

Advances in Ferritin Physiology and Possible Implications in Bacterial Infection
 , , , and
, , , and
Abstract
1. Introduction
1.1. Mammalian Ferritin’s Morphology and Function
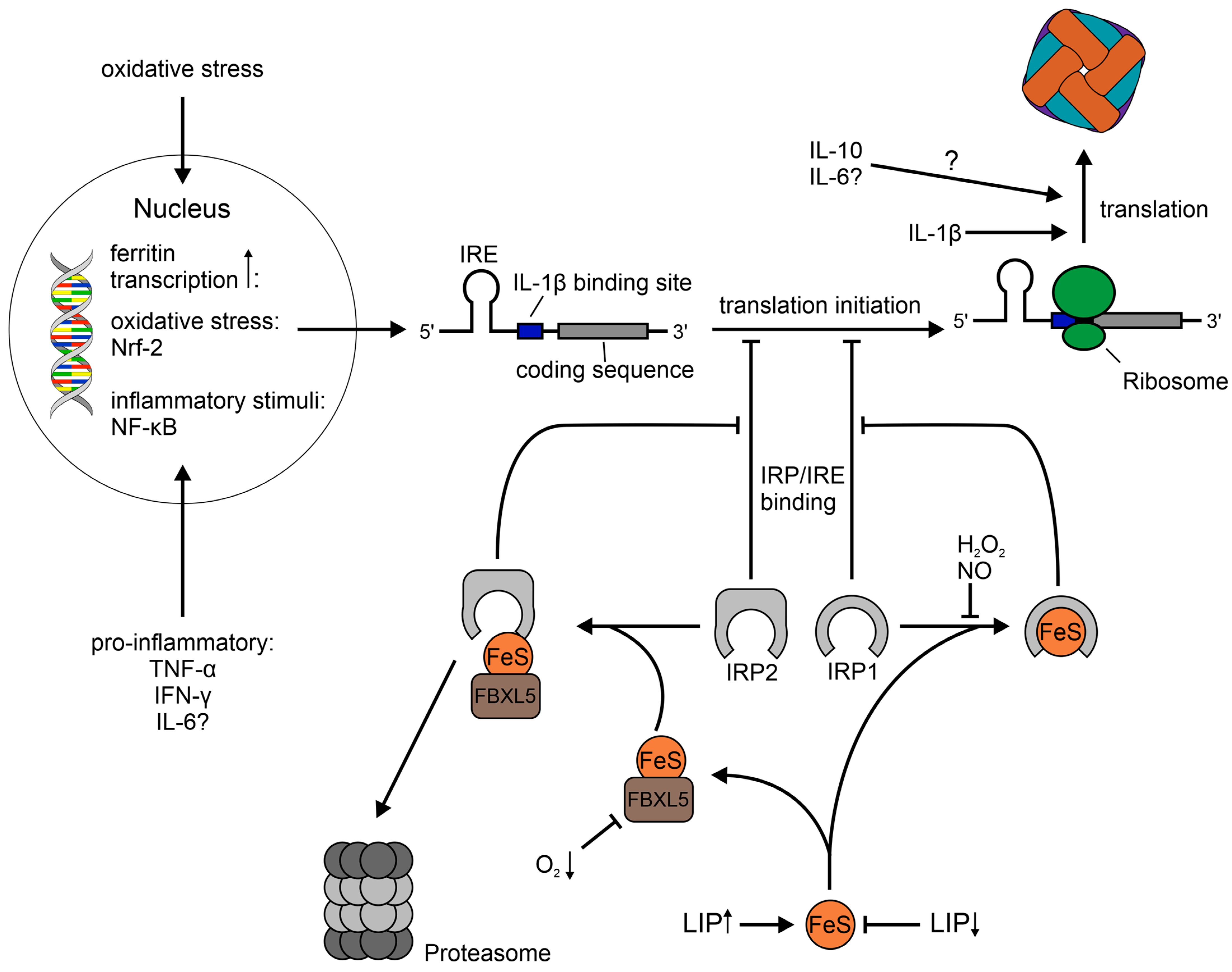
1.2. Regulation of Ferritin
1.3. Cellular Location, Trafficking and Utilization of Ferritin
1.4. General Aspects of Nutritional Immunity
2. Main Part
2.1. Ferritin as a Bacterial Iron Source

{kind=link}
{kind=link}
{kind=link}
| Bacterial Species Investigated | Phylum | Ferritin Used as a Sole Iron Source | Proposed Mechanisms for Pathogen-Ferritin Interaction | Sources |
|---|---|---|---|---|
| Mycobacterium tuberculosis | Actinomycetota | yes | siderophores | [173] |
| Bacillus cereus | Bacillota | yes | binding of ferritin via IlsA, siderophores | [169] |
| Listeria monocytogenes | Bacillota | yes | surface-associated ferrireductases | [165,166] |
| Streptococcus pneumonia | Bacillota | ? | proteolysis | [174] |
| Streptococcus pyogenes | Bacillota | yes | ? | [170] |
| Burkholderia cenocepacia | Pseudomonadota | yes | proteolysis | [167] |
| Escherichia coli | Pseudomonadota | yes | siderophores | [163,164] |
| Pseudomonas aeruginosa | Pseudomonadota | yes | siderophores, reduction of ferritin-bound iron, proteolysis | [168] |
| Salmonella enterica serovar Typhimurium | Pseudomonadota | yes | siderophores, ferrous iron uptake systems | [164] |
| Vibrio parahaemolyticus | Pseudomonadota | yes | ? | [172] |
| Vibrio vulnificus | Pseudomonadota | yes | ? | [171] |
| Yersinia pestis | Pseudomonadota | yes | ? | [162] |
| Investigations of cell infection | ||||
| Mycobacterium bovis | Actinomycetota | ? | colocalization with ferritin a | [178] |
| Helicobacter pylori | Campylobacterota | ? | colocalization with ferritin b | [175] |
| Chlamydia pneumoniae | Chlamydiota | ? | colocalization with ferritin a | [176] |
| Chlamydia trachomatis | Chlamydiota | ? | colocalization with ferritin a | [176] |
| Ehrlichia chaffeensis | Pseudomonadota | ? | induction of ferritin via Etf-3 | [180] |
| Neisseria meningitidis | Pseudomonadota | no | induction of ferritinophagy via iron starvation of the host, colocalization with ferritin | [179] |
| Uropathogenic Escherichia coli | Pseudomonadota | yes | iron acquisition via ferritinophagy | [177] |
2.2. Infection and Establishment of the Intraellular Replication Niche of Salmonella enterica serovar Typhimurium
2.3. Implications on Ferritin Metabolism upon Infection with Salmonella Typhimurium
2.4. Outlook on Future Research
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2019 Antimicrobial Resistance Collaborators. Global mortality associated with 33 bacterial pathogens in 2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248. [Google Scholar] [CrossRef] [PubMed]
- Abbafati, C.; Machado, D.B.; Cislaghi, B.; Salman, O.M.; Karanikolos, M.; McKee, M.; Abbas, K.M.; Brady, O.J.; Larson, H.J.; Trias-Llimós, S.; et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Kalia, V.C.; Patel, S.K.S.; Kang, Y.C.; Lee, J.K. Quorum sensing inhibitors as antipathogens: Biotechnological applications. Biotechnol. Adv. 2019, 37, 68–90. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Guilliams, M.; Haskó, G. Quorum sensing in the immune system. Nat. Rev. Immunol. 2018, 18, 537–538. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Núñez, G.; Sakamoto, K.; Soares, M.P. Innate Nutritional Immunity. J. Immunol. 2018, 201, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Cassat, J.E.; Skaar, E.P. Iron in infection and immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef]
- Wessels, I.; Fischer, H.J.; Rink, L. Dietary and Physiological Effects of Zinc on the Immune System. Annu. Rev. Nutr. 2021, 41, 133–175. [Google Scholar] [CrossRef]
- Weiss, G.; Carver, P.L. Role of divalent metals in infectious disease susceptibility and outcome. Clin. Microbiol. Infect. 2018, 24, 16–23. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef]
- Bradley, J.M.; Svistunenko, D.A.; Wilson, M.T.; Hemmings, A.M.; Moore, G.R.; Le Brun, N.E. Bacterial iron detoxification at the molecular level. J. Biol. Chem. 2020, 295, 17602–17623. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Adelman, T.G.; Drysdale, J.W. On ferritin heterogeneity. Further evidence for heteropolymers. J. Biol. Chem. 1978, 253, 4451–4458. [Google Scholar] [CrossRef] [PubMed]
- Bomford, A.; Conlon-Hollingshead, C.; Munro, H.N. Adaptive responses of rat tissue isoferritins to iron administration. Changes in subunit synthesis, isoferritin abundance, and capacity for iron storage. J. Biol. Chem. 1981, 256, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Galazka-Friedman, J.; Bauminger, E.R.; Friedman, A.; Koziorowski, D.; Szlachta, K. Human nigral and liver iron—Comparison by Mössbauer spectroscopy, electron microscopy and ELISA. Hyperfine Interact. 2005, 165, 285–288. [Google Scholar] [CrossRef]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta (BBA)-Bioenerg. 1996, 1275, 161–203. [Google Scholar] [CrossRef]
- Powell, L.W.; Alpert, E.; Isselbacher, K.J.; Drysdale, J.W. Human isoferritins: Organ specific iron and apoferritin distribution. Br. J. Haematol. 1975, 30, 47–55. [Google Scholar] [CrossRef]
- Watanabe, K.; Yamashita, Y.; Ohgawara, H.; Sekiguchi, M.; Satake, N.; Orino, K.; Yamamoto, S. Iron content of rat serum ferritin. J. Vet. Med. Sci. 2001, 63, 587–589. [Google Scholar] [CrossRef]
- Worwood, M.; Dawkins, S.; Wagstaff, M.; Jacobs, A. The purification and properties of ferritin from human serum. Biochem. J. 1976, 157, 97–103. [Google Scholar] [CrossRef]
- Arosio, P.; Yokota, M.; Drysdale, J.W. Characterization of serum ferritin in iron overload: Possible identity to natural apoferritin. Br. J. Haematol. 1977, 36, 199–207. [Google Scholar] [CrossRef]
- Wang, W.; Knovich, M.A.; Coffman, L.G.; Torti, F.M.; Torti, S.V. Serum ferritin: Past, present and future. Biochim. Biophys. Acta 2010, 1800, 760–769. [Google Scholar] [CrossRef]
