cellular Potency of HDAC6is Is on Average 100-Fold Lower Compared to In Vitro Data
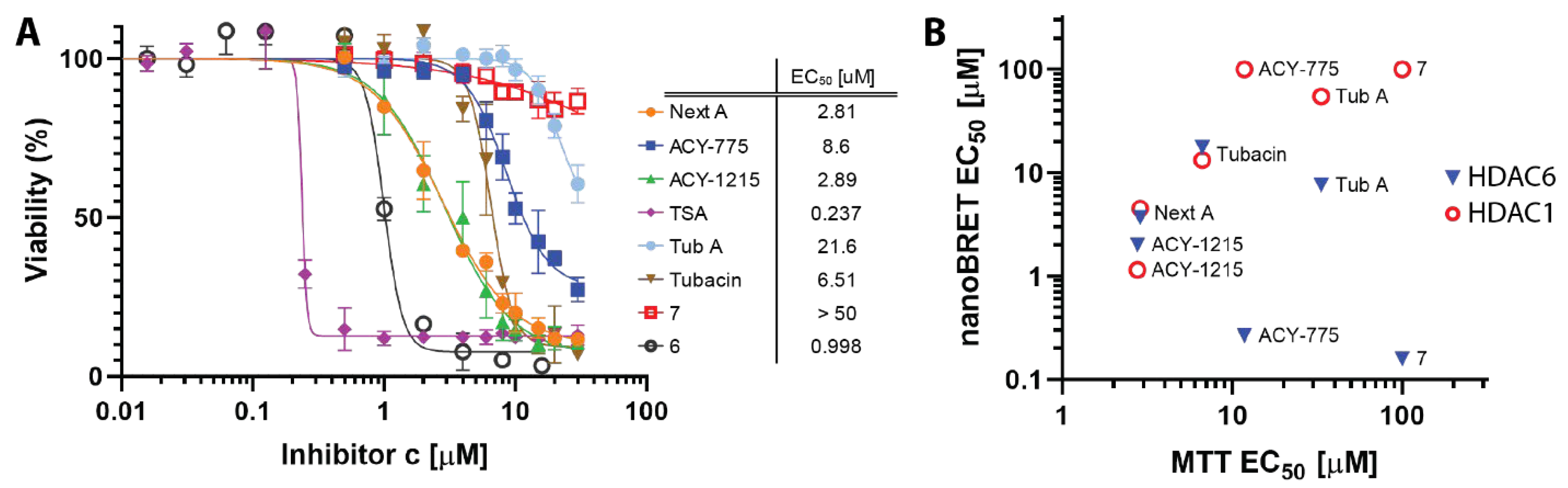
4.4. Cellular Toxicity of HDACis Correlates with Class I HDAC Off-Targeting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Boyault, C.; Sadoul, K.; Pabion, M.; Khochbin, S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007, 26, 5468–5476. [Google Scholar] [CrossRef] [PubMed]
- Matthias, P.; Yoshida, M.; Khochbin, S. HDAC6 a new cellular stress surveillance factor. Cell Cycle 2008, 7, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Perdiz, D.; Mackeh, R.; Pous, C.; Baillet, A. The ins and outs of tubulin acetylation: More than just a post-translational modification? Cell. Signal. 2011, 23, 763–771. [Google Scholar] [CrossRef]
- Arce, C.A.; Casale, C.H.; Barra, H.S. Submembraneous microtubule cytoskeleton: Regulation of ATPases by interaction with acetylated tubulin. FEBS J. 2008, 275, 4664–4674. [Google Scholar] [CrossRef]
- Hammond, J.W.; Huang, C.F.; Kaech, S.; Jacobson, C.; Banker, G.; Verhey, K.J. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell 2010, 21, 572–583. [Google Scholar] [CrossRef]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef]
- Gardiner, J.; Barton, D.; Marc, J.; Overall, R. Potential role of tubulin acetylation and microtubule-based protein trafficking in familial dysautonomia. Traffic 2007, 8, 1145–1149. [Google Scholar] [CrossRef]
- Liu, X.A.; Rizzo, V.; Puthanveettil, S.V. Pathologies of Axonal Transport in Neurodegenerative Diseases. Transl. Neurosci. 2012, 3, 355–372. [Google Scholar] [CrossRef]
- Jochems, J.; Boulden, J.; Lee, B.G.; Blendy, J.A.; Jarpe, M.; Mazitschek, R.; Van Duzer, J.H.; Jones, S.; Berton, O. Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacology 2014, 39, 389–400. [Google Scholar] [CrossRef]
- Wang, Z.; Leng, Y.; Wang, J.; Liao, H.M.; Bergman, J.; Leeds, P.; Kozikowski, A.; Chuang, D.M. Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: Potential roles of alpha-tubulin acetylation and FGF-21 up-regulation. Sci. Rep. 2016, 6, 19626. [Google Scholar] [CrossRef]
- Pinho, B.R.; Reis, S.D.; Guedes-Dias, P.; Leitao-Rocha, A.; Quintas, C.; Valentao, P.; Andrade, P.B.; Santos, M.M.; Oliveira, J.M. Pharmacological modulation of HDAC1 and HDAC6 in vivo in a zebrafish model: Therapeutic implications for Parkinson’s disease. Pharmacol. Res. 2016, 103, 328–339. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, C.; Wu, J.; Tao, J.J.; Sui, X.L.; Yao, Z.G.; Xu, Y.F.; Huang, L.; Zhu, H.; Sheng, S.L.; et al. Tubastatin A/ACY-1215 improves cognition in Alzheimer’s disease transgenic mice. J. Alzheimer’s Dis. JAD 2014, 41, 1193–1205. [Google Scholar] [CrossRef]
