
Using the Proteomics Toolbox to Resolve Topology and Dynamics of Compartmentalized cAMP Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
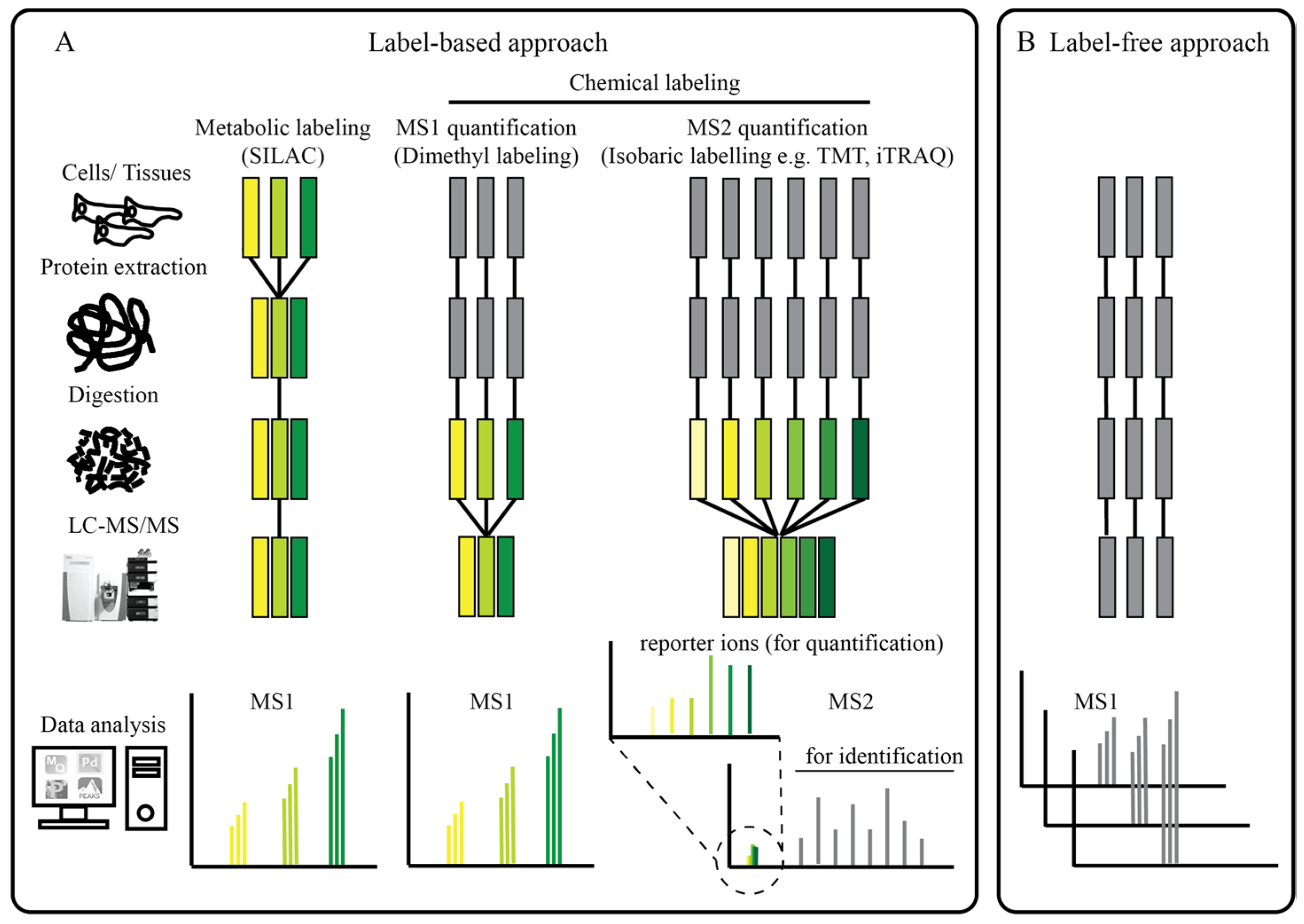
2. The Proteomics Toolbox

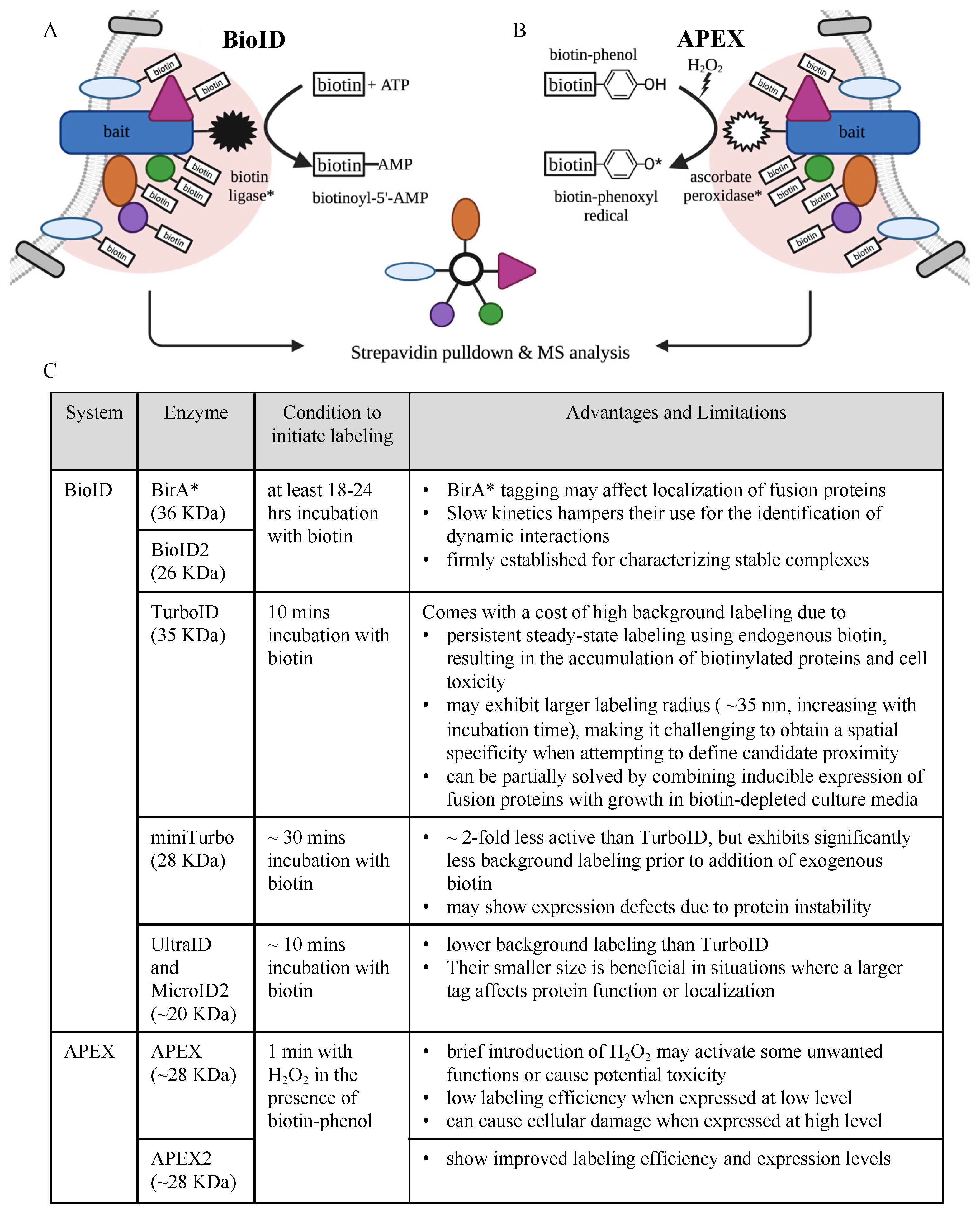
2.1. Protein Interaction and Proximity Profiling—Basic Principles
2.2. Phosphoproteomics—An Overview
3. Application of Proteomics Methodologies to Study Compartmentalized cAMP Signaling
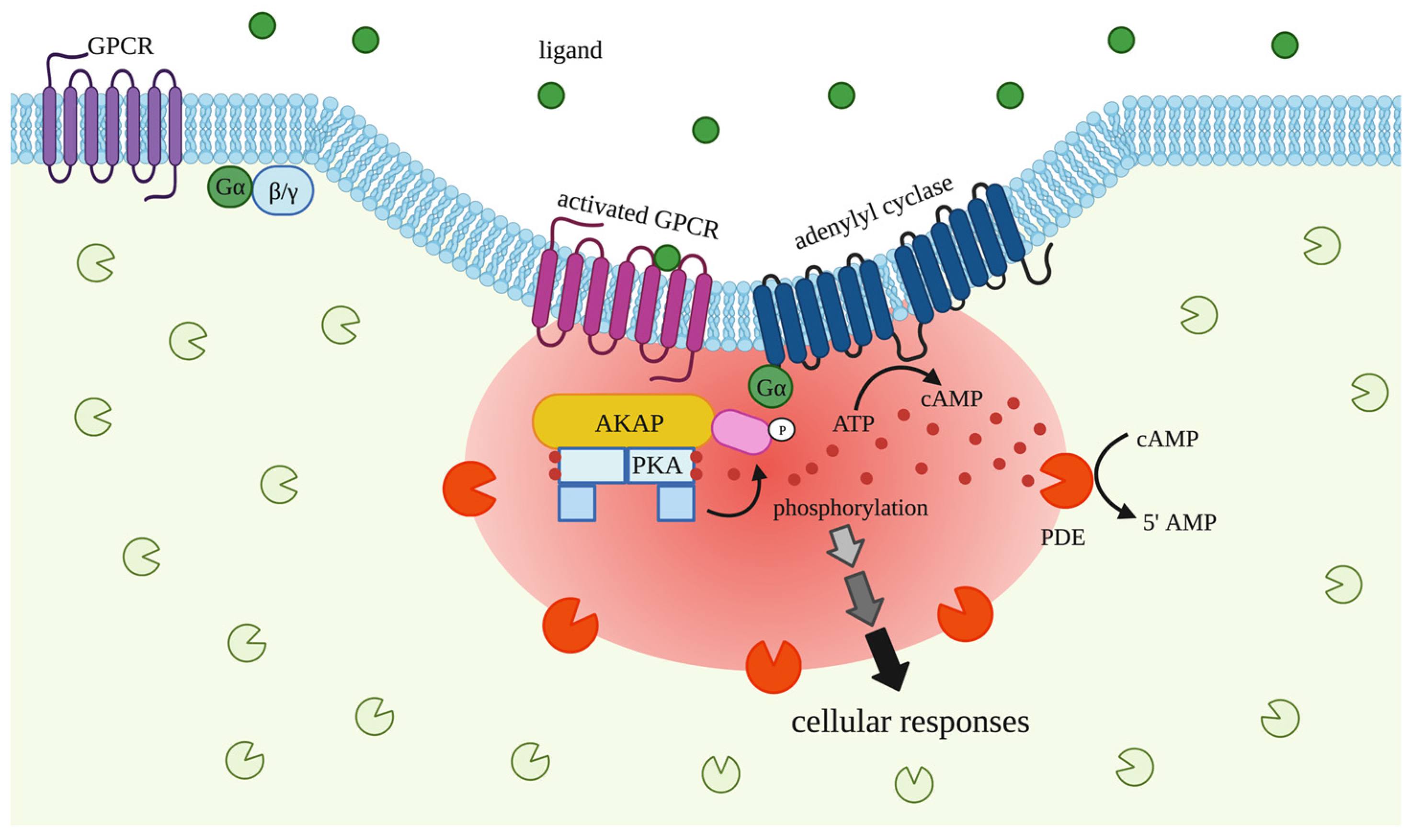
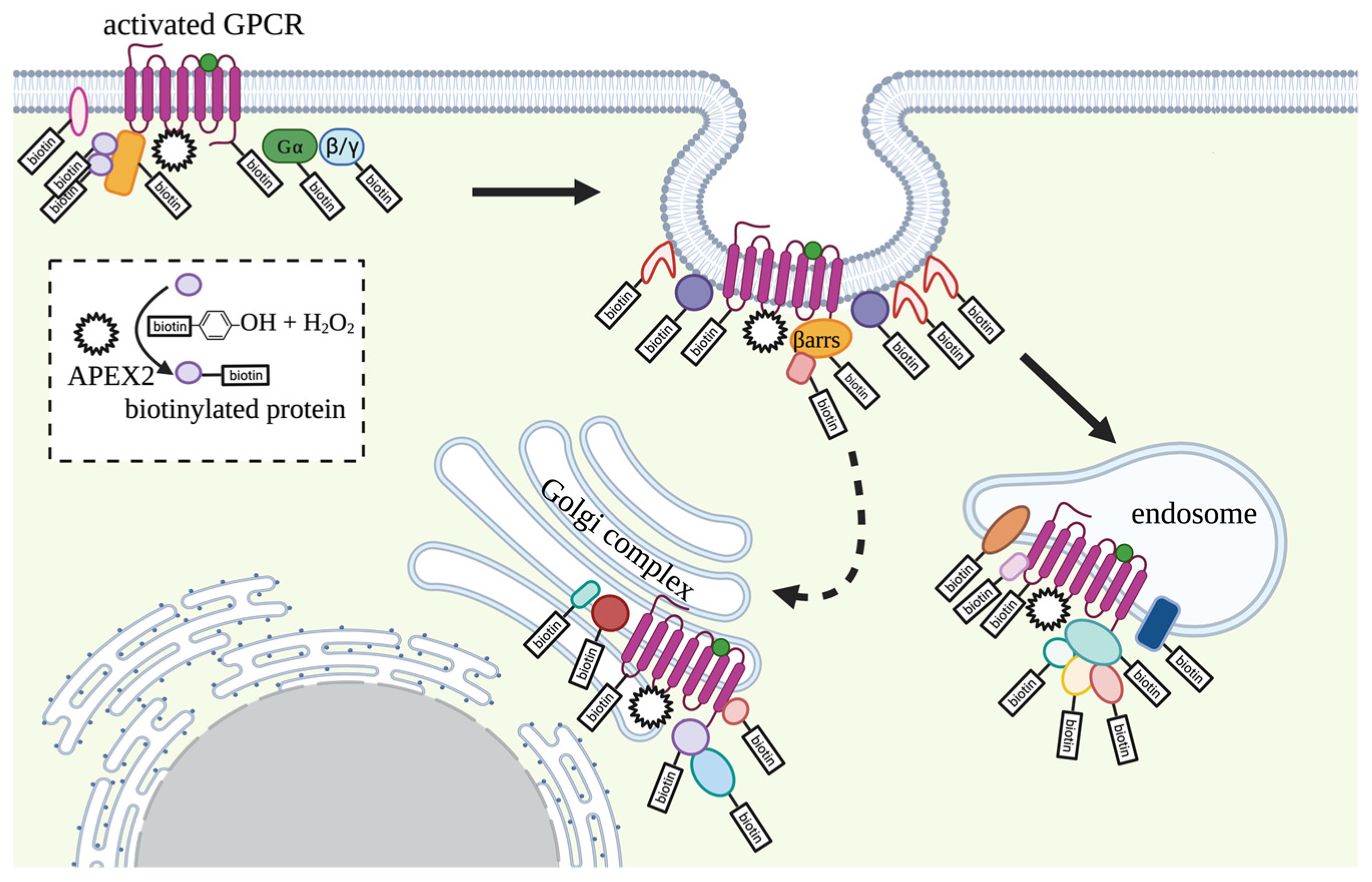
3.1. G Proteins Coupled Receptors (GPCRs)
3.2. A-Kinase Anchoring Proteins (AKAPs)
3.3. cAMP-Dependent Protein Kinase (PKA)
3.4. Phosphodiesterases (PDEs)
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Sriram, K.; Insel, P.A.G. Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP–PKA–CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the cAMP Signaling Pathway. Pharmacol. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef]
- Buxton, I.L.; Brunton, L.L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 1983, 258, 10233–10239. [Google Scholar] [CrossRef]
- Theurkauf, W.E.; Vallee, R.B. Molecular characterization of the cAMP-dependent protein kinase bound to microtubule-associated protein 2. J. Biol. Chem. 1982, 257, 3284–3290. [Google Scholar] [CrossRef]
- Lohmann, S.M.; DeCamilli, P.; Einig, I.; Walter, U. High-affinity binding of the regulatory subunit (RII) of cAMP-dependent protein kinase to microtubule-associated and other cellular proteins. Proc. Natl. Acad. Sci. USA 1984, 81, 6723–6727. [Google Scholar] [CrossRef] [PubMed]
- Omar, M.H.; Scott, J.D. AKAP Signaling Islands: Venues for Precision Pharmacology. Trends Pharmacol. Sci. 2020, 41, 933–946. [Google Scholar] [CrossRef]
- Anton, S.E.; Kayser, C.; Maiellaro, I.; Nemec, K.; Möller, J.; Koschinski, A.; Zaccolo, M.; Annibale, P.; Falcke, M.; Lohse, M.J.; et al. Receptor-associated independent cAMP nanodomains mediate spatiotemporal specificity of GPCR signaling. Cell 2022, 185, 1130–1142.e1111. [Google Scholar] [CrossRef]
- Bock, A.; Annibale, P.; Konrad, C.; Hannawacker, A.; Anton, S.E.; Maiellaro, I.; Zabel, U.; Sivaramakrishnan, S.; Falcke, M.; Lohse, M.J. Optical Mapping of cAMP Signaling at the Nanometer Scale. Cell 2020, 182, 1519–1530.e1517. [Google Scholar] [CrossRef]
- Surdo, N.C.; Berrera, M.; Koschinski, A.; Brescia, M.; Machado, M.R.; Carr, C.; Wright, P.; Gorelik, J.; Morotti, S.; Grandi, E.; et al. FRET biosensor uncovers cAMP nano-domains at β-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 2017, 8, 15031. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Day, P.; Agrawal, R.; Bruss, M.D.; Granier, S.; Wang, Y.L.; Rasmussen, S.G.; Horner, K.; Wang, P.; Lei, T.; et al. Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 2008, 27, 384–393. [Google Scholar] [CrossRef]
- Berthouze-Duquesnes, M.; Lucas, A.; Saulière, A.; Sin, Y.Y.; Laurent, A.C.; Galés, C.; Baillie, G.; Lezoualc’h, F. Specific interactions between Epac1, β-arrestin2 and PDE4D5 regulate β-adrenergic receptor subtype differential effects on cardiac hypertrophic signaling. Cell. Signal. 2013, 25, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Mika, D.; Blanchard, E.; Day, P.; Conti, M. β1-adrenergic receptor antagonists signal via PDE4 translocation. EMBO Rep. 2013, 14, 276–283. [Google Scholar] [CrossRef]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef]
- Duong, V.A.; Park, J.M.; Lee, H. Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics. Int. J. Mol. Sci. 2020, 21, 1524. [Google Scholar] [CrossRef]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Krijgsveld, J.; Ketting, R.F.; Mahmoudi, T.; Johansen, J.; Artal-Sanz, M.; Verrijzer, C.P.; Plasterk, R.H.; Heck, A.J. Metabolic labeling of C. elegans and D. melanogaster for quantitative proteomics. Nat. Biotechnol. 2003, 21, 927–931. [Google Scholar] [CrossRef]
- Yi, E.C.; Li, X.-j.; Cooke, K.; Lee, H.; Raught, B.; Page, A.; Aneliunas, V.; Hieter, P.; Goodlett, D.R.; Aebersold, R. Increased quantitative proteome coverage with 13C/12C-based, acid-cleavable isotope-coded affinity tag reagent and modified data acquisition scheme. Proteomics 2005, 5, 380–387. [Google Scholar] [CrossRef]
- Xiang, F.; Ye, H.; Chen, R.; Fu, Q.; Li, L. N,N-Dimethyl Leucines as Novel Isobaric Tandem Mass Tags for Quantitative Proteomics and Peptidomics. Anal. Chem. 2010, 82, 2817–2825. [Google Scholar] [CrossRef]