- Cohen, L.A.; Gutierrez, L.; Weiss, A.; Leichtmann-Bardoogo, Y.; Zhang, D.L.; Crooks, D.R.; Sougrat, R.; Morgenstern, A.; Galy, B.; Hentze, M.W.; et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010, 116, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.; Bolisetty, S. Ferritins in Kidney Disease. Semin. Nephrol. 2020, 40, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.M.; Artymiuk, P.J.; Yewdall, S.J.; Smith, J.M.; Livingstone, J.C.; Treffry, A.; Luzzago, A.; Levi, S.; Arosio, P.; Cesareni, G.; et al. Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts. Nature 1991, 349, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Levi, S. Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 2002, 33, 457–463. [Google Scholar] [CrossRef]
- Pozzi, C.; Ciambellotti, S.; Bernacchioni, C.; Di Pisa, F.; Mangani, S.; Turano, P. Chemistry at the protein-mineral interface in L-ferritin assists the assembly of a functional (μ(3)-oxo)Tris[(μ(2)-peroxo)] triiron(III) cluster. Proc. Natl. Acad. Sci. USA 2017, 114, 2580–2585. [Google Scholar] [CrossRef]
- Ciambellotti, S.; Pozzi, C.; Mangani, S.; Turano, P. Iron Biomineral Growth from the Initial Nucleation Seed in L-Ferritin. Chem. A Eur. J. 2020, 26, 5770–5773. [Google Scholar] [CrossRef]
- Mehlenbacher, M.; Poli, M.; Arosio, P.; Santambrogio, P.; Levi, S.; Chasteen, N.D.; Bou-Abdallah, F. Iron Oxidation and Core Formation in Recombinant Heteropolymeric Human Ferritins. Biochemistry 2017, 56, 3900–3912. [Google Scholar] [CrossRef]
- Yang, X.; Arosio, P.; Chasteen, N.D. Molecular diffusion into ferritin: Pathways, temperature dependence, incubation time, and concentration effects. Biophys. J. 2000, 78, 2049–2059. [Google Scholar] [CrossRef]
- Takahashi, T.; Kuyucak, S. Functional properties of threefold and fourfold channels in ferritin deduced from electrostatic calculations. Biophys. J. 2003, 84, 2256–2263. [Google Scholar] [CrossRef]
- Levi, S.; Santambrogio, P.; Corsi, B.; Cozzi, A.; Arosio, P. Evidence that residues exposed on the three-fold channels have active roles in the mechanism of ferritin iron incorporation. Biochem. J. 1996, 317 Pt 2, 467–473. [Google Scholar] [CrossRef]
- Treffry, A.; Bauminger, E.R.; Hechel, D.; Hodson, N.W.; Nowik, I.; Yewdall, S.J.; Harrison, P.M. Defining the roles of the threefold channels in iron uptake, iron oxidation and iron-core formation in ferritin: A study aided by site-directed mutagenesis. Biochem. J. 1993, 296 Pt 3, 721–728. [Google Scholar] [CrossRef]
- Levi, S.; Luzzago, A.; Cesareni, G.; Cozzi, A.; Franceschinelli, F.; Albertini, A.; Arosio, P. Mechanism of ferritin iron uptake: Activity of the H-chain and deletion mapping of the ferro-oxidase site. A study of iron uptake and ferro-oxidase activity of human liver, recombinant H-chain ferritins, and of two H-chain deletion mutants. J. Biol. Chem. 1988, 263, 18086–18092. [Google Scholar] [CrossRef]
- Ingrassia, R.; Gerardi, G.; Biasiotto, G.; Arosio, P. Mutations of ferritin H chain C-terminus produced by nucleotide insertions have altered stability and functional properties. J. Biochem. 2006, 139, 881–885. [Google Scholar] [CrossRef]
- Levi, S.; Luzzago, A.; Franceschinelli, F.; Santambrogio, P.; Cesareni, G.; Arosio, P. Mutational analysis of the channel and loop sequences of human ferritin H-chain. Biochem. J. 1989, 264, 381–388. [Google Scholar] [CrossRef]
- Douglas, T.; Ripoll, D.R. Calculated electrostatic gradients in recombinant human H-chain ferritin. Protein Sci. A Publ. Protein Soc. 1998, 7, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Stillman, T.J.; Hempstead, P.D.; Artymiuk, P.J.; Andrews, S.C.; Hudson, A.J.; Treffry, A.; Guest, J.R.; Harrison, P.M. The high-resolution X-ray crystallographic structure of the ferritin (EcFtnA) of Escherichia coli; comparison with human H ferritin (HuHF) and the structures of the Fe3+ and Zn2+ derivatives11Edited by R. Huber. J. Mol. Biol. 2001, 307, 587–603. [Google Scholar] [CrossRef] [PubMed]
- Ciacchi, L.C.; Payne, M.C. The entry pathway of O2 into human ferritin. Chem. Phys. Lett. 2004, 390, 491–495. [Google Scholar] [CrossRef]
- Bou-Abdallah, F.; Zhao, G.; Biasiotto, G.; Poli, M.; Arosio, P.; Chasteen, N.D. Facilitated Diffusion of Iron(II) and Dioxygen Substrates into Human H-Chain Ferritin. A Fluorescence and Absorbance Study Employing the Ferroxidase Center Substitution Y34W. J. Am. Chem. Soc. 2008, 130, 17801–17811. [Google Scholar] [CrossRef]
- Honarmand Ebrahimi, K.; Hagedoorn, P.L.; Hagen, W.R. Unity in the biochemistry of the iron-storage proteins ferritin and bacterioferritin. Chem. Rev. 2015, 115, 295–326. [Google Scholar] [CrossRef]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef]
- Behera, R.K.; Theil, E.C. Moving Fe2+ from ferritin ion channels to catalytic OH centers depends on conserved protein cage carboxylates. Proc. Natl. Acad. Sci. USA 2014, 111, 7925–7930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wilson, P.E.; Watt, G.D. Ferritin-catalyzed consumption of hydrogen peroxide by amine buffers causes the variable Fe2+ to O2 stoichiometry of iron deposition in horse spleen ferritin. JBIC J. Biol. Inorg. Chem. 2006, 11, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chasteen, N.D. Iron oxidation chemistry in ferritin. Increasing Fe/O2 stoichiometry during core formation. J. Biol. Chem. 1991, 266, 19965–19970. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Firlar, E.; Rasul, M.G.; Foroozan, T.; Farajpour, N.; Covnot, L.; Shahbazian-Yassar, R.; Shokuhfar, T. On the structure and chemistry of iron oxide cores in human heart and human spleen ferritins using graphene liquid cell electron microscopy. Nanoscale 2019, 11, 16868–16878. [Google Scholar] [CrossRef]
- Bolann, B.J.; Ulvik, R.J. Decay of superoxide catalyzed by ferritin. FEBS Lett. 1993, 318, 149–152. [Google Scholar] [CrossRef]
- Ruddell, R.G.; Hoang-Le, D.; Barwood, J.M.; Rutherford, P.S.; Piva, T.J.; Watters, D.J.; Santambrogio, P.; Arosio, P.; Ramm, G.A. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology 2009, 49, 887–900. [Google Scholar] [CrossRef]
- Broxmeyer, H.E.; Williams, D.E.; Geissler, K.; Hangoc, G.; Cooper, S.; Bicknell, D.C.; Levi, S.; Arosio, P. Suppressive effects in vivo of purified recombinant human H-subunit (acidic) ferritin on murine myelopoiesis. Blood 1989, 73, 74–79. [Google Scholar] [CrossRef]
- Fargion, S.; Fracanzani, A.L.; Brando, B.; Arosio, P.; Levi, S.; Fiorelli, G. Specific binding sites for H-ferritin on human lymphocytes: Modulation during cellular proliferation and potential implication in cell growth control. Blood 1991, 78, 1056–1061. [Google Scholar] [CrossRef]