- Zhang, L.; Sheng, S.; Qin, C. The role of HDAC6 in Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2013, 33, 283–295. [Google Scholar] [CrossRef]
- Selenica, M.L.; Benner, L.; Housley, S.B.; Manchec, B.; Lee, D.C.; Nash, K.R.; Kalin, J.; Bergman, J.A.; Kozikowski, A.; Gordon, M.N.; et al. Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimer’s Res. Ther. 2014, 6, 12. [Google Scholar] [CrossRef]
- d’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Vanden Berghe, P.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef]
- Kim, J.Y.; Woo, S.Y.; Hong, Y.B.; Choi, H.; Kim, J.; Choi, H.; Mook-Jung, I.; Ha, N.; Kyung, J.; Koo, S.K.; et al. HDAC6 Inhibitors Rescued the Defective Axonal Mitochondrial Movement in Motor Neurons Derived from the Induced Pluripotent Stem Cells of Peripheral Neuropathy Patients with HSPB1 Mutation. Stem Cells Int. 2016, 2016, 9475981. [Google Scholar] [CrossRef]
- Gold, W.A.; Lacina, T.A.; Cantrill, L.C.; Christodoulou, J. MeCP2 deficiency is associated with reduced levels of tubulin acetylation and can be restored using HDAC6 inhibitors. J. Mol. Med. 2015, 93, 63–72. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovas, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Shen, S.; Pardo, M.; Tavares, M.T.; Szarics, D.; Benoy, V.; Zimprich, C.A.; Kutil, Z.; Zhang, G.; Barinka, C.; et al. Brain Penetrable Histone Deacetylase 6 Inhibitor SW-100 Ameliorates Memory and Learning Impairments in a Mouse Model of Fragile X Syndrome. ACS Chem. Neurosci. 2019, 10, 1679–1695. [Google Scholar] [CrossRef]
- Langley, B.; D’Annibale, M.A.; Suh, K.; Ayoub, I.; Tolhurst, A.; Bastan, B.; Yang, L.; Ko, B.; Fisher, M.; Cho, S.; et al. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J. Neurosci. 2008, 28, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Rivieccio, M.A.; Brochier, C.; Willis, D.E.; Walker, B.A.; D’Annibale, M.A.; McLaughlin, K.; Siddiq, A.; Kozikowski, A.P.; Jaffrey, S.R.; Twiss, J.L.; et al. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 19599–19604. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Wan, R.Z.; Liu, Z.P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 2018, 143, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Clinical Trials for ACY-1215. Available online: https://clinicaltrials.gov/ct2/results?term=ACY-1215&Search=Search (accessed on 28 September 2017).