- Boersema, P.J.; Raijmakers, R.; Lemeer, S.; Mohammed, S.; Heck, A.J.R. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009, 4, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents*. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Imami, K.; Sugiyama, N.; Tomita, M.; Ishihama, Y. Quantitative proteome and phosphoproteome analyses of cultured cells based on SILAC labeling without requirement of serum dialysis. Mol. BioSyst. 2010, 6, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Overmyer, K.A.; Tyanova, S.; Hebert, A.S.; Westphall, M.S.; Cox, J.; Coon, J.J. Multiplexed proteome analysis with neutron-encoded stable isotope labeling in cells and mice. Nat. Protoc. 2018, 13, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Kovanich, D.; Cappadona, S.; Raijmakers, R.; Mohammed, S.; Scholten, A.; Heck, A.J.R. Applications of stable isotope dimethyl labeling in quantitative proteomics. Anal. Bioanal. Chem. 2012, 404, 991–1009. [Google Scholar] [CrossRef]
- Li, J.; Van Vranken, J.G.; Pontano Vaites, L.; Schweppe, D.K.; Huttlin, E.L.; Etienne, C.; Nandhikonda, P.; Viner, R.; Robitaille, A.M.; Thompson, A.H.; et al. TMTpro reagents: A set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 2020, 17, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Choe, L.; D’Ascenzo, M.; Relkin, N.R.; Pappin, D.; Ross, P.; Williamson, B.; Guertin, S.; Pribil, P.; Lee, K.H. 8-plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer’s disease. Proteomics 2007, 7, 3651–3660. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially Modified Protein Abundance Index (emPAI) for Estimation of Absolute Protein Amount in Proteomics by the Number of Sequenced Peptides per Protein. Mol. Cell. Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Palomba, A.; Abbondio, M.; Fiorito, G.; Uzzau, S.; Pagnozzi, D.; Tanca, A. Comparative Evaluation of MaxQuant and Proteome Discoverer MS1-Based Protein Quantification Tools. J. Proteome Res. 2021, 20, 3497–3507. [Google Scholar] [CrossRef]
- Lazar, C.; Gatto, L.; Ferro, M.; Bruley, C.; Burger, T. Accounting for the Multiple Natures of Missing Values in Label-Free Quantitative Proteomics Data Sets to Compare Imputation Strategies. J. Proteome Res. 2016, 15, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Tabb, D.L.; Vega-Montoto, L.; Rudnick, P.A.; Variyath, A.M.; Ham, A.J.; Bunk, D.M.; Kilpatrick, L.E.; Billheimer, D.D.; Blackman, R.K.; Cardasis, H.L.; et al. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2010, 9, 761–776. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- Meier, F.; Geyer, P.E.; Virreira Winter, S.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448. [Google Scholar] [CrossRef]
- Collins, B.C.; Hunter, C.L.; Liu, Y.; Schilling, B.; Rosenberger, G.; Bader, S.L.; Chan, D.W.; Gibson, B.W.; Gingras, A.-C.; Held, J.M.; et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017, 8, 291. [Google Scholar] [CrossRef]
- Low, T.Y.; Syafruddin, S.E.; Mohtar, M.A.; Vellaichamy, A.; NS, A.R.; Pung, Y.F.; Tan, C.S.H. Recent progress in mass spectrometry-based strategies for elucidating protein-protein interactions. Cell. Mol. Life Sci. 2021, 78, 5325–5339. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-C.; Bååth, J.A.; Bastle, R.M.; Bhattacharjee, S.; Cantoria, M.J.; Dornan, M.; Gamero-Estevez, E.; Ford, L.; Halova, L.; Kernan, J.; et al. Impact of detergents on membrane protein complex isolation. J. Proteome. Res. 2018, 17, 348–358. [Google Scholar] [CrossRef] [PubMed]
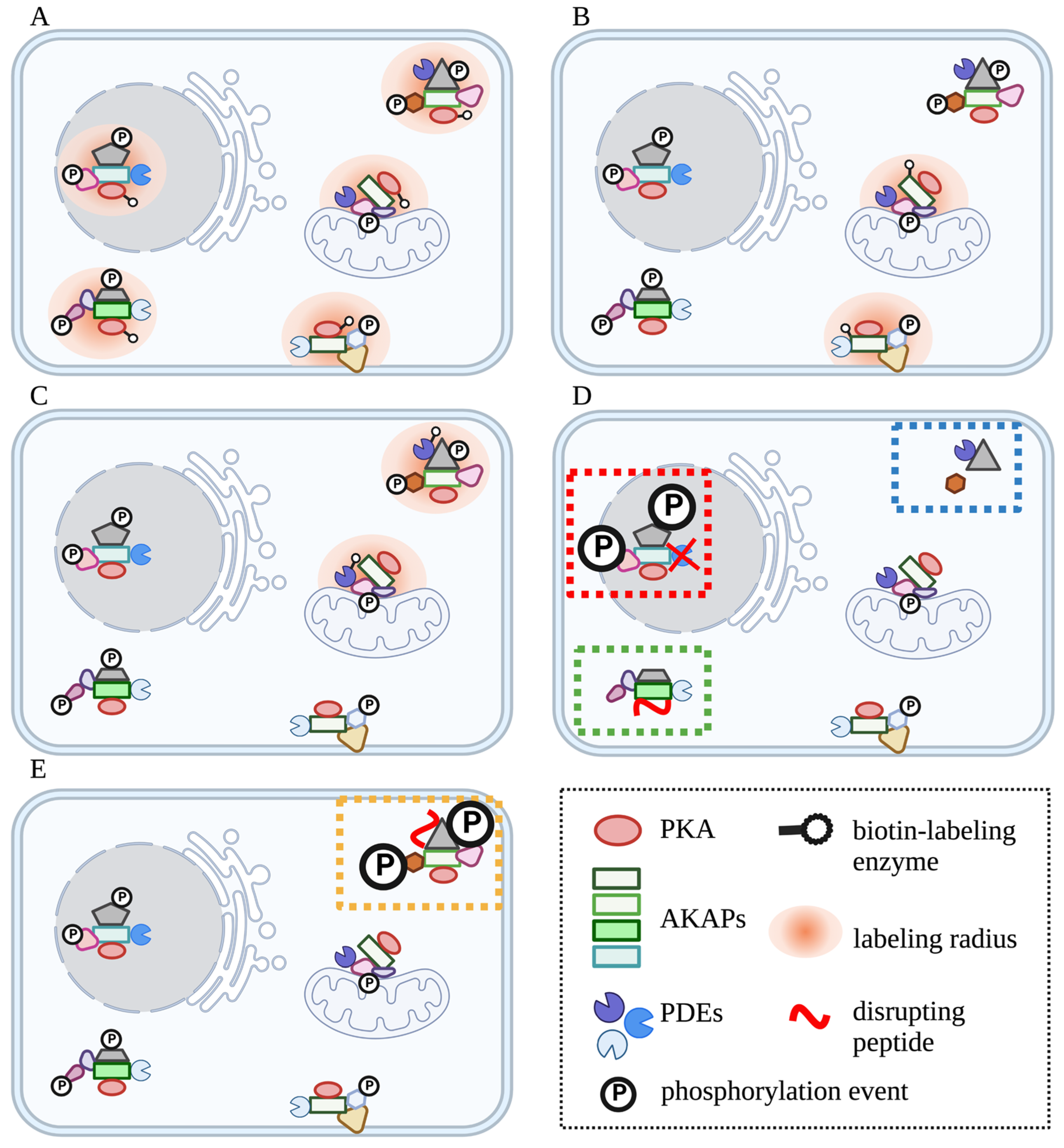
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Samavarchi-Tehrani, P.; Abdouni, H.; Samson, R.; Gingras, A.-C. A Versatile Lentiviral Delivery Toolkit for Proximity-dependent Biotinylation in Diverse Cell Types. Mol. Cell. Proteom. 2018, 17, 2256–2269. [Google Scholar] [CrossRef]
- Vandemoortele, G.; De Sutter, D.; Moliere, A.; Pauwels, J.; Gevaert, K.; Eyckerman, S. A Well-Controlled BioID Design for Endogenous Bait Proteins. J. Proteome Res. 2019, 18, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Kc, B.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Kubitz, L.; Bitsch, S.; Zhao, X.; Schmitt, K.; Deweid, L.; Roehrig, A.; Barazzone, E.C.; Valerius, O.; Kolmar, H.; Béthune, J. Engineering of ultraID, a compact and hyperactive enzyme for proximity-dependent biotinylation in living cells. Commun. Biol. 2022, 5, 657. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.S.; Chafin, L.; Farkas, D.; Adair, J.; Elhance, A.; Farkas, L.; Bednash, J.S.; Londino, J.D. MicroID2: A Novel Biotin Ligase Enables Rapid Proximity-Dependent Proteomics. Mol. Cell. Proteom. 2022, 21, 100256. [Google Scholar] [CrossRef]
