
Schmidtea mediterranea as a Model Organism to Study the Molecular Background of Human Motile Ciliopathies



Abstract
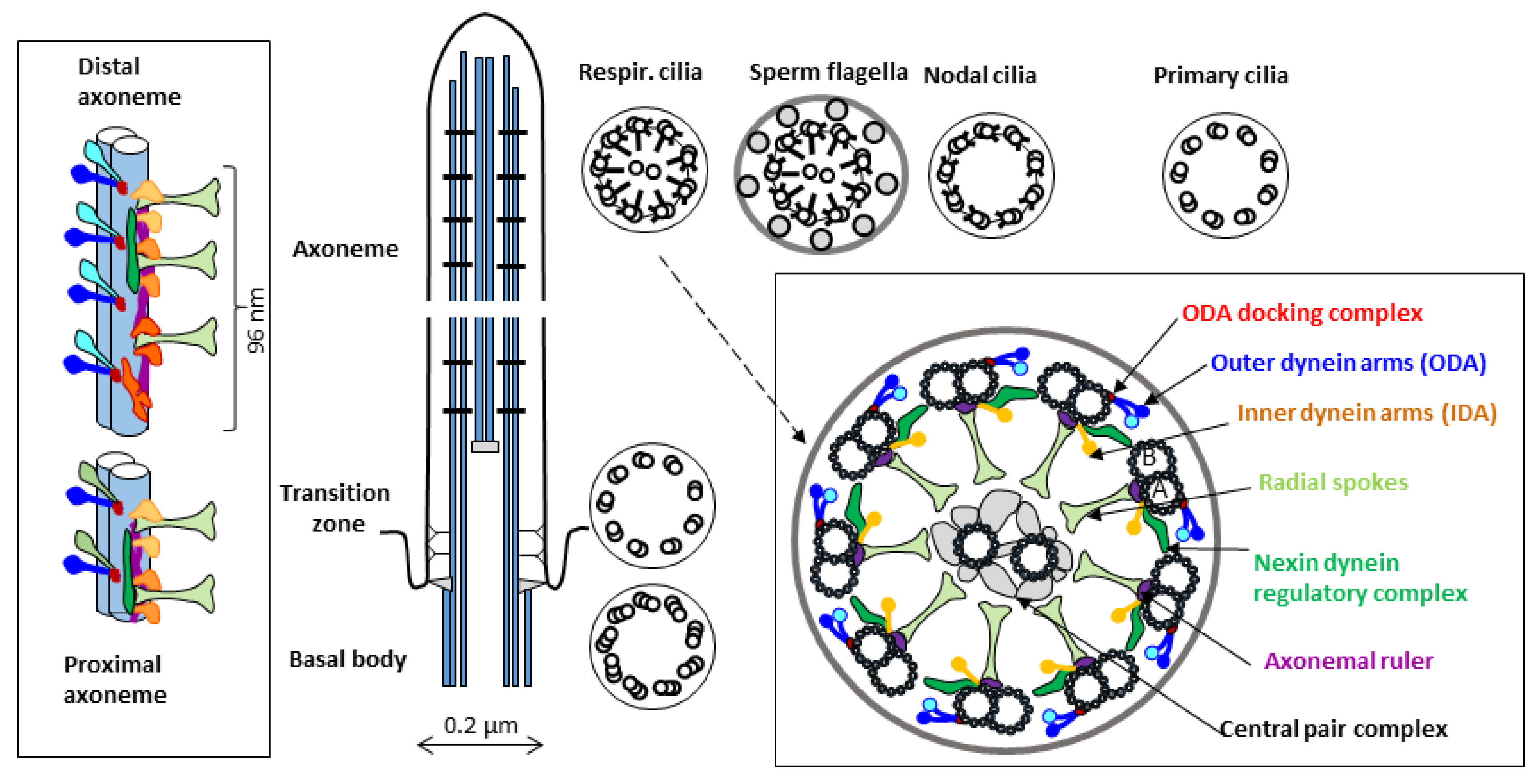
1. Introduction–Cilia and Ciliopathies
1.1. Primary Ciliary Dyskinesia
1.2. The Validation of New Genes Underlying PCD and PCD-like Ciliopathies—The Role of Model Organisms
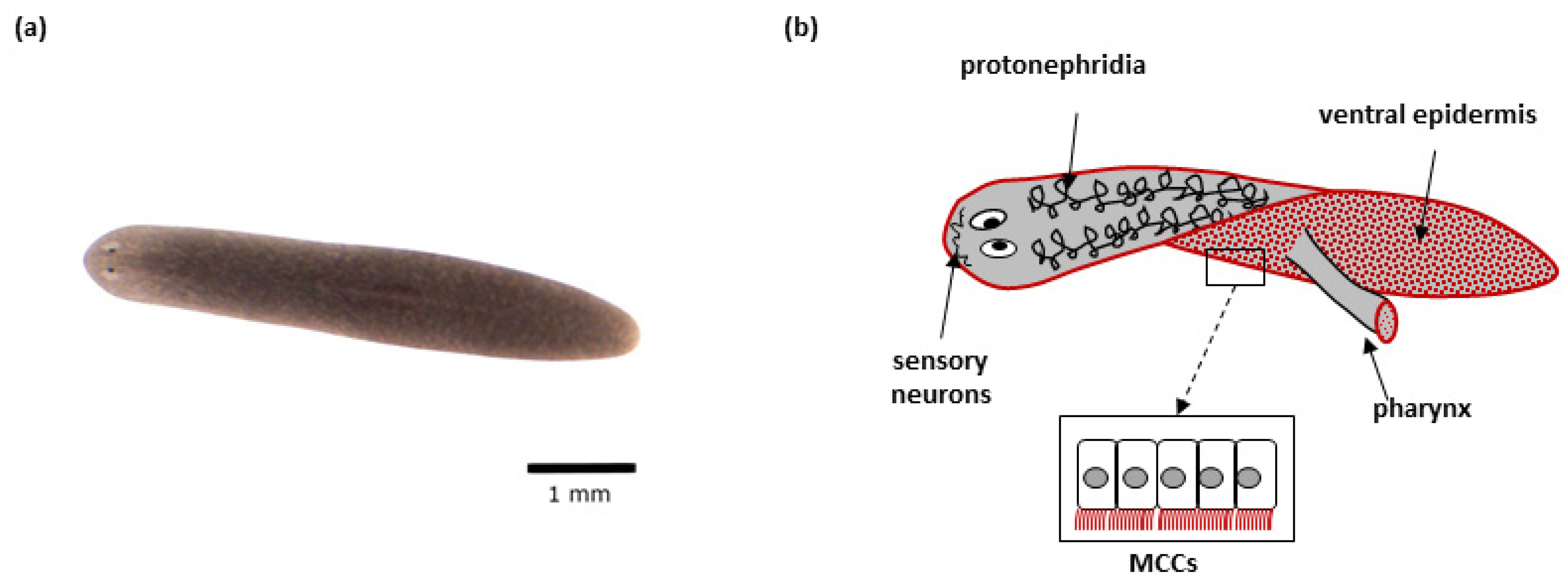
2. Schmidtea mediterranea
2.1. Advantages of the S. mediterranea as a Model Organism
2.2. S. mediterranea Model in the Context of Studying Cilia Biology
2.3. S. mediterranea Model in the Context of Studying PCD and PCD-like Ciliopathies
3. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carvalho-Santos, Z.; Azimzadeh, J.; Pereira-Leal, J.B.; Bettencourt-Dias, M. Evolution: Tracing the origins of centrioles, cilia, and flagella. J. Cell Biol. 2011, 194, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.R. Evolution of cilia. Cold Spring Harb. Perspect. Biol. 2017, 9, a028290. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Scholey, J.M. Assembly, functions and evolution of archaella, flagella and cilia. Curr. Biol. 2018, 28, R278–R292. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Reiter, J.F. Open sesame: How transition fibers and the transition zone control ciliary composition. Cold Spring Harb. Perspect. Biol. 2017, 9, a028134. [Google Scholar] [CrossRef]
- Takeda, S.; Narita, K. Structure and function of vertebrate cilia, towards a new taxonomy. Differentiation 2012, 83, S4–S11. [Google Scholar] [CrossRef]
- Kempeneers, C.; Chilvers, M.A. To beat, or not to beat, that is question! The spectrum of ciliopathies. Pediatr. Pulmonol. 2018, 53, 1122–1129. [Google Scholar] [CrossRef]
- Shah, A.S.; Ben-Shahar, Y.; Moninger, T.O.; Kline, J.N.; Welsh, M.J. Motile cilia of human airway epithelia are chemosensory. Science 2009, 325, 1131–1134. [Google Scholar] [CrossRef]
- Jain, R.; Javidan-Nejad, C.; Alexander-Brett, J.; Horani, A.; Cabellon, M.C.; Walter, M.J.; Brody, S.L. Sensory functions of motile cilia and implication for bronchiectasis. Front. Biosci. 2012, 4, 1088–1098. [Google Scholar] [CrossRef][Green Version]
- Brooks, E.R.; Wallingford, J.B. Multiciliated cells. Curr. Biol. 2014, 24, R973–R982. [Google Scholar] [CrossRef]