- Clinical Trials for ACY-241. Available online: https://clinicaltrials.gov/ct2/results?term=ACY-241&Search=Search (accessed on 28 September 2017).
- Lechner, S.; Malgapo, M.I.P.; Gratz, C.; Steimbach, R.R.; Baron, A.; Ruther, P.; Nadal, S.; Stumpf, C.; Loos, C.; Ku, X.; et al. Target deconvolution of HDAC pharmacopoeia reveals MBLAC2 as common off-target. Nat. Chem. Biol. 2022, 18, 812–820. [Google Scholar] [CrossRef]
- Liang, Y.Y.; Zhang, C.M.; Liu, Z.P. Evaluation of WO2017018805: 1,3,4-oxadiazole sulfamide derivatives as selective HDAC6 inhibitors. Expert Opin. Ther. Pat. 2018, 28, 647–651. [Google Scholar] [CrossRef]
- Cellupica, E.; Caprini, G.; Cordella, P.; Cukier, C.; Fossati, G.; Marchini, M.; Rocchio, I.; Sandrone, G.; Vanoni, M.A.; Vergani, B.; et al. Difluoromethyl-1,3,4-oxadiazoles are slow-binding substrate analog inhibitors of histone deacetylase 6 with unprecedented isotype selectivity. J. Biol. Chem. 2022, 299, 102800. [Google Scholar] [CrossRef]
- Shen, S.; Kozikowski, A.P. A patent review of histone deacetylase 6 inhibitors in neurodegenerative diseases (2014–2019). Expert Opin. Ther. Pat. 2020, 30, 121–136. [Google Scholar] [CrossRef]
- Tan, B.Y.H.; Teo, Y.C.; Seow, A.H. Low Catalyst Loadings for Ligand-Free Copper(I)-Oxide-Catalyzed N-Arylation of Methanesulfonamide in Water. Eur. J. Org. Chem. 2014, 2014, 1541–1546. [Google Scholar] [CrossRef]
- Humphreys, K.J.; Karlin, K.D.; Rokita, S.E. Efficient and specific strand scission of DNA by a dinuclear copper complex: Comparative reactivity of complexes with linked tris(2-pyridylmethyl)amine moieties. J. Am. Chem. Soc. 2002, 124, 6009–6019. [Google Scholar] [CrossRef]
- Skultetyova, L.; Ustinova, K.; Kutil, Z.; Novakova, Z.; Pavlicek, J.; Mikesova, J.; Trapl, D.; Baranova, P.; Havlinova, B.; Hubalek, M.; et al. Human histone deacetylase 6 shows strong preference for tubulin dimers over assembled microtubules. Sci. Rep. 2017, 7, 11547. [Google Scholar] [CrossRef]
- Noonepalle, S.; Shen, S.; Ptacek, J.; Tavares, M.T.; Zhang, G.; Stransky, J.; Pavlicek, J.; Ferreira, G.M.; Hadley, M.; Pelaez, G.; et al. Rational Design of Suprastat: A Novel Selective Histone Deacetylase 6 Inhibitor with the Ability to Potentiate Immunotherapy in Melanoma Models. J. Med. Chem. 2020, 63, 10246–10262. [Google Scholar] [CrossRef]
- Mikesova, J.; Ondrakova, M.; Jelinkova, I.; Ptacek, J.; Novakova, Z.; Barinka, C. Determining Potency of Inhibitors Targeting Histone Deacetylase 6 by Quantification of Acetylated Tubulin in Cells. Methods Mol. Biol. 2023, 2589, 455–466. [Google Scholar] [CrossRef]
- Bergman, J.A.; Woan, K.; Perez-Villarroel, P.; Villagra, A.; Sotomayor, E.M.; Kozikowski, A.P. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J. Med. Chem. 2012, 55, 9891–9899. [Google Scholar] [CrossRef]