- May, D.G.; Scott, K.L.; Campos, A.R.; Roux, K.J. Comparative Application of BioID and TurboID for Protein-Proximity Biotinylation. Cells 2020, 9, 1070. [Google Scholar] [CrossRef]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef]
- Trinkle-Mulcahy, L. Recent advances in proximity-based labeling methods for interactome mapping. F1000Res 2019, 8, F1000. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.A.; Chen, C.L.; Perrimon, N. Proximity-dependent labeling methods for proteomic profiling in living cells: An update. Wiley Interdiscip. Rev. Dev. Biol. 2021, 10, e392. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.-C.; Abe, K.T.; Raught, B. Getting to know the neighborhood: Using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol. 2019, 48, 44–54. [Google Scholar] [CrossRef]
- Chua, X.Y.; Aballo, T.; Elnemer, W.; Tran, M.; Salomon, A. Quantitative Interactomics of Lck-TurboID in Living Human T Cells Unveils T Cell Receptor Stimulation-Induced Proximal Lck Interactors. J. Proteome Res. 2021, 20, 715–726. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, E.L.; Giansanti, P.; Altelaar, A.F.; Heck, A.J. Single-step enrichment by Ti4+-IMAC and label-free quantitation enables in-depth monitoring of phosphorylation dynamics with high reproducibility and temporal resolution. Mol. Cell. Proteom. 2014, 13, 2426–2434. [Google Scholar] [CrossRef]
- Humphrey, S.J.; Azimifar, S.B.; Mann, M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol. 2015, 33, 990–995. [Google Scholar] [CrossRef]
- Dephoure, N.; Gould, K.L.; Gygi, S.P.; Kellogg, D.R. Mapping and analysis of phosphorylation sites: A quick guide for cell biologists. Mol. Biol. Cell 2013, 24, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Low, T.Y.; Mohtar, M.A.; Lee, P.Y.; Omar, N.; Zhou, H.; Ye, M. Widening the bottleneck of phosphoproteomics: Evolving strategies for phosphopeptide enrichment. Mass Spectrom. Rev. 2021, 40, 309–333. [Google Scholar] [CrossRef]
- Gauci, S.; Helbig, A.O.; Slijper, M.; Krijgsveld, J.; Heck, A.J.; Mohammed, S. Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal. Chem. 2009, 81, 4493–4501. [Google Scholar] [CrossRef]
- Villén, J.; Gygi, S.P. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 2008, 3, 1630–1638. [Google Scholar] [CrossRef]
- Zhou, H.; Low, T.Y.; Hennrich, M.L.; van der Toorn, H.; Schwend, T.; Zou, H.; Mohammed, S.; Heck, A.J. Enhancing the identification of phosphopeptides from putative basophilic kinase substrates using Ti (IV) based IMAC enrichment. Mol. Cell. Proteom. 2011, 10, M110.006452. [Google Scholar] [CrossRef]
- Humphrey, S.J.; Karayel, O.; James, D.E.; Mann, M. High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat. Protoc. 2018, 13, 1897–1916. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef]
- Pinkse, M.W.; Uitto, P.M.; Hilhorst, M.J.; Ooms, B.; Heck, A.J. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal. Chem. 2004, 76, 3935–3943. [Google Scholar] [CrossRef]
- Matheron, L.; van den Toorn, H.; Heck, A.J.; Mohammed, S. Characterization of biases in phosphopeptide enrichment by Ti(4+)-immobilized metal affinity chromatography and TiO2 using a massive synthetic library and human cell digests. Anal. Chem. 2014, 86, 8312–8320. [Google Scholar] [CrossRef]
- Yue, X.; Schunter, A.; Hummon, A.B. Comparing multistep immobilized metal affinity chromatography and multistep TiO2 methods for phosphopeptide enrichment. Anal. Chem. 2015, 87, 8837–8844. [Google Scholar] [CrossRef]
- Giansanti, P.; Stokes, M.P.; Silva, J.C.; Scholten, A.; Heck, A.J. Interrogating cAMP-dependent kinase signaling in Jurkat T cells via a protein kinase A targeted immune-precipitation phosphoproteomics approach. Mol. Cell. Proteom. 2013, 12, 3350–3359. [Google Scholar] [CrossRef] [PubMed]