- Sironen, A.; Shoemark, A.; Patel, M.; Loebinger, M.R.; Mitchison, H.M. Sperm defects in primary ciliary dyskinesia and related causes of male infertility. Cell. Mol. Life Sci. 2020, 77, 2029–2048. [Google Scholar] [CrossRef]
- Shinohara, K.; Hamada, H. Cilia in left-right symmetry breaking. Cold Spring Harb. Perspect. Biol. 2017, 9, a028282. [Google Scholar] [CrossRef] [PubMed]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Nishimura, Y.; Kasahara, K.; Shiromizu, T.; Watanabe, M.; Inagaki, M. Primary cilia as signaling hubs in health and disease. Adv. Sci. 2019, 6, 1801138. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Mick, D.U. Establishing and regulating the composition of cilia for signal transduction. Nat. Rev. Mol. Cell Biol. 2019, 20, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Samsel, Z.; Sekretarska, J.; Osinka, A.; Wloga, D.; Joachimiak, E. Central apparatus, the molecular kickstarter of ciliary and flagellar nanomachines. Int. J. Mol. Sci. 2021, 22, 3013. [Google Scholar] [CrossRef]
- Kurkowiak, M.; Ziętkiewicz, E.; Witt, M. Recent advances in primary ciliary dyskinesia genetics. J. Med. Genet. 2015, 52, 1–9. [Google Scholar] [CrossRef]
- Osinka, A.; Poprzeczko, M.; Zielinska, M.M.; Fabczak, H.; Joachimiak, E.; Wloga, D. Ciliary proteins: Filling the gaps. Recent advances in deciphering the protein composition of motile ciliary complexes. Cells 2019, 8, 730. [Google Scholar] [CrossRef]
- Nicastro, D.; Schwartz, C.; Pierson, J.; Gaudette, R.; Porter, M.E.; McIntosh, J.R. The molecular architecture of axonemes revealed by cryoelectron tomography. Science 2006, 313, 944–948. [Google Scholar] [CrossRef]
- Oda, T.; Yanagisawa, H.; Kamiya, R.; Kikkawa, M. A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science 2014, 346, 857–860. [Google Scholar] [CrossRef]
- Whitfield, M.; Thomas, L.; Bequignon, E.; Schmitt, A.; Stouvenel, L.; Montantin, G.; Tissier, S.; Duquesnoy, P.; Copin, B.; Chantot, S.; et al. Mutations in DNAH17, encoding a sperm-specific axonemal outer dynein arm heavy chain, cause isolated male infertility due to asthenozoospermia. Am. J. Hum. Genet. 