- Yamashita, M.; Harada, G.; Matsumoto, S.E.; Aiba, Y.; Ichikawa, A.; Fujiki, T.; Udono, M.; Kabayama, S.; Yoshida, T.; Zhang, P.; et al. Suppression of immunoglobulin production in human peripheral blood mononuclear cells by monocytes via secretion of heavy-chain ferritin. Immunobiology 2014, 219, 149–157. [Google Scholar] [CrossRef]
- Gray, C.P.; Arosio, P.; Hersey, P. Heavy chain ferritin activates regulatory T cells by induction of changes in dendritic cells. Blood 2002, 99, 3326–3334. [Google Scholar] [CrossRef]
- Zarjou, A.; Black, L.M.; McCullough, K.R.; Hull, T.D.; Esman, S.K.; Boddu, R.; Varambally, S.; Chandrashekar, D.S.; Feng, W.; Arosio, P.; et al. Ferritin Light Chain Confers Protection Against Sepsis-Induced Inflammation and Organ Injury. Front. Immunol. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Carlos, A.R.; Moita, M.R.; Singh, S.; Blankenhaus, B.; Cardoso, S.; Larsen, R.; Rebelo, S.; Schauble, S.; Del Barrio, L.; et al. Metabolic Adaptation Establishes Disease Tolerance to Sepsis. Cell 2017, 169, 1263–1275.e1214. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Wang, M.; Meng, J.; Ma, Y.; Wang, Y.; Miao, N.; Teng, J.; Zhu, D.; Shi, H.; Sun, Y.; et al. Ferritin triggers neutrophil extracellular trap-mediated cytokine storm through Msr1 contributing to adult-onset Still’s disease pathogenesis. Nat. Commun. 2022, 13, 6804. [Google Scholar] [CrossRef] [PubMed]
- Haschka, D.; Tymoszuk, P.; Petzer, V.; Hilbe, R.; Heeke, S.; Dichtl, S.; Skvortsov, S.; Demetz, E.; Berger, S.; Seifert, M.; et al. Ferritin H deficiency deteriorates cellular iron handling and worsens Salmonella typhimurium infection by triggering hyperinflammation. JCI Insight 2021, 6, e141760. [Google Scholar] [CrossRef] [PubMed]
- Torti, F.M.; Torti, S.V. Regulation of ferritin genes and protein. Blood 2002, 99, 3505–3516. [Google Scholar] [CrossRef]
- Rogers, J.; Munro, H. Translation of ferritin light and heavy subunit mRNAs is regulated by intracellular chelatable iron levels in rat hepatoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 2277–2281. [Google Scholar] [CrossRef]
- Guo, B.; Yu, Y.; Leibold, E.A. Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J. Biol. Chem. 1994, 269, 24252–24260. [Google Scholar] [CrossRef]
- Leibold, E.A.; Munro, H.N. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5’ untranslated region of ferritin heavy- and light-subunit mRNAs. Proc. Natl. Acad. Sci. USA 1988, 85, 2171–2175. [Google Scholar] [CrossRef]
- Hentze, M.W.; Caughman, S.W.; Rouault, T.A.; Barriocanal, J.G.; Dancis, A.; Harford, J.B.; Klausner, R.D. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 1987, 238, 1570–1573. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef]
- Rouault, T.A. Mammalian iron-sulphur proteins: Novel insights into biogenesis and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 45–55. [Google Scholar] [CrossRef]
- Gray, N.K.; Hentze, M.W. Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. EMBO J. 1994, 13, 3882–3891. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.; Gray, N.K.; Hentze, M.W. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol. Cell 1998, 2, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Kaptain, S.; Downey, W.E.; Tang, C.; Philpott, C.; Haile, D.; Orloff, D.G.; Harford, J.B.; Rouault, T.A.; Klausner, R.D. A regulated RNA binding protein also possesses aconitase activity. Proc. Natl. Acad. Sci. USA 1991, 88, 10109–10113. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, H.; Rajan, M.; Canarie, E.R.; Hong, S.; Simoneschi, D.; Pagano, M.; Bush, M.F.; Stoll, S.; Leibold, E.A.; et al. FBXL5 Regulates IRP2 Stability in Iron Homeostasis via an Oxygen-Responsive [2Fe2S] Cluster. Mol. Cell 2020, 78, 31–41.e35. [Google Scholar] [CrossRef]
- Pulos-Holmes, M.C.; Srole, D.N.; Juarez, M.G.; Lee, A.S.; McSwiggen, D.T.; Ingolia, N.T.; Cate, J.H. Repression of ferritin light chain translation by human eIF3. eLife 2019, 8, e48193. [Google Scholar] [CrossRef]
- Cairo, G.; Recalcati, S. Iron-regulatory proteins: Molecular biology and pathophysiological implications. Expert Rev. Mol. Med. 2007, 9, 1–13. [Google Scholar] [CrossRef]
- Pantopoulos, K.; Hentze, M.W. Activation of iron regulatory protein-1 by oxidative stress in vitro. Proc. Natl. Acad. Sci. 1998, 95, 10559–10563. [Google Scholar] [CrossRef]
- Pantopoulos, K.; Mueller, S.; Atzberger, A.; Ansorge, W.; Stremmel, W.; Hentze, M.W. Differences in the regulation of iron regulatory protein-1 (IRP-1) by extra- and intracellular oxidative stress. J. Biol. Chem. 1997, 272, 9802–9808. [Google Scholar] [CrossRef]
- Weiss, G.; Goossen, B.; Doppler, W.; Fuchs, D.; Pantopoulos, K.; Werner-Felmayer, G.; Wachter, H.; Hentze, M.W. Translational regulation via iron-responsive elements by the nitric oxide/NO-synthase pathway. EMBO J. 1993, 12, 3651–3657. [Google Scholar] [CrossRef]
- Weiss, G.; Bogdan, C.; Hentze, M.W. Pathways for the regulation of macrophage iron metabolism by the anti-inflammatory cytokines IL-4 and IL-13. J. Immunol. 1997, 158, 420–425. [Google Scholar] [CrossRef]
- Lu, Q.; Harris, V.A.; Rafikov, R.; Sun, X.; Kumar, S.; Black, S.M. Nitric oxide induces hypoxia ischemic injury in the neonatal brain via the disruption of neuronal iron metabolism. Redox Biol. 2015, 6, 112–121. [Google Scholar] [CrossRef]
- Recalcati, S.; Taramelli, D.; Conte, D.; Cairo, G. Nitric oxide-mediated induction of ferritin synthesis in J774 macrophages by inflammatory cytokines: Role of selective iron regulatory protein-2 downregulation. Blood 1998, 91, 1059–1066. [Google Scholar] [CrossRef]
- Kim, S.; Ponka, P. Effects of interferon-gamma and lipopolysaccharide on macrophage iron metabolism are mediated by nitric oxide-induced degradation of iron regulatory protein 2. J. Biol. Chem. 2000, 275, 6220–6226. [Google Scholar] [CrossRef]
- Soum, E.; Drapier, J.C. Nitric oxide and peroxynitrite promote complete disruption of the [4Fe-4S] cluster of recombinant human iron regulatory protein 1. J. Biol. Inorg. Chem. JBIC A Publ. Soc. Biol. Inorg. Chem. 2003, 8, 226–232. [Google Scholar] [CrossRef]
- Wang, J.; Chen, G.; Pantopoulos, K. Nitric oxide inhibits the degradation of IRP2. Mol. Cell. Biol. 2005, 25, 1347–1353. [Google Scholar] [CrossRef]