- Potel, C.M.; Lemeer, S.; Heck, A.J.R. Phosphopeptide Fragmentation and Site Localization by Mass Spectrometry: An Update. Anal. Chem. 2019, 91, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Locard-Paulet, M.; Bouyssié, D.; Froment, C.; Burlet-Schiltz, O.; Jensen, L.J. Comparing 22 Popular Phosphoproteomics Pipelines for Peptide Identification and Site Localization. J. Proteome Res. 2020, 19, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Savage, S.R.; Zhang, B. Using phosphoproteomics data to understand cellular signaling: A comprehensive guide to bioinformatics resources. Clin. Proteom. 2020, 17, 27. [Google Scholar] [CrossRef] [PubMed]
- Golkowski, M.; Shimizu-Albergine, M.; Suh, H.W.; Beavo, J.A.; Ong, S.E. Studying mechanisms of cAMP and cyclic nucleotide phosphodiesterase signaling in Leydig cell function with phosphoproteomics. Cell. Signal. 2016, 28, 764–778. [Google Scholar] [CrossRef]
- Beltejar, M.-C.G.; Lau, H.-T.; Golkowski, M.G.; Ong, S.E.; Beavo, J.A. Analyses of PDE-regulated phosphoproteomes reveal unique and specific cAMP-signaling modules in T cells. Proc. Natl. Acad. Sci. USA 2017, 114, E6240–E6249. [Google Scholar] [CrossRef]
- Ellisdon, A.M.; Halls, M.L. Compartmentalization of GPCR signalling controls unique cellular responses. Biochem. Soc. Trans. 2016, 44, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Eason, M.G.; Jacinto, M.T.; Liggett, S.B. Contribution of ligand structure to activation of alpha 2-adrenergic receptor subtype coupling to Gs. Mol. Pharmacol. 1994, 45, 696–702. [Google Scholar]
- Hoffmann, C.; Ziegler, N.; Reiner, S.; Krasel, C.; Lohse, M.J. Agonist-selective, receptor-specific interaction of human P2Y receptors with beta-arrestin-1 and -2. J. Biol. Chem. 2008, 283, 30933–30941. [Google Scholar] [CrossRef] [PubMed]
- Strachan, R.T.; Sun, J.P.; Rominger, D.H.; Violin, J.D.; Ahn, S.; Rojas Bie Thomsen, A.; Zhu, X.; Kleist, A.; Costa, T.; Lefkowitz, R.J. Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J. Biol. Chem. 2014, 289, 14211–14224. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; DeWire, S.M.; Yamashita, D.; Rominger, D.H.; Nguyen, L.; Schiller, K.; Whalen, E.J.; Gowen, M.; Lark, M.W. Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 2010, 335, 572–579. [Google Scholar] [CrossRef]
- Peña, K.A. Endosomal parathyroid hormone receptor signaling. Am. J. Physiol. Cell Physiol. 2022, 323, C783–C790. [Google Scholar] [CrossRef] [PubMed]
- Godbole, A.; Lyga, S.; Lohse, M.J.; Calebiro, D. Internalized TSH receptors en route to the TGN induce local G(s)-protein signaling and gene transcription. Nat. Commun. 2017, 8, 443. [Google Scholar] [CrossRef]
- Peng, G.E.; Pessino, V.; Huang, B.; von Zastrow, M. Spatial decoding of endosomal cAMP signals by a metastable cytoplasmic PKA network. Nat. Chem. Biol. 2021, 17, 558–566. [Google Scholar] [CrossRef]
- Mohammad Nezhady, M.A.; Rivera, J.C.; Chemtob, S. Location Bias as Emerging Paradigm in GPCR Biology and Drug Discovery. iScience 2020, 23, 101643. [Google Scholar] [CrossRef] [PubMed]
- Head, B.P.; Patel, H.H.; Roth, D.M.; Lai, N.C.; Niesman, I.R.; Farquhar, M.G.; Insel, P.A. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J. Biol. Chem. 2005, 280, 31036–31044. [Google Scholar] [CrossRef]
- Liang, W.; Curran, P.K.; Hoang, Q.; Moreland, R.T.; Fishman, P.H. Differences in endosomal targeting of human (beta)1- and (beta)2-adrenergic receptors following clathrin-mediated endocytosis. J. Cell. Sci. 2004, 117, 723–734. [Google Scholar] [CrossRef]
- Irannejad, R.; Tomshine, J.C.; Tomshine, J.R.; Chevalier, M.; Mahoney, J.P.; Steyaert, J.; Rasmussen, S.G.; Sunahara, R.K.; El-Samad, H.; Huang, B.; et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature 2013, 495, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.B.; Filardo, E.J. Trans-Golgi Network (TGN) as a regulatory node for β1-adrenergic receptor (β1AR) down-modulation and recycling. J. Biol. Chem. 2012, 287, 14178–14191. [Google Scholar] [CrossRef]
- Plouffe, B.; Thomsen, A.R.B.; Irannejad, R. Emerging Role of Compartmentalized G Protein-Coupled Receptor Signaling in the Cardiovascular Field. ACS Pharmacol. Transl. Sci. 2020, 3, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef]
- Schleicher, K.; Zaccolo, M. Axelrod Symposium 2019: Phosphoproteomic Analysis of G-Protein–Coupled Pathways. Mol. Pharmacol. 2021, 99, 383. [Google Scholar] [CrossRef]
- Lobingier, B.T.; Hüttenhain, R.; Eichel, K.; Miller, K.B.; Ting, A.Y.; von Zastrow, M.; Krogan, N.J. An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 2017, 169, 350–360.e312. [Google Scholar] [CrossRef]