- Furumai, R.; Komatsu, Y.; Nishino, N.; Khochbin, S.; Yoshida, M.; Horinouchi, S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc. Natl. Acad. Sci. USA 2001, 98, 87–92. [Google Scholar] [CrossRef]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef]
- Steimbach, R.R.; Herbst-Gervasoni, C.J.; Lechner, S.; Stewart, T.M.; Klinke, G.; Ridinger, J.; Geraldy, M.N.E.; Tihanyi, G.; Foley, J.R.; Uhrig, U.; et al. Aza-SAHA Derivatives Are Selective Histone Deacetylase 10 Chemical Probes That Inhibit Polyamine Deacetylation and Phenocopy HDAC10 Knockout. J. Am. Chem. Soc. 2022, 144, 18861–18875. [Google Scholar] [CrossRef]
- Herp, D.; Ridinger, J.; Robaa, D.; Shinsky, S.A.; Schmidtkunz, K.; Yesiloglu, T.Z.; Bayer, T.; Steimbach, R.R.; Herbst-Gervasoni, C.J.; Merz, A.; et al. First Fluorescent Acetylspermidine Deacetylation Assay for HDAC10 Identifies Selective Inhibitors with Cellular Target Engagement. Chembiochem 2022, 23, e202200180. [Google Scholar] [CrossRef]
- Zeyen, P.; Zeyn, Y.; Herp, D.; Mahmoudi, F.; Yesiloglu, T.Z.; Erdmann, F.; Schmidt, M.; Robaa, D.; Romier, C.; Ridinger, J.; et al. Identification of histone deacetylase 10 (HDAC10) inhibitors that modulate autophagy in transformed cells. Eur. J. Med. Chem. 2022, 234, 114272. [Google Scholar] [CrossRef]
- Geraldy, M.; Morgen, M.; Sehr, P.; Steimbach, R.R.; Moi, D.; Ridinger, J.; Oehme, I.; Witt, O.; Malz, M.; Nogueira, M.S.; et al. Selective Inhibition of Histone Deacetylase 10: Hydrogen Bonding to the Gatekeeper Residue is Implicated. J. Med. Chem. 2019, 62, 4426–4443. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Shinsky, S.A.; Porter, N.J.; Christianson, D.W. Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun. 2017, 8, 15368. [Google Scholar] [CrossRef] [PubMed]
- Herbst-Gervasoni, C.J.; Steimbach, R.R.; Morgen, M.; Miller, A.K.; Christianson, D.W. Structural Basis for the Selective Inhibition of HDAC10, the Cytosolic Polyamine Deacetylase. ACS Chem. Biol. 2020, 15, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- Herbst-Gervasoni, C.J.; Christianson, D.W. X-ray Crystallographic Snapshots of Substrate Binding in the Active Site of Histone Deacetylase 10. Biochemistry 2021, 60, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Shon, S.; Yoo, H.J.; Suh, D.H.; Bae, D.; Shin, J.; Jun, J.H.; Ha, N.; Song, H.; Choi, Y.I.; et al. Inhibition of histone deacetylase 6 suppresses inflammatory responses and invasiveness of fibroblast-like-synoviocytes in inflammatory arthritis. Arthritis Res. Ther. 2021, 23, 177. [Google Scholar] [CrossRef]
- Huang, W.J.; Chen, C.C.; Chao, S.W.; Yu, C.C.; Yang, C.Y.; Guh, J.H.; Lin, Y.C.; Kuo, C.I.; Yang, P.; Chang, C.I. Synthesis and evaluation of aliphatic-chain hydroxamates capped with osthole derivatives as histone deacetylase inhibitors. Eur. J. Med. Chem. 2011, 46, 4042–4049. [Google Scholar] [CrossRef]
- Shen, S.; Kozikowski, A.P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors—What Some May Have Forgotten or Would Rather Forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef]