- Kim, S.; Wing, S.S.; Ponka, P. S-nitrosylation of IRP2 regulates its stability via the ubiquitin-proteasome pathway. Mol. Cell. Biol. 2004, 24, 330–337. [Google Scholar] [CrossRef]
- Richardson, D.R.; Neumannova, V.; Nagy, E.; Ponka, P. The effect of redox-related species of nitrogen monoxide on transferrin and iron uptake and cellular proliferation of erythroleukemia (K562) cells. Blood 1995, 86, 3211–3219. [Google Scholar] [CrossRef]
- Tsuji, Y.; Ayaki, H.; Whitman, S.P.; Morrow, C.S.; Torti, S.V.; Torti, F.M. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol. Cell. Biol. 2000, 20, 5818–5827. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef]
- Pietsch, E.C.; Chan, J.Y.; Torti, F.M.; Torti, S.V. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 2003, 278, 2361–2369. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, I.M.; Bailey, K.; Flowers, C.H.; Garrigues, N.W.; Wesselius, L.J. Effects of TNF-alpha and IL-1beta on iron metabolism by A549 cells and influence on cytotoxicity. Am. J. Physiol. 1999, 277, L257–L263. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Bridges, K.R.; Durmowicz, G.P.; Glass, J.; Auron, P.E.; Munro, H.N. Translational control during the acute phase response. Ferritin synthesis in response to interleukin-1. J. Biol. Chem. 1990, 265, 14572–14578. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Andriotakis, J.L.; Lacroix, L.; Durmowicz, G.P.; Kasschau, K.D.; Bridges, K.R. Translational enhancement of H-ferritin mRNA by interleukin-1 beta acts through 5’ leader sequences distinct from the iron responsive element. Nucleic Acids Res. 1994, 22, 2678–2686. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, M.; Kohgo, Y.; Kondo, H.; Shintani, N.; Fujikawa, K.; Sasaki, K.; Kato, J.; Niitsu, Y. Regulation of iron metabolism in HepG2 cells: A possible role for cytokines in the hepatic deposition of iron. Hepatology 1993, 18, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, M.; Young, S.P. Modulation of iron metabolism in monocyte cell line U937 by inflammatory cytokines: Changes in transferrin uptake, iron handling and ferritin mRNA. Biochem. J. 1993, 296, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Ulmer, H.; Kaser, A.; Weiss, G. Role of IL-10 for induction of anemia during inflammation. J. Immunol. 2002, 169, 2204–2209. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Larochelle, D.A.; Beaumont, C.; Torti, S.V.; Torti, F.M. Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J. Biol. Chem. 1995, 270, 15285–15293. [Google Scholar] [CrossRef]
- Byrd, T.F.; Horwitz, M.A. Regulation of transferrin receptor expression and ferritin content in human mononuclear phagocytes. Coordinate upregulation by iron transferrin and downregulation by interferon gamma. J. Clin. Investig. 1993, 91, 969–976. [Google Scholar] [CrossRef]
- Nairz, M.; Theurl, I.; Ludwiczek, S.; Theurl, M.; Mair, S.M.; Fritsche, G.; Weiss, G. The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell. Microbiol. 2007, 9, 2126–2140. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Fritsche, G.; Brunner, P.; Talasz, H.; Hantke, K.; Weiss, G. Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur. J. Immunol. 2008, 38, 1923–1936. [Google Scholar] [CrossRef]
- Ohshima, T.; Yamamoto, H.; Sakamaki, Y.; Saito, C.; Mizushima, N. NCOA4 drives ferritin phase separation to facilitate macroferritinophagy and microferritinophagy. J. Cell Biol. 2022, 221, e202203102. [Google Scholar] [CrossRef] [PubMed]
- Heynen, M.J.; Verwilghen, R.L. A quantitative ultrastructural study of normal rat erythroblasts and reticulocytes. Cell Tissue Res. 1982, 224, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Takano-Ohmuro, H.; Mukaida, M.; Kominami, E.; Morioka, K. Autophagy in embryonic erythroid cells: Its role in maturation. Eur. J. Cell Biol. 2000, 79, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014, 127, 4089–4102. [Google Scholar] [CrossRef]
- Goodwin, J.M.; Dowdle, W.E.; DeJesus, R.; Wang, Z.; Bergman, P.; Kobylarz, M.; Lindeman, A.; Xavier, R.J.; McAllister, G.; Nyfeler, B.; et al. Autophagy-Independent Lysosomal Targeting Regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 2017, 20, 2341–2356. [Google Scholar] [CrossRef]
- Ohnstad, A.E.; Delgado, J.M.; North, B.J.; Nasa, I.; Kettenbach, A.N.; Schultz, S.W.; Shoemaker, C.J. Receptor-mediated clustering of FIP200 bypasses the role of LC3 lipidation in autophagy. EMBO J. 2020, 39, e104948. [Google Scholar] [CrossRef]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Vaisman, B.; Fibach, E.; Konijn, A.M. Utilization of intracellular ferritin iron for hemoglobin synthesis in developing human erythroid precursors. Blood 1997, 90, 831–838. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Srivastava, A.K.; Reutovich, A.A.; Hunter, N.J.; Arosio, P.; Melman, A.; Bou-Abdallah, F. Iron Mobilization from Ferritin in Yeast Cell Lysate and Physiological Implications. Int. J. Mol. Sci. 2022, 23, 6100. [Google Scholar] [CrossRef] [PubMed]
- Bou-Abdallah, F.; Paliakkara, J.J.; Melman, G.; Melman, A. Reductive Mobilization of Iron from Intact Ferritin: Mechanisms and Physiological Implication. Pharmaceuticals 2018, 11, 120. [Google Scholar] [CrossRef]
- De Domenico, I.; Vaughn, M.B.; Li, L.; Bagley, D.; Musci, G.; Ward, D.M.; Kaplan, J. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 2006, 25, 5396–5404. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; Ward, D.M.; Kaplan, J. Specific iron chelators determine the route of ferritin degradation. Blood 2009, 114, 4546–4551. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, S.; Poole, B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA 1978, 75, 3327–3331. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol. Cell. Biol. 2011, 31, 2040–2052. [Google Scholar] [CrossRef]
- Meng, F.; Fleming, B.A.; Jia, X.; Rousek, A.A.; Mulvey, M.A.; Ward, D.M. Lysosomal iron recycling in mouse macrophages is dependent upon both LcytB and Steap3 reductases. Blood Adv. 2022, 6, 1692–1707. [Google Scholar] [CrossRef]
- La, A.; Nguyen, T.; Tran, K.; Sauble, E.; Tu, D.; Gonzalez, A.; Kidane, T.Z.; Soriano, C.; Morgan, J.; Doan, M.; et al. Mobilization of iron from ferritin: New steps and details. Met. Integr. Biometal Sci. 2018, 10, 154–168. [Google Scholar] [CrossRef]