- Paek, J.; Kalocsay, M.; Staus, D.P.; Wingler, L.; Pascolutti, R.; Paulo, J.A.; Gygi, S.P.; Kruse, A.C. Multidimensional Tracking of GPCR Signaling via Peroxidase-Catalyzed Proximity Labeling. Cell 2017, 169, 338–349.e311. [Google Scholar] [CrossRef]
- Pfeiffer, C.T.; Wang, J.; Paulo, J.A.; Jiang, X.; Gygi, S.P.; Rockman, H.A. Mapping Angiotensin II Type 1 Receptor-Biased Signaling Using Proximity Labeling and Proteomics Identifies Diverse Actions of Biased Agonists. J. Proteome Res. 2021, 20, 3256–3267. [Google Scholar] [CrossRef]
- Polacco, B.J.; Lobingier, B.T.; Blythe, E.E.; Abreu, N.; Xu, J.; Li, Q.; Naing, Z.Z.C.; Shoichet, B.K.; Levitz, J.; Krogan, N.J.; et al. Profiling the diversity of agonist-selective effects on the proximal proteome environment of G protein-coupled receptors. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ribeiro-Oliveira, R.; Vojtek, M.; Gonçalves-Monteiro, S.; Vieira-Rocha, M.S.; Sousa, J.B.; Gonçalves, J.; Diniz, C. Nuclear G-protein-coupled receptors as putative novel pharmacological targets. Drug. Discov. Today 2019, 24, 2192–2201. [Google Scholar] [CrossRef]
- Clister, T.; Greenwald, E.C.; Baillie, G.S.; Zhang, J. AKAP95 Organizes a Nuclear Microdomain to Control Local cAMP for Regulating Nuclear PKA. Cell Chem. Biol. 2019, 26, 885–891.e884. [Google Scholar] [CrossRef]
- Newlon, M.G.; Roy, M.; Morikis, D.; Carr, D.W.; Westphal, R.; Scott, J.D.; Jennings, P.A. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 2001, 20, 1651–1662. [Google Scholar] [CrossRef]
- Kammerer, S.; Burns-Hamuro, L.L.; Ma, Y.; Hamon, S.C.; Canaves, J.M.; Shi, M.M.; Nelson, M.R.; Sing, C.F.; Cantor, C.R.; Taylor, S.S.; et al. Amino acid variant in the kinase binding domain of dual-specific A kinase-anchoring protein 2: A disease susceptibility polymorphism. Proc. Natl. Acad. Sci. USA 2003, 100, 4066–4071. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, S.V.; Jadhav, S.M.; McConnell, B.K. Polymorphisms/Mutations in A-Kinase Anchoring Proteins (AKAPs): Role in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.D.; Omar, M.H.; Nygren, P.J.; Soughayer, J.; Hoshi, N.; Lau, H.T.; Snyder, C.G.; Branon, T.C.; Ghosh, D.; Langeberg, L.K.; et al. Single nucleotide polymorphisms alter kinase anchoring and the subcellular targeting of A-kinase anchoring proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E11465–E11474. [Google Scholar] [CrossRef]
- Kjällquist, U.; Erlandsson, R.; Tobin, N.P.; Alkodsi, A.; Ullah, I.; Stålhammar, G.; Karlsson, E.; Hatschek, T.; Hartman, J.; Linnarsson, S.; et al. Exome sequencing of primary breast cancers with paired metastatic lesions reveals metastasis-enriched mutations in the A-kinase anchoring protein family (AKAPs). BMC Cancer 2018, 18, 174. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Marquardt, M.L.; Tester, D.J.; Sampson, K.J.; Ackerman, M.J.; Kass, R.S. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 20990–20995. [Google Scholar] [CrossRef]
- Aye, T.T.; Soni, S.; van Veen, T.A.B.; van der Heyden, M.A.G.; Cappadona, S.; Varro, A.; de Weger, R.A.; de Jonge, N.; Vos, M.A.; Heck, A.J.R.; et al. Reorganized PKA-AKAP associations in the failing human heart. J. Mol. Cell. Cardiol. 2012, 52, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Wild, A.R.; Dell’Acqua, M.L. Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol. Ther. 2018, 185, 99–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, D.; Tu, C.; Meng, L.; Tan, Y.; Ji, Z.; Cheng, J.; Lu, G.; Lin, G.; Zhang, H.; et al. Loss-of-function missense variant of AKAP4 induced male infertility through reduced interaction with QRICH2 during sperm flagella development. Hum. Mol. Genet. 2021, 31, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Scholten, A.; Poh, M.K.; van Veen, T.A.; van Breukelen, B.; Vos, M.A.; Heck, A.J. Analysis of the cGMP/cAMP interactome using a chemical proteomics approach in mammalian heart tissue validates sphingosine kinase type 1-interacting protein as a genuine and highly abundant AKAP. J. Proteome Res. 2006, 5, 1435–1447. [Google Scholar] [CrossRef]
- Scholten, A.; van Veen, T.A.; Vos, M.A.; Heck, A.J. Diversity of cAMP-dependent protein kinase isoforms and their anchoring proteins in mouse ventricular tissue. J. Proteome Res. 2007, 6, 1705–1717. [Google Scholar] [CrossRef]