2019, 105, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Bazan, R.; Schröfel, A.; Joachimiak, E.; Poprzeczko, M.; Pigino, G.; Wloga, D. Ccdc113/Ccdc96 complex, a novel regulator of ciliary beating that connects radial spoke 3 to dynein g and the nexin link. PLoS Genet. 2021, 17, e1009388. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Stoyanova, M.; Rademacher, G.; Dutcher, S.K.; Brown, A.; Zhang, R. Structure of the decorated ciliary doublet microtubule. Cell 2019, 179, 909–922.e12. [Google Scholar] [CrossRef]
- Owa, M.; Uchihashi, T.; Yanagisawa, H.-A.; Yamano, T.; Iguchi, H.; Fukuzawa, H.; Wakabayashi, K.-I.; Ando, T.; Kikkawa, M. Inner lumen proteins stabilize doublet microtubules in cilia and flagella. Nat. Commun. 2019, 10, 1143. [Google Scholar] [CrossRef] [PubMed]
- Joachimiak, E.; Osinka, A.; Farahat, H.; Świderska, B.; Sitkiewicz, E.; Poprzeczko, M.; Fabczak, H.; Wloga, D. Composition and function of the C1b/C1f region in the ciliary central apparatus. Sci. Rep. 2021, 11, 11760. [Google Scholar] [CrossRef]
- Satir, P.; Heuser, T.; Sale, W.S. A structural basis for how motile cilia beat. Bioscience 2014, 64, 1073–1083. [Google Scholar] [CrossRef]
- Pigino, G.; Ishikawa, T. Axonemal radial spokes: 3D structure, function and assembly. Bioarchitecture 2012, 2, 50–58. [Google Scholar] [CrossRef]
- Choksi, S.P.; Lauter, G.; Swoboda, P.; Roy, S. Switching on cilia: Transcriptional networks regulating ciliogenesis. Development 2014, 141, 1427–1441. [Google Scholar] [CrossRef]
- Boon, M.; Wallmeier, J.; Ma, L.; Loges, N.T.; Jaspers, M.; Olbrich, H.; Dougherty, G.W.; Raidt, J.; Werner, C.; Amirav, I.; et al. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Commun. 2014, 5, 4418. [Google Scholar] [CrossRef]
- Wallmeier, J.; Al-Mutairi, D.A.; Chen, C.-T.; Loges, N.T.; Pennekamp, P.; Menchen, T.; Ma, L.; Shamseldin, H.E.; Olbrich, H.; Dougherty, G.W.; et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Genet. 2014, 46, 646–651. [Google Scholar] [CrossRef]
- Wallmeier, J.; Frank, D.; Shoemark, A.; Nöthe-Menchen, T.; Cindric, S.; Olbrich, H.; Loges, N.T.; Aprea, I.; Dougherty, G.W.; Pennekamp, P.; et al. De novo mutations in FOXJ1 result in a motile ciliopathy with hydrocephalus and randomization of left/right body asymmetry. Am. J. Hum. Genet. 2019, 105, 1030–1039. [Google Scholar] [CrossRef]