- Dong, X.P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef]
- Lam-Yuk-Tseung, S.; Gros, P. Distinct targeting and recycling properties of two isoforms of the iron transporter DMT1 (NRAMP2, Slc11A2). Biochemistry 2006, 45, 2294–2301. [Google Scholar] [CrossRef]
- Searle, S.; Bright, N.A.; Roach, T.I.; Atkinson, P.G.; Barton, C.H.; Meloen, R.H.; Blackwell, J.M. Localisation of Nramp1 in macrophages: Modulation with activation and infection. J. Cell Sci. 1998, 111 Pt 19, 2855–2866. [Google Scholar] [CrossRef]
- Truman-Rosentsvit, M.; Berenbaum, D.; Spektor, L.; Cohen, L.A.; Belizowsky-Moshe, S.; Lifshitz, L.; Ma, J.; Li, W.; Kesselman, E.; Abutbul-Ionita, I.; et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018, 131, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hevi, S.; Chuck, S.L. Regulated secretion of glycosylated human ferritin from hepatocytes. Blood 2004, 103, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Yanatori, I.; Richardson, D.R.; Dhekne, H.S.; Toyokuni, S.; Kishi, F. CD63 is regulated by iron via the IRE-IRP system and is important for ferritin secretion by extracellular vesicles. Blood 2021, 138, 1490–1503. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Kato, K.; Yanatori, I.; Murohara, T.; Toyokuni, S. Ferroptosis-dependent extracellular vesicles from macrophage contribute to asbestos-induced mesothelial carcinogenesis through loading ferritin. Redox Biol. 2021, 47, 102174. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jia, J.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; Farzam, F.; et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017, 36, 42–60. [Google Scholar] [CrossRef]
- Chiou, B.; Lucassen, E.; Sather, M.; Kallianpur, A.; Connor, J. Semaphorin4A and H-ferritin utilize Tim-1 on human oligodendrocytes: A novel neuro-immune axis. Glia 2018, 66, 1317–1330. [Google Scholar] [CrossRef]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebrón, J.A.; Björkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Jorge, L.; Ramos, D.; Valença, A.; López-Luppo, M.; Pires, V.M.; Catita, J.; Nacher, V.; Navarro, M.; Carretero, A.; Rodriguez-Baeza, A.; et al. L-ferritin binding to scara5: A new iron traffic pathway potentially implicated in retinopathy. PLoS ONE 2014, 9, e106974. [Google Scholar] [CrossRef]
- Yu, B.; Cheng, C.; Wu, Y.; Guo, L.; Kong, D.; Zhang, Z.; Wang, Y.; Zheng, E.; Liu, Y.; He, Y. Interactions of ferritin with scavenger receptor class A members. J. Biol. Chem. 2020, 295, 15727–15741. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.T.; Li, L.; Chung, D.H.; Allen, C.D.; Torti, S.V.; Torti, F.M.; Cyster, J.G.; Chen, C.Y.; Brodsky, F.M.; Niemi, E.C.; et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J. Exp. Med. 2005, 202, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Paragas, N.; Ned, R.M.; Qiu, A.; Viltard, M.; Leete, T.; Drexler, I.R.; Chen, X.; Sanna-Cherchi, S.; Mohammed, F.; et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev. Cell 2009, 16, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Hessvik, N.P.; Sandvig, K.; Llorente, A. Exosomal lipid composition and the role of ether lipids and phosphoinositides in exosome biology. J. Lipid Res. 2019, 60, 9–18. [Google Scholar] [CrossRef]
- Miyanishi, M.; Tada, K.; Koike, M.; Uchiyama, Y.; Kitamura, T.; Nagata, S. Identification of Tim4 as a phosphatidylserine receptor. Nature 2007, 450, 435–439. [Google Scholar] [CrossRef]
- Gonda, A.; Kabagwira, J.; Senthil, G.N.; Wall, N.R. Internalization of Exosomes through Receptor-Mediated Endocytosis. Mol. Cancer Res. MCR 2019, 17, 337–347. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef]
- ten Kate, J.; Wolthuis, A.; Westerhuis, B.; van Deursen, C. The iron content of serum ferritin: Physiological importance and diagnostic value. Eur. J. Clin. Chem. Clin. Biochem. J. Forum Eur. Clin. Chem. Soc. 1997, 35, 53–56. [Google Scholar] [CrossRef]
- Konz, T.; Añón Alvarez, E.; Montes-Bayon, M.; Sanz-Medel, A. Antibody labeling and elemental mass spectrometry (inductively coupled plasma-mass spectrometry) using isotope dilution for highly sensitive ferritin determination and iron-ferritin ratio measurements. Anal. Chem. 2013, 85, 8334–8340. [Google Scholar] [CrossRef]
- Herbert, V.; Jayatilleke, E.; Shaw, S.; Rosman, A.S.; Giardina, P.; Grady, R.W.; Bowman, B.; Gunter, E.W. Serum ferritin iron, a new test, measures human body iron stores unconfounded by inflammation. Stem Cells 1997, 15, 291–296. [Google Scholar] [CrossRef]
- Ferreira, C.; Bucchini, D.; Martin, M.E.; Levi, S.; Arosio, P.; Grandchamp, B.; Beaumont, C. Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem. 2000, 275, 3021–3024. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Garringer, H.J.; Goodwin, C.B.; Richine, B.; Acton, A.; VanDuyn, N.; Muhoberac, B.B.; Irimia-Dominguez, J.; Chan, R.J.; Peacock, M.; et al. Systemic and cerebral iron homeostasis in ferritin knock-out mice. PLoS ONE 2015, 10, e0117435. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, E.D. The Lactobacillus anomaly: Total iron abstinence. Perspect Biol. Med. 1997, 40, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E. Iron and bacterial virulence—A brief overview. Biol. Met. 1991, 4, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Parrow, N.L.; Fleming, R.E.; Minnick, M.F.; Maurelli, A.T. Sequestration and Scavenging of Iron in Infection. Infect. Immun. 2013, 81, 3503–3514. [Google Scholar] [CrossRef]
- Nairz, M.; Weiss, G. Iron in infection and immunity. Mol. Asp. Med. 2020, 75, 100864. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood J. Am. Soc. Hematol. 2003, 101, 2461–2463. [Google Scholar] [CrossRef]
- Nairz, M.; Haschka, D.; Demetz, E.; Weiss, G. Iron at the interface of immunity and infection. Front. Pharmacol. 2014, 5, 152. [Google Scholar] [CrossRef]
- Soares, M.P.; Weiss, G. The Iron age of host–microbe interactions. EMBO Rep. 2015, 16, 1482–1500. [Google Scholar] [CrossRef]