- Day, M.E.; Gaietta, G.M.; Sastri, M.; Koller, A.; Mackey, M.R.; Scott, J.D.; Perkins, G.A.; Ellisman, M.H.; Taylor, S.S. Isoform-specific targeting of PKA to multivesicular bodies. J. Cell. Biol. 2011, 193, 347–363. [Google Scholar] [CrossRef]
- Raslan, Z.; Magwenzi, S.; Aburima, A.; Taskén, K.; Naseem, K.M. Targeting of type I protein kinase A to lipid rafts is required for platelet inhibition by the 3′,5′-cyclic adenosine monophosphate-signaling pathway. J. Thromb. Haemost. 2015, 13, 1721–1734. [Google Scholar] [CrossRef]
- Aye, T.T.; Mohammed, S.; van den Toorn, H.W.P.; van Veen, T.A.B.; van der Heyden, M.A.G.; Scholten, A.; Heck, A.J.R. Selectivity in Enrichment of cAMP-dependent Protein Kinase Regulatory Subunits Type I and Type II and Their Interactors Using Modified cAMP Affinity Resins. Mol. Cell. Proteom. 2009, 8, 1016–1028. [Google Scholar] [CrossRef]
- Kovanich, D.; Aye, T.T.; Heck, A.J.; Scholten, A. Probing the specificity of protein-protein interactions by quantitative chemical proteomics. Methods Mol. Biol. 2012, 803, 167–181. [Google Scholar]
- Margarucci, L.; Roest, M.; Preisinger, C.; Bleijerveld, O.B.; van Holten, T.C.; Heck, A.J.R.; Scholten, A. Collagen stimulation of platelets induces a rapid spatial response of cAMP and cGMP signaling scaffolds. Mol. BioSyst. 2011, 7, 2311–2319. [Google Scholar] [CrossRef]
- Soni, S.; Corradini, E.; Van Der Nagel, R.; Boulaksil, M.; Heck, A.J.R.; Scholten, A.J.R.; Vos, M.A.; Van Veen, T.A.B. Anchored cAMP signalling in progression from hypertrophy to heart failure in a rat model of pressure overload. Eur. Heart J. 2013, 34, P5715. [Google Scholar] [CrossRef]
- Schwarz, U.R.; Walter, U.; Eigenthaler, M. Taming platelets with cyclic nucleotides. Biochem. Pharmacol. 2001, 62, 1153–1161. [Google Scholar] [CrossRef]
- Machackova, J.; Barta, J.; Dhalla, N.S. Myofibrillar remodeling in cardiac hypertrophy, heart failure and cardiomyopathies. Can. J. Cardiol. 2006, 22, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Kovanich , D.; van der Heyden, M.A.G.; Aye, T.T.; van Veen, T.A.B.; Heck , A.J.R.; Scholten , A. Sphingosine Kinase Interacting Protein is an A-Kinase Anchoring Protein Specific for Type I cAMP-Dependent Protein Kinase. ChemBioChem 2010, 11, 963–971. [Google Scholar] [CrossRef]
- Means, C.K.; Lygren, B.; Langeberg, L.K.; Jain, A.; Dixon, R.E.; Vega, A.L.; Gold, M.G.; Petrosyan, S.; Taylor, S.S.; Murphy, A.N.; et al. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, E1227–E1235. [Google Scholar] [CrossRef] [PubMed]
- Akabane, S.; Uno, M.; Tani, N.; Shimazaki, S.; Ebara, N.; Kato, H.; Kosako, H.; Oka, T. PKA Regulates PINK1 Stability and Parkin Recruitment to Damaged Mitochondria through Phosphorylation of MIC60. Mol. Cell. 2016, 62, 371–384. [Google Scholar] [CrossRef]
- Lygren, B.; Carlson, C.R.; Santamaria, K.; Lissandron, V.; McSorley, T.; Litzenberg, J.; Lorenz, D.; Wiesner, B.; Rosenthal, W.; Zaccolo, M.; et al. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007, 8, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Henn, V.; Edemir, B.; Stefan, E.; Wiesner, B.; Lorenz, D.; Theilig, F.; Schmitt, R.; Vossebein, L.; Tamma, G.; Beyermann, M.; et al. Identification of a Novel A-kinase Anchoring Protein 18 Isoform and Evidence for Its Role in the Vasopressin-induced Aquaporin-2 Shuttle in Renal Principal Cells. J. Biol. Chem. 2004, 279, 26654–26665. [Google Scholar] [CrossRef]
- Brown, R.L.; August, S.L.; Williams, C.J.; Moss, S.B. AKAP7gamma is a nuclear RI-binding AKAP. Biochem. Biophys. Res. Commun. 2003, 306, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Benz, P.M.; Ding, Y.; Stingl, H.; Loot, A.E.; Zink, J.; Wittig, I.; Popp, R.; Fleming, I. AKAP12 deficiency impairs VEGF-induced endothelial cell migration and sprouting. Acta Physiol. 2020, 228, e13325. [Google Scholar] [CrossRef]
- Yu, H.; Yuan, C.; Westenbroek, R.E.; Catterall, W.A. The AKAP Cypher/Zasp contributes to β-adrenergic/PKA stimulation of cardiac Ca(V)1.2 calcium channels. J. Gen. Physiol. 2018, 150, 883–889. [Google Scholar] [CrossRef]
- Lv, J.; Pan, Z.; Chen, J.; Xu, R.; Wang, D.; Huang, J.; Dong, Y.; Jiang, J.; Yin, X.; Cheng, H.; et al. Phosphoproteomic Analysis Reveals Downstream PKA Effectors of AKAP Cypher/ZASP in the Pathogenesis of Dilated Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 753072. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.L.; Jensen, L.J.; Diella, F.; Jørgensen, C.; Tinti, M.; Li, L.; Hsiung, M.; Parker, S.A.; Bordeaux, J.; Sicheritz-Ponten, T.; et al. Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal. 2008, 1, ra2. [Google Scholar] [CrossRef]