- Summers, J.B.; Gunn, B.P.; Mazdiyasni, H.; Goetze, A.M.; Young, P.R.; Bouska, J.B.; Dyer, R.D.; Brooks, D.W.; Carter, G.W. In vivo characterization of hydroxamic acid inhibitors of 5-lipoxygenase. J. Med. Chem. 1987, 30, 2121–2126. [Google Scholar] [CrossRef]
- Ginsel, C.; Plitzko, B.; Froriep, D.; Stolfa, D.A.; Jung, M.; Kubitza, C.; Scheidig, A.J.; Havemeyer, A.; Clement, B. The Involvement of the Mitochondrial Amidoxime Reducing Component (mARC) in the Reductive Metabolism of Hydroxamic Acids. Drug Metab. Dispos. 2018, 46, 1396–1402. [Google Scholar] [CrossRef]
- Dalvie, D.; Cosker, T.; Boyden, T.; Zhou, S.; Schroeder, C.; Potchoiba, M.J. Metabolism distribution and excretion of a matrix metalloproteinase-13 inhibitor, 4-[4-(4-fluorophenoxy)-benzenesulfonylamino]tetrahydropyran-4-carboxylic acid hydroxyamide (CP-544439), in rats and dogs: Assessment of the metabolic profile of CP-544439 in plasma and urine of humans. Drug Metab. Dispos. 2008, 36, 1869–1883. [Google Scholar] [CrossRef]
- Lee, M.S.; Isobe, M. Metabolic activation of the potent mutagen, 2-naphthohydroxamic acid, in Salmonella typhimurium TA98. Cancer Res. 1990, 50, 4300–4307. [Google Scholar]
- Fruhauf, A.; Meyer-Almes, F.J. Non-Hydroxamate Zinc-Binding Groups as Warheads for Histone Deacetylases. Molecules 2021, 26, 5151. [Google Scholar] [CrossRef]
- Zha, Y.L.; Zhang, G.; Li, J.J. Medicinal chemistry insights into non-hydroxamate HDAC6 selective inhibitors. Med. Chem. Res. 2023, 32, 1–14. [Google Scholar] [CrossRef]
- Lee, J.; Younghue, H.; Kim, Y.; Choi, D.; Min, J.; Miseon, B.; Yang, H.; Kim, D. 1,3,4-Oxadiazole Sulfonamide Derivative Compounds as Histone Deacetylase 6 Inhibitor, and the Pharmaceutical Composition Comprising the Same. WO2017018803A1, 2 February 2017. [Google Scholar]
- Kim, Y.; Lee, C.S.; Oh, J.T.; Hyeseung, S.; Choi, J.; Lee, J. Oxadiazole Amine Derivative Compounds as Histone Deacetylase 6 Inhibitor, and the Pharmaceutical Composition Comprising the Same. WO2017065473, 20 April 2017. [Google Scholar]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Keuler, T.; Konig, B.; Buckreiss, N.; Kraft, F.B.; Konig, P.; Schaker-Hubner, L.; Steinebach, C.; Bendas, G.; Gutschow, M.; Hansen, F.K. Development of the first non-hydroxamate selective HDAC6 degraders. Chem. Commun. 2022, 58, 11087–11090. [Google Scholar] [CrossRef]
- Maciolek, C.M.; Ma, B.; Menzel, K.; Laliberte, S.; Bateman, K.; Krolikowski, P.; Gibson, C.R. Novel cytochrome P450-mediated ring opening of the 1,3,4-oxadiazole in setileuton, a 5-lipoxygenase inhibitor. Drug Metab. Dispos. 2011, 39, 763–770. [Google Scholar] [CrossRef]
- Machleidt, T.; Woodroofe, C.C.; Schwinn, M.K.; Mendez, J.; Robers, M.B.; Zimmerman, K.; Otto, P.; Daniels, D.L.; Kirkland, T.A.; Wood, K.V. NanoBRET—A Novel BRET Platform for the Analysis of Protein-Protein Interactions. ACS Chem. Biol. 2015, 10, 1797–1804. [Google Scholar] [CrossRef]