- Grubwieser, P.; Hoffmann, A.; Hilbe, R.; Seifert, M.; Sonnweber, T.; Böck, N.; Theurl, I.; Weiss, G.; Nairz, M. Airway Epithelial Cells Differentially Adapt Their Iron Metabolism to Infection With Klebsiella pneumoniae and Escherichia coli In Vitro. Front. Cell. Infect. Microbiol. 2022, 12, 619. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Prentice, A.M. Hepcidin and the iron-infection axis. Science 2012, 338, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Arezes, J.; Jung, G.; Gabayan, V.; Valore, E.; Ruchala, P.; Gulig, P.A.; Ganz, T.; Nemeth, E.; Bulut, Y. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe 2015, 17, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; De Domenico, I.; Durchfort, N.; Zohn, I.; Kaplan, J.; Ward, D.M. Iron depletion limits intracellular bacterial growth in macrophages. Blood 2008, 112, 866–874. [Google Scholar] [CrossRef]
- Pan, X.; Tamilselvam, B.; Hansen, E.J.; Daefler, S. Modulation of iron homeostasis in macrophages by bacterial intracellular pathogens. BMC Microbiol. 2010, 10, 64. [Google Scholar] [CrossRef]
- Schmidt, I.H.E.; Gildhorn, C.; Böning, M.A.L.; Kulow, V.A.; Steinmetz, I.; Bast, A. Burkholderia pseudomallei modulates host iron homeostasis to facilitate iron availability and intracellular survival. PLoS Negl. Trop. Dis. 2018, 12, e0006096. [Google Scholar] [CrossRef]
- Hoffmann, A.; Haschka, D.; Valente de Souza, L.; Tymoszuk, P.; Seifert, M.; von Raffay, L.; Hilbe, R.; Petzer, V.; Moser, P.L.; Nairz, M.; et al. Baseline iron status and presence of anaemia determine the course of systemic Salmonella infection following oral iron supplementation in mice. eBioMedicine 2021, 71, 103568. [Google Scholar] [CrossRef]
- Cunnington, A.J.; de Souza, J.B.; Walther, M.; Riley, E.M. Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat. Med. 2011, 18, 120–127. [Google Scholar] [CrossRef]
- Silva-Gomes, S.; Vale-Costa, S.; Appelberg, R.; Gomes, M.S. Iron in intracellular infection: To provide or to deprive? Front. Cell. Infect. Microbiol. 2013, 3, 96. [Google Scholar] [CrossRef]
- Johnson, E.E.; Sandgren, A.; Cherayil, B.J.; Murray, M.; Wessling-Resnick, M. Role of Ferroportin in Macrophage-Mediated Immunity. Infect. Immun. 2010, 78, 5099–5106. [Google Scholar] [CrossRef]
- Abreu, R.; Essler, L.; Giri, P.; Quinn, F. Interferon-gamma promotes iron export in human macrophages to limit intracellular bacterial replication. PLoS ONE 2020, 15, e0240949. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Schleicher, U.; Schroll, A.; Sonnweber, T.; Theurl, I.; Ludwiczek, S.; Talasz, H.; Brandacher, G.; Moser, P.L.; Muckenthaler, M.U.; et al. Nitric oxide-mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J. Exp. Med. 2013, 210, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Grander, M.; Hoffmann, A.; Seifert, M.; Demetz, E.; Grubwieser, P.; Pfeifhofer-Obermair, C.; Haschka, D.; Weiss, G. DMT1 Protects Macrophages from Salmonella Infection by Controlling Cellular Iron Turnover and Lipocalin 2 Expression. Int. J. Mol. Sci. 2022, 23, 6789. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.R.; Gros, P. Iron, manganese, and cobalt transport by Nramp1 (Slc11a1) and Nramp2 (Slc11a2) expressed at the plasma membrane. Blood 2003, 102, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Fritsche, G.; Crouch, M.L.; Barton, H.C.; Fang, F.C.; Weiss, G. Slc11a1 limits intracellular growth of Salmonella enterica sv. Typhimurium by promoting macrophage immune effector functions and impairing bacterial iron acquisition. Cell. Microbiol. 2009, 11, 1365–1381. [Google Scholar] [CrossRef]
- Levay, P.F.; Viljoen, M. Lactoferrin: A general review. Haematologica 1995, 80, 252–267. [Google Scholar] [CrossRef]
- Zagulski, T.; Lipiński, P.; Zagulska, A.; Broniek, S.; Jarzabek, Z. Lactoferrin can protect mice against a lethal dose of Escherichia coli in experimental infection in vivo. Br. J. Exp. Pathol. 1989, 70, 697–704. [Google Scholar]
- Miethke, M.; Marahiel, M.A. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [Google Scholar] [CrossRef]
- Jaberi, S.A.; Cohen, A.; D’Souza, C.; Abdulrazzaq, Y.M.; Ojha, S.; Bastaki, S.; Adeghate, E.A. Lipocalin-2: Structure, function, distribution and role in metabolic disorders. Biomed. Pharmacother. 2021, 142, 112002. [Google Scholar] [CrossRef]
- Behnsen, J.; Raffatellu, M. Siderophores: More than Stealing Iron. mBio 2016, 7, e01906-16. [Google Scholar] [CrossRef]
- Kramer, J.; Özkaya, Ö.; Kümmerli, R. Bacterial siderophores in community and host interactions. Nat. Rev. Microbiol. 2020, 18, 152–163. [Google Scholar] [CrossRef]
- Bachman, M.A.; Oyler, J.E.; Burns, S.H.; Caza, M.; Lépine, F.; Dozois, C.M.; Weiser, J.N.; Bäumler, A.J. Klebsiella pneumoniae Yersiniabactin Promotes Respiratory Tract Infection through Evasion of Lipocalin 2. Infect. Immun. 2011, 79, 3309–3316. [Google Scholar] [CrossRef] [PubMed]
- Sikkema, D.J.; Brubaker, R.R. Outer membrane peptides of Yersinia pestis mediating siderophore-independent assimilation of iron. Biol. Met. 1989, 2, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Brock, J.H.; Williams, P.H.; Licéaga, J.; Wooldridge, K.G. Relative availability of transferrin-bound iron and cell-derived iron to aerobactin-producing and enterochelin-producing strains of Escherichia coli and to other microorganisms. Infect. Immun. 1991, 59, 3185–3190. [Google Scholar] [CrossRef] [PubMed]
- Gehrer, C.M.; Hoffmann, A.; Hilbe, R.; Grubwieser, P.; Mitterstiller, A.M.; Talasz, H.; Fang, F.C.; Meyron-Holtz, E.G.; Atkinson, S.H.; Weiss, G.; et al. Availability of Ferritin-Bound Iron to Enterobacteriaceae. Int. J. Mol. Sci. 2022, 23, 13087. [Google Scholar] [CrossRef]
- Deneer, H.G.; Healey, V.; Boychuk, I. Reduction of exogenous ferric iron by a surface-associated ferric reductase of Listeria spp. Microbiology 1995, 141 Pt 8, 1985–1992. [Google Scholar] [CrossRef]
- Jin, B.; Newton, S.M.; Shao, Y.; Jiang, X.; Charbit, A.; Klebba, P.E. Iron acquisition systems for ferric hydroxamates, haemin and haemoglobin in Listeria monocytogenes. Mol. Microbiol. 2006, 59, 1185–1198. [Google Scholar] [CrossRef]
- Whitby, P.W.; VanWagoner, T.M.; Springer, J.M.; Morton, D.J.; Seale, T.W.; Stull, T.L. Burkholderia cenocepacia utilizes ferritin as an iron source. J. Med. Microbiol. 2006, 55, 661–668. [Google Scholar] [CrossRef]
- Dehner, C.; Morales-Soto, N.; Behera, R.K.; Shrout, J.; Theil, E.C.; Maurice, P.A.; Dubois, J.L. Ferritin and ferrihydrite nanoparticles as iron sources for Pseudomonas aeruginosa. J. Biol. Inorg. Chem. JBIC A Publ. Soc. Biol. Inorg. Chem. 2013, 18, 371–381. [Google Scholar] [CrossRef]