- Hennrich, M.L.; Marino, F.; Groenewold, V.; Kops, G.J.; Mohammed, S.; Heck, A.J. Universal quantitative kinase assay based on diagonal SCX chromatography and stable isotope dimethyl labeling provides high-definition kinase consensus motifs for PKA and human Mps1. J. Proteome Res. 2013, 12, 2214–2224. [Google Scholar] [CrossRef]
- Tillo, S.E.; Xiong, W.H.; Takahashi, M.; Miao, S.; Andrade, A.L.; Fortin, D.A.; Yang, G.; Qin, M.; Smoody, B.F.; Stork, P.J.S.; et al. Liberated PKA Catalytic Subunits Associate with the Membrane via Myristoylation to Preferentially Phosphorylate Membrane Substrates. Cell Rep. 2017, 19, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.D.; Esseltine, J.L.; Nygren, P.J.; Veesler, D.; Byrne, D.P.; Vonderach, M.; Strashnov, I.; Eyers, C.E.; Eyers, P.A.; Langeberg, L.K.; et al. Local protein kinase A action proceeds through intact holoenzymes. Science 2017, 356, 1288–1293. [Google Scholar] [CrossRef]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed]
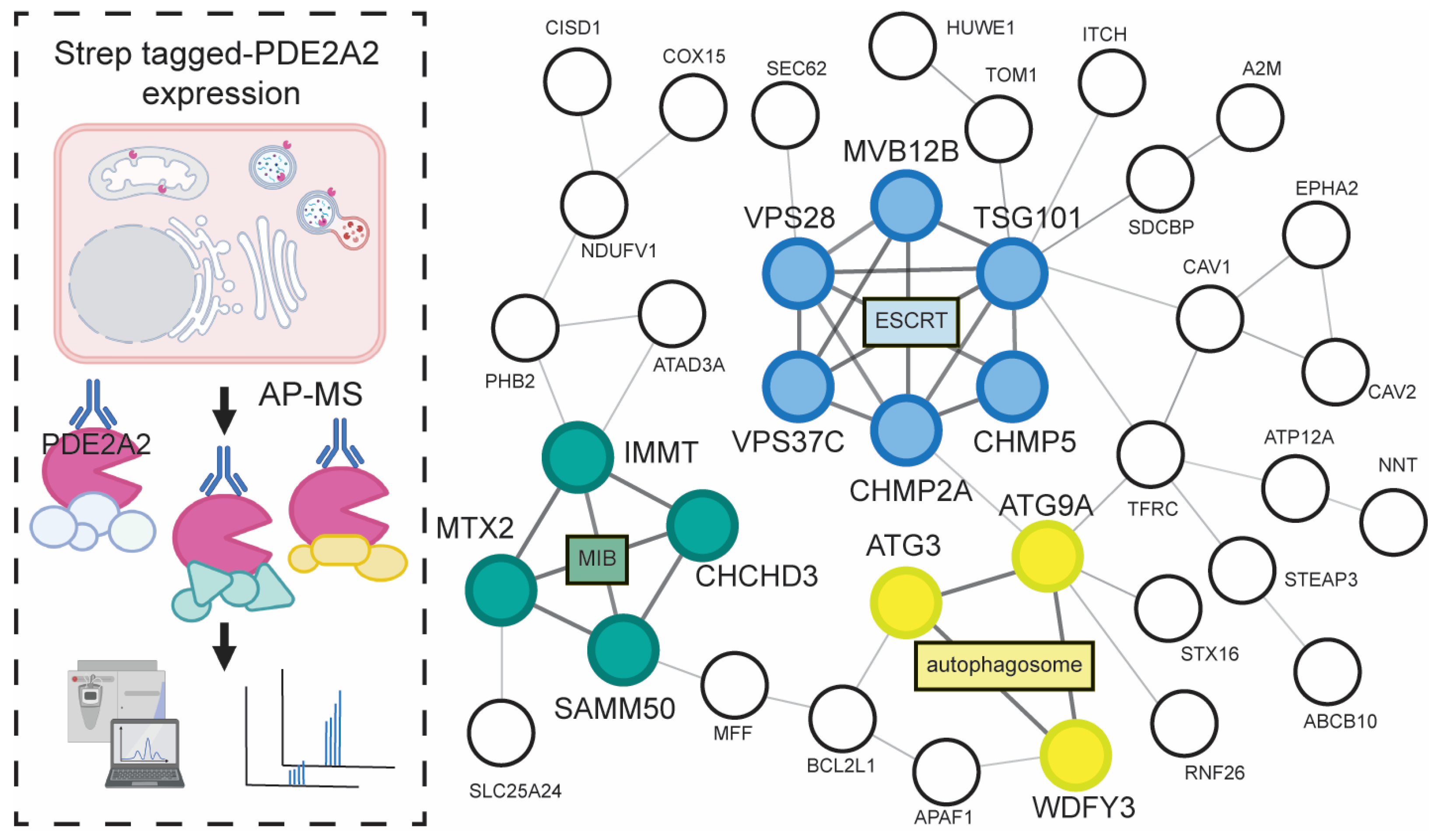
- Monterisi, S.; Lobo, M.J.; Livie, C.; Castle, J.C.; Weinberger, M.; Baillie, G.; Surdo, N.C.; Musheshe, N.; Stangherlin, A.; Gottlieb, E.; et al. PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. eLife 2017, 6, e21374. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, L.; Qi, Y.; Xu, H. Mitochondrial cAMP signaling. Cell. Mol. Life Sci. 2016, 73, 4577–4590. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Lefkimmiatis, K.; Pozzan, T. The basics of mitochondrial cAMP signalling: Where, when, why. Cell Calcium 2021, 93, 102320. [Google Scholar] [CrossRef]
- Ould Amer, Y.; Hebert-Chatelain, E. Insight into the Interactome of Intramitochondrial PKA Using Biotinylation-Proximity Labeling. Int. J. Mol. Sci. 2020, 21, 8283. [Google Scholar] [CrossRef] [PubMed]
- Omar, M.H.; Byrne, D.P.; Jones, K.N.; Lakey, T.M.; Collins, K.B.; Lee, K.-S.; Daly, L.A.; Forbush, K.A.; Lau, H.-T.; Golkowski, M.; et al. Mislocalization of protein kinase A drives pathology in Cushing’s syndrome. Cell Rep. 2021, 40, 111073. [Google Scholar] [CrossRef] [PubMed]
- Salhadar, K.; Matthews, A.; Raghuram, V.; Limbutara, K.; Yang, C.-R.; Datta, A.; Chou, C.-L.; Knepper, M.A. Phosphoproteomic Identification of Vasopressin/cAMP/Protein Kinase A–Dependent Signaling in Kidney. Mol. Pharmacol. 2021, 99, 358. [Google Scholar] [CrossRef]
- Isobe, K.; Jung, H.J.; Yang, C.R.; Claxton, J.; Sandoval, P.; Burg, M.B.; Raghuram, V.; Knepper, M.A. Systems-level identification of PKA-dependent signaling in epithelial cells. Proc. Natl. Acad. Sci. USA 2017, 114, E8875–E8884. [Google Scholar] [CrossRef]
- Taylor, S.S.; Wallbott, M.; Machal, E.M.F.; Søberg, K.; Ahmed, F.; Bruystens, J.; Vu, L.; Baker, B.; Wu, J.; Raimondi, F.; et al. PKA Cβ: A forgotten catalytic subunit of cAMP-dependent protein kinase opens new windows for PKA signaling and disease pathologies. Biochem. J. 2021, 478, 2101–2119. [Google Scholar] [CrossRef]
- Raghuram, V.; Salhadar, K.; Limbutara, K.; Park, E.; Yang, C.-R.; Knepper, M.A. Protein kinase A catalytic-α and catalytic-β proteins have nonredundant regulatory functions. Am. J. Physiol. Ren. Physiol. 2020, 319, F848–F862. [Google Scholar] [CrossRef]
- Noda, Y.; Sasaki, S. Updates and Perspectives on Aquaporin-2 and Water Balance Disorders. Int. J. Mol. Sci. 2021, 22, 12950. [Google Scholar] [CrossRef] [PubMed]
- Leo, K.T.; Chou, C.-L.; Yang, C.-R.; Park, E.; Raghuram, V.; Knepper, M.A. Bayesian analysis of dynamic phosphoproteomic data identifies protein kinases mediating GPCR responses. Cell Commun. Signal. 2022, 20, 80. [Google Scholar] [CrossRef]
- Deshpande, V.; Kao, A.; Raghuram, V.; Datta, A.; Chou, C.-L.; Knepper, M.A. Phosphoproteomic identification of vasopressin V2 receptor-dependent signaling in the renal collecting duct. Am. J. Physiol. Ren. Physiol. 2019, 317, F789–F804. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Murata, T.; Shimizu, K.; Degerman, E.; Maurice, D.; Manganiello, V. Cyclic nucleotide phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2015, 21, e25–e50. [Google Scholar] [CrossRef]