- Robers, M.B.; Dart, M.L.; Woodroofe, C.C.; Zimprich, C.A.; Kirkland, T.A.; Machleidt, T.; Kupcho, K.R.; Levin, S.; Hartnett, J.R.; Zimmerman, K.; et al. Target engagement and drug residence time can be observed in living cells with BRET. Nat. Commun. 2015, 6, 10091. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef]
- Schrump, D.S. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: Mechanisms and potential clinical implications. Clin. Cancer Res. 2009, 15, 3947–3957. [Google Scholar] [CrossRef]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 [nM] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| HDAC6 GAK | HDAC6 Literature * | HDAC1 GAK | HDAC1 RHKK | HDAC1 Literature * | SI (HD6/HD1) | SI (HD6/HD1) | HDAC10 | SI (HD6/HD10) | |
| GAK | RHKK | ||||||||
| Next A | 3.8 | 5.02 | 362 | 4959 | 3020 | 95.3 | 1305 | 11 | 2.9 |
| ACY-775 | 1.11 | 7.5 | 1194 | 21,700 | 2123 | 1076 | 19,550 | 34 | 30.6 |
| ACY-1215 | 2.37 | 4.7 | 106 | 625 | 58 | 44.7 | 263.6 | 18.2 | 7.7 |
| TSA | 0.41 | 8.6 | 0.71 | 8.13 | 6 | 1.73 | 19.84 | 4.3 | 10.5 |
| Tub A | 4.65 | 15 | 2288 | 24,680 | 16,400 | 492 | 5308 | 3.8 | 0.82 |
| Tubacin | 35.9 | 4 | 102 | 15,070 | 1400 | 2.84 | 420 | 678 | 18.9 |
| 6 | 0.35 | NA | 49.2 | 281 | NA | 141 | 803 | 68.3 | 195 |
| 7 | 2.06 | NA | >50,000 | >50,000 | NA | >25,000 | >25,000 | >50,000 | >25,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ptacek, J.; Snajdr, I.; Schimer, J.; Kutil, Z.; Mikesova, J.; Baranova, P.; Havlinova, B.; Tueckmantel, W.; Majer, P.; Kozikowski, A.; et al. Selectivity of Hydroxamate- and Difluoromethyloxadiazole-Based Inhibitors of Histone Deacetylase 6 In Vitro and in Cells. Int. J. Mol. Sci. 2023, 24, 4720. https://doi.org/10.3390/ijms24054720
Ptacek J, Snajdr I, Schimer J, Kutil Z, Mikesova J, Baranova P, Havlinova B, Tueckmantel W, Majer P, Kozikowski A, et al. Selectivity of Hydroxamate- and Difluoromethyloxadiazole-Based Inhibitors of Histone Deacetylase 6 In Vitro and in Cells. International Journal of Molecular Sciences. 2023; 24(5):4720. https://doi.org/10.3390/ijms24054720
Chicago/Turabian StylePtacek, Jakub, Ivan Snajdr, Jiri Schimer, Zsofia Kutil, Jana Mikesova, Petra Baranova, Barbora Havlinova, Werner Tueckmantel, Pavel Majer, Alan Kozikowski, and et al. 2023. "Selectivity of Hydroxamate- and Difluoromethyloxadiazole-Based Inhibitors of Histone Deacetylase 6 In Vitro and in Cells" International Journal of Molecular Sciences 24, no. 5: 4720. https://doi.org/10.3390/ijms24054720
APA StylePtacek, J., Snajdr, I., Schimer, J., Kutil, Z., Mikesova, J., Baranova, P., Havlinova, B., Tueckmantel, W., Majer, P., Kozikowski, A., & Barinka, C. (2023). Selectivity of Hydroxamate- and Difluoromethyloxadiazole-Based Inhibitors of Histone Deacetylase 6 In Vitro and in Cells. International Journal of Molecular Sciences, 24(5), 4720. https://doi.org/10.3390/ijms24054720