- Segond, D.; Abi Khalil, E.; Buisson, C.; Daou, N.; Kallassy, M.; Lereclus, D.; Arosio, P.; Bou-Abdallah, F.; Nielsen Le Roux, C. Iron acquisition in Bacillus cereus: The roles of IlsA and bacillibactin in exogenous ferritin iron mobilization. PLoS Pathog. 2014, 10, e1003935. [Google Scholar] [CrossRef]
- Eichenbaum, Z.; Muller, E.; Morse, S.A.; Scott, J.R. Acquisition of iron from host proteins by the group A streptococcus. Infect. Immun. 1996, 64, 5428–5429. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.M.; Oliver, J.D. Ability ofVibrio vulnificus to obtain iron from transferrin and other iron-binding proteins. Curr. Microbiol. 1987, 15, 155–157. [Google Scholar] [CrossRef]
- Wong, H.C.; Liu, C.C.; Yu, C.M.; Lee, Y.S. Utilization of iron sources and its possible roles in the pathogenesis of Vibrio parahaemolyticus. Microbiol. Immunol. 1996, 40, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Gobin, J.; Horwitz, M.A. Exochelins of Mycobacterium tuberculosis remove iron from human iron-binding proteins and donate iron to mycobactins in the M. tuberculosis cell wall. J. Exp. Med. 1996, 183, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Kheirandish, M.; Motlagh, B.; Afshar, D. Ferritin Degradation by Pneumococcal HtrA, RadA and ClpP Serine Proteases: A Probable Way For Releasing and Acquisition Of Iron. Infect. Drug Resist. 2020, 13, 3145–3152. [Google Scholar] [CrossRef] [PubMed]
- Flores, S.E.; Aitchison, A.; Day, A.S.; Keenan, J.I. Helicobacter pylori infection perturbs iron homeostasis in gastric epithelial cells. PLoS ONE 2017, 12, e0184026. [Google Scholar] [CrossRef]
- Al-Younes, H.M.; Meyer, T.F. Host Ferritin Translocates into the Chlamydial Inclusion: A Clear Alteration in its Subcellular Distribution as a Result of Infection. Dirasat Pure Sci. 2011, 38, 4–33. [Google Scholar]
- Bauckman, K.A.; Mysorekar, I.U. Ferritinophagy drives uropathogenic Escherichia coli persistence in bladder epithelial cells. Autophagy 2016, 12, 850–863. [Google Scholar] [CrossRef]
- Abreu, R.; Essler, L.; Loy, A.; Quinn, F.; Giri, P. Heparin inhibits intracellular Mycobacterium tuberculosis bacterial replication by reducing iron levels in human macrophages. Sci. Rep. 2018, 8, 7296. [Google Scholar] [CrossRef]
- Larson, J.A.; Howie, H.L.; So, M. Neisseria meningitidis accelerates ferritin degradation in host epithelial cells to yield an essential iron source. Mol. Microbiol. 2004, 53, 807–820. [Google Scholar] [CrossRef]
- Yan, Q.; Zhang, W.; Lin, M.; Teymournejad, O.; Budachetri, K.; Lakritz, J.; Rikihisa, Y. Iron robbery by intracellular pathogen via bacterial effector-induced ferritinophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2026598118. [Google Scholar] [CrossRef] [PubMed]
- Olakanmi, O.; Schlesinger, L.S.; Ahmed, A.; Britigan, B.E. Intraphagosomal Mycobacterium tuberculosis acquires iron from both extracellular transferrin and intracellular iron pools. Impact of interferon-gamma and hemochromatosis. J. Biol. Chem. 2002, 277, 49727–49734. [Google Scholar] [CrossRef] [PubMed]
- Archibald, F.S.; DeVoe, I.W. Iron acquisition by Neisseria meningitidis in vitro. Infect. Immun. 1980, 27, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Calver, G.A.; Kenny, C.P.; Kushner, D.J. Inhibition of the growth of Neisseria meningitidis by reduced ferritin and other iron-binding agents. Infect. Immun. 1979, 25, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Ferring-Appel, D.; Casarrubea, D.; Sonnweber, T.; Viatte, L.; Schroll, A.; Haschka, D.; Fang, F.C.; Hentze, M.W.; Weiss, G.; et al. Iron Regulatory Proteins Mediate Host Resistance to Salmonella Infection. Cell Host Microbe 2015, 18, 254–261. [Google Scholar] [CrossRef]
- Knuff, K.; Finlay, B.B. What the SIF Is Happening-The Role of Intracellular Salmonella-Induced Filaments. Front. Cell. Infect. Microbiol. 2017, 7, 335. [Google Scholar] [CrossRef]
- Drecktrah, D.; Knodler, L.A.; Ireland, R.; Steele-Mortimer, O. The mechanism of Salmonella entry determines the vacuolar environment and intracellular gene expression. Traffic 2006, 7, 39–51. [Google Scholar] [CrossRef]
- Garcia-del Portillo, F.; Foster, J.W.; Maguire, M.E.; Finlay, B.B. Characterization of the micro-environment of Salmonella typhimurium-containing vacuoles within MDCK epithelial cells. Mol. Microbiol. 1992, 6, 3289–3297. [Google Scholar] [CrossRef]
- Lou, L.; Zhang, P.; Piao, R.; Wang, Y. Salmonella Pathogenicity Island 1 (SPI-1) and Its Complex Regulatory Network. Front. Cell. Infect. Microbiol. 2019, 9, 270. [Google Scholar] [CrossRef]
- Steele-Mortimer, O.; Méresse, S.; Gorvel, J.P.; Toh, B.H.; Finlay, B.B. Biogenesis of Salmonella typhimurium-containing vacuoles in epithelial cells involves interactions with the early endocytic pathway. Cell. Microbiol. 1999, 1, 33–49. [Google Scholar] [CrossRef]
- Mallo, G.V.; Espina, M.; Smith, A.C.; Terebiznik, M.R.; Alemán, A.; Finlay, B.B.; Rameh, L.E.; Grinstein, S.; Brumell, J.H. SopB promotes phosphatidylinositol 3-phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J. Cell Biol. 2008, 182, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Cirulis, J.T.; Casanova, J.E.; Scidmore, M.A.; Brumell, J.H. Interaction of the Salmonella-containing vacuole with the endocytic recycling system. J. Biol. Chem. 2005, 280, 24634–24641. [Google Scholar] [CrossRef]
- Birmingham, C.L.; Smith, A.C.; Bakowski, M.A.; Yoshimori, T.; Brumell, J.H. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 2006, 281, 11374–11383. [Google Scholar] [CrossRef] [PubMed]
- Thurston, T.L.; Wandel, M.P.; von Muhlinen, N.; Foeglein, A.; Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012, 482, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Kreibich, S.; Emmenlauer, M.; Fredlund, J.; Rämö, P.; Münz, C.; Dehio, C.; Enninga, J.; Hardt, W.-D. Autophagy Proteins Promote Repair of Endosomal Membranes Damaged by the Salmonella Type Three Secretion System 1. Cell Host Microbe 2015, 18, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Méresse, S.; Steele-Mortimer, O.; Finlay, B.B.; Gorvel, J.P. The rab7 GTPase controls the maturation of Salmonella typhimurium-containing vacuoles in HeLa cells. EMBO J. 1999, 18, 4394–4403. [Google Scholar] [CrossRef]
- Beuzón, C.R.; Méresse, S.; Unsworth, K.E.; Ruíz-Albert, J.; Garvis, S.; Waterman, S.R.; Ryder, T.A.; Boucrot, E.; Holden, D.W. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 2000, 19, 3235–3249. [Google Scholar] [CrossRef]