- Ercu, M.; Klussmann, E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 14. [Google Scholar] [CrossRef]
- Lorigo, M.; Oliveira, N.; Cairrao, E. PDE-Mediated Cyclic Nucleotide Compartmentation in Vascular Smooth Muscle Cells: From Basic to a Clinical Perspective. J. Cardiovasc. Dev. Dis. 2021, 9, 4. [Google Scholar] [CrossRef]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Lu, T.-W.; Stolerman, L.M.; Tenner, B.; Yang, J.R.; Zhang, J.-F.; Falcke, M.; Rangamani, P.; Taylor, S.S.; Mehta, S.; et al. Phase Separation of a PKA Regulatory Subunit Controls cAMP Compartmentation and Oncogenic Signaling. Cell 2020, 182, 1531–1544.e1515. [Google Scholar] [CrossRef]
- Lohse, C.; Bock, A.; Maiellaro, I.; Hannawacker, A.; Schad, L.R.; Lohse, M.J.; Bauer, W.R. Experimental and mathematical analysis of cAMP nanodomains. PLoS ONE 2017, 12, e0174856. [Google Scholar] [CrossRef]
- Amsallem, E.; Kasparian, C.; Haddour, G.; Boissel, J.P.; Nony, P. Phosphodiesterase III inhibitors for heart failure. Cochrane Database Syst. Rev. 2005, 2005, Cd002230. [Google Scholar] [CrossRef]
- Movsesian, M.A.; Kukreja, R.C. Phosphodiesterase inhibition in heart failure. Handb. Exp. Pharmacol. 2011, 204, 237–249. [Google Scholar]
- Beavo, J.A.; Golkowski, M.; Shimizu-Albergine, M.; Beltejar, M.-C.G.; Bornfeldt, K.E.; Ong, S.E. Phosphoproteomic Analysis as an Approach for Understanding Molecular Mechanisms of cAMP-Dependent Actions. Mol. Pharmacol. 2021, 99, 342. [Google Scholar] [CrossRef]
- Lobo, M.J.; Reverte-Salisa, L.; Chao, Y.-C.; Koschinski, A.; Gesellchen, F.; Subramaniam, G.; Jiang, H.; Pace, S.; Larcom, N.; Paolocci, E.; et al. Phosphodiesterase 2A2 regulates mitochondria clearance through Parkin-dependent mitophagy. Commun. Biol. 2020, 3, 596. [Google Scholar] [CrossRef]
- Wang, Y.; Ho, T.G.; Franz, E.; Hermann, J.S.; Smith, F.D.; Hehnly, H.; Esseltine, J.L.; Hanold, L.E.; Murph, M.M.; Bertinetti, D.; et al. PKA-type I selective constrained peptide disruptors of AKAP complexes. ACS Chem. Biol. 2015, 10, 1502–1510. [Google Scholar] [CrossRef]
- Bendzunas, N.G.; Dörfler, S.; Autenrieth, K.; Bertinetti, D.; Machal, E.M.F.; Kennedy, E.J.; Herberg, F.W. Investigating PKA-RII specificity using analogs of the PKA:AKAP peptide inhibitor STAD-2. Bioorg. Med. Chem. 2018, 26, 1174–1178. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ho, T.G.; Bertinetti, D.; Neddermann, M.; Franz, E.; Mo, G.C.H.; Schendowich, L.P.; Sukhu, A.; Spelts, R.C.; Zhang, J.; et al. Isoform-Selective Disruption of AKAP-Localized PKA Using Hydrocarbon Stapled Peptides. ACS Chem. Biol. 2014, 9, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Faruque, O.M.; Le-Nguyen, D.; Lajoix, A.-D.; Vives, E.; Petit, P.; Bataille, D.; Hani, E.-H. Cell-permeable peptide-based disruption of endogenous PKA-AKAP complexes: A tool for studying the molecular roles of AKAP-mediated PKA subcellular anchoring. Am. J. Physiol. Cell Physiol. 2009, 296, C306–C316. [Google Scholar] [CrossRef] [PubMed]
- Sin, Y.Y.; Edwards, H.V.; Li, X.; Day, J.P.; Christian, F.; Dunlop, A.J.; Adams, D.R.; Zaccolo, M.; Houslay, M.D.; Baillie, G.S. Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)-HSP20 complex attenuates the β-agonist induced hypertrophic response in cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 872–883. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovanich, D.; Low, T.Y.; Zaccolo, M. Using the Proteomics Toolbox to Resolve Topology and Dynamics of Compartmentalized cAMP Signaling. Int. J. Mol. Sci. 2023, 24, 4667. https://doi.org/10.3390/ijms24054667
Kovanich D, Low TY, Zaccolo M. Using the Proteomics Toolbox to Resolve Topology and Dynamics of Compartmentalized cAMP Signaling. International Journal of Molecular Sciences. 2023; 24(5):4667. https://doi.org/10.3390/ijms24054667
Chicago/Turabian StyleKovanich, Duangnapa, Teck Yew Low, and Manuela Zaccolo. 2023. "Using the Proteomics Toolbox to Resolve Topology and Dynamics of Compartmentalized cAMP Signaling" International Journal of Molecular Sciences 24, no. 5: 4667. https://doi.org/10.3390/ijms24054667
APA StyleKovanich, D., Low, T. Y., & Zaccolo, M. (2023). Using the Proteomics Toolbox to Resolve Topology and Dynamics of Compartmentalized cAMP Signaling. International Journal of Molecular Sciences, 24(5), 4667. https://doi.org/10.3390/ijms24054667