- Garcia-del Portillo, F.; Zwick, M.B.; Leung, K.Y.; Finlay, B.B. Salmonella induces the formation of filamentous structures containing lysosomal membrane glycoproteins in epithelial cells. Proc. Natl. Acad. Sci. USA 1993, 90, 10544–10548. [Google Scholar] [CrossRef]
- Liss, V.; Swart, A.L.; Kehl, A.; Hermanns, N.; Zhang, Y.; Chikkaballi, D.; Böhles, N.; Deiwick, J.; Hensel, M. Salmonella enterica Remodels the Host Cell Endosomal System for Efficient Intravacuolar Nutrition. Cell Host Microbe 2017, 21, 390–402. [Google Scholar] [CrossRef]
- Green, E.R.; Mecsas, J. Bacterial Secretion Systems: An Overview. Microbiol. Spectr. 2016, 4, 215–239. [Google Scholar] [CrossRef]
- Beuzón, C.R.; Salcedo, S.P.; Holden, D.W. Growth and killing of a Salmonella enterica serovar Typhimurium sifA mutant strain in the cytosol of different host cell lines. Microbiology 2002, 148, 2705–2715. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, J.L.; Berche, P.; Mounier, J.; Richard, S.; Sansonetti, P. In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2. Infect. Immun. 1987, 55, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, B.; Abu Kwaik, Y. Idiosyncratic Biogenesis of Intracellular Pathogens-Containing Vacuoles. Front. Cell. Infect. Microbiol. 2021, 11, 722433. [Google Scholar] [CrossRef] [PubMed]
- Gruenheid, S.; Canonne-Hergaux, F.; Gauthier, S.; Hackam, D.J.; Grinstein, S.; Gros, P. The iron transport protein NRAMP2 is an integral membrane glycoprotein that colocalizes with transferrin in recycling endosomes. J. Exp. Med. 1999, 189, 831–841. [Google Scholar] [CrossRef]
- Wang, D.; Wang, L.H.; Zhao, Y.; Lu, Y.P.; Zhu, L. Hypoxia regulates the ferrous iron uptake and reactive oxygen species level via divalent metal transporter 1 (DMT1) Exon1B by hypoxia-inducible factor-1. IUBMB Life 2010, 62, 629–636. [Google Scholar] [CrossRef]
- Erra Díaz, F.; Dantas, E.; Geffner, J. Unravelling the Interplay between Extracellular Acidosis and Immune Cells. Mediat. Inflamm. 2018, 2018, 1218297. [Google Scholar] [CrossRef]
- Ludwiczek, S.; Theurl, I.; Muckenthaler, M.U.; Jakab, M.; Mair, S.M.; Theurl, M.; Kiss, J.; Paulmichl, M.; Hentze, M.W.; Ritter, M.; et al. Ca2+ channel blockers reverse iron overload by a new mechanism via divalent metal transporter-1. Nat. Med. 2007, 13, 448–454. [Google Scholar] [CrossRef]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- Mair, S.M.; Nairz, M.; Bellmann-Weiler, R.; Muehlbacher, T.; Schroll, A.; Theurl, I.; Moser, P.L.; Talasz, H.; Fang, F.C.; Weiss, G. Nifedipine affects the course of Salmonella enterica serovar Typhimurium infection by modulating macrophage iron homeostasis. J. Infect. Dis. 2011, 204, 685–694. [Google Scholar] [CrossRef]
- Yang, S.; Deng, Q.; Sun, L.; Dong, K.; Li, Y.; Wu, S.; Huang, R. Salmonella effector SpvB interferes with intracellular iron homeostasis via regulation of transcription factor NRF2. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 13450–13464. [Google Scholar] [CrossRef]
- Kuang, H.; Dou, G.; Cheng, L.; Wang, X.; Xu, H.; Liu, X.; Ding, F.; Yang, X.; Liu, S.; Bao, L.; et al. Humoral regulation of iron metabolism by extracellular vesicles drives antibacterial response. Nat. Metab. 2023, 5, 111–128. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Conlon, M.M.; Rider, M.A.; Brownstein, N.C.; Meckes, D.G., Jr. Nanoparticle analysis sheds budding insights into genetic drivers of extracellular vesicle biogenesis. J. Extracell. Vesicles 2016, 5, 31295. [Google Scholar] [CrossRef]
- Tumbarello, D.A.; Manna, P.T.; Allen, M.; Bycroft, M.; Arden, S.D.; Kendrick-Jones, J.; Buss, F. The Autophagy Receptor TAX1BP1 and the Molecular Motor Myosin VI Are Required for Clearance of Salmonella Typhimurium by Autophagy. PLoS Pathog. 2015, 11, e1005174. [Google Scholar] [CrossRef]
- Zeng, M.; Sang, W.; Chen, S.; Chen, R.; Zhang, H.; Xue, F.; Li, Z.; Liu, Y.; Gong, Y.; Zhang, H.; et al. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol. Lett. 2017, 271, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Alshareef, M.H.; Hartland, E.L.; McCaffrey, K. Effectors Targeting the Unfolded Protein Response during Intracellular Bacterial Infection. Microorganisms 2021, 9, 705. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Van de Leur, E.; Meurer, S.K.; Buhl, E.M.; Weiskirchen, R. N-Glycosylation of Lipocalin 2 Is Not Required for Secretion or Exosome Targeting. Front. Pharmacol. 2018, 9, 426. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Suazo, C.; Boltansky, A.; Ursu, M.; Carvajal, D.; Innocenti, G.; Vukusich, A.; Hurtado, M.; Villanueva, S.; Carreño, J.E.; et al. Urinary exosomes as a source of kidney dysfunction biomarker in renal transplantation. Transplant. Proc. 2013, 45, 3719–3723. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Pokrishevsky, E.; Silverman, J.M.; Cashman, N.R. Exosome-dependent and independent mechanisms are involved in prion-like transmission of propagated Cu/Zn superoxide dismutase misfolding. Prion 2014, 8, 331–335. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gehrer, C.M.; Mitterstiller, A.-M.; Grubwieser, P.; Meyron-Holtz, E.G.; Weiss, G.; Nairz, M. Advances in Ferritin Physiology and Possible Implications in Bacterial Infection. Int. J. Mol. Sci. 2023, 24, 4659. https://doi.org/10.3390/ijms24054659
Gehrer CM, Mitterstiller A-M, Grubwieser P, Meyron-Holtz EG, Weiss G, Nairz M. Advances in Ferritin Physiology and Possible Implications in Bacterial Infection. International Journal of Molecular Sciences. 2023; 24(5):4659. https://doi.org/10.3390/ijms24054659
Chicago/Turabian StyleGehrer, Clemens M., Anna-Maria Mitterstiller, Philipp Grubwieser, Esther G. Meyron-Holtz, Günter Weiss, and Manfred Nairz. 2023. "Advances in Ferritin Physiology and Possible Implications in Bacterial Infection" International Journal of Molecular Sciences 24, no. 5: 4659. https://doi.org/10.3390/ijms24054659
APA StyleGehrer, C. M., Mitterstiller, A.-M., Grubwieser, P., Meyron-Holtz, E. G., Weiss, G., & Nairz, M. (2023). Advances in Ferritin Physiology and Possible Implications in Bacterial Infection. International Journal of Molecular Sciences, 24(5), 4659. https://doi.org/10.3390/ijms24054659