- Lechtreck, K.F. IFT-Cargo interactions and protein transport in cilia. Trends Biochem. Sci. 2015, 40, 765–778. [Google Scholar] [CrossRef]
- Ishikawa, H.; Marshall, W.F. Intraflagellar transport and ciliary dynamics. Cold Spring Harb. Perspect. Biol. 2017, 9, a021998. [Google Scholar] [CrossRef] [PubMed]
- Wallmeier, J.; Nielsen, K.G.; Kuehni, C.E.; Lucas, J.S.; Leigh, M.W.; Zariwala, M.A.; Omran, H. Motile ciliopathies. Nat. Rev. Dis. Prim. 2020, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Zaragosi, L.-E.; Mitchison, H.M. Motile cilia and airway disease. Semin. Cell Dev. Biol. 2021, 110, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Focșa, I.O.; Budișteanu, M.; Bălgrădean, M. Clinical and genetic heterogeneity of primary ciliopathies (review). Int. J. Mol. Med. 2021, 48, 176. [Google Scholar] [CrossRef] [PubMed]
- Tobin, J.L.; Beales, P.L. The nonmotile ciliopathies. Genet. Med. 2009, 11, 386–402. [Google Scholar] [CrossRef]
- Horani, A.; Ferkol, T.W.; Dutcher, S.K.; Brody, S.L. Genetics and biology of primary ciliary dyskinesia. Paediatr. Respir. Rev. 2016, 18, 18–24. [Google Scholar] [CrossRef]
- Mirra, V.; Werner, C.; Santamaria, F. Primary ciliary dyskinesia: An update on clinical aspects, genetics, diagnosis, and future treatment strategies. Front. Pediatr. 2017, 5, 135. [Google Scholar] [CrossRef]
- Damseh, N.; Quercia, N.; Rumman, N.; Dell, S.D.; Kim, R.H. Primary ciliary dyskinesia: Mechanisms and management. Appl. Clin. Genet. 2017, 10, 67–74. [Google Scholar] [CrossRef]
- Lucas, J.S.; Davis, S.D.; Omran, H.; Shoemark, A. Primary ciliary dyskinesia in the genomics age. Lancet Respir. Med. 2020, 8, 202–216. [Google Scholar] [CrossRef]
- Horani, A.; Ferkol, T.W. Understanding primary ciliary dyskinesia and other ciliopathies. J. Pediatr. 2021, 230, 15–22. [Google Scholar] [CrossRef]
- Wheway, G.; Thomas, N.S.; Carroll, M.; Coles, J.; Doherty, R.; Genomics England Research Consortium; Goggin, P.; Green, B.; Harris, A.; Hunt, D.; et al. Whole genome sequencing in the diagnosis of primary ciliary dyskinesia. BMC Med. Genom. 2021, 14, 234. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.K.; Ferkol, T.W.; Davis, S.D. Emerging genotype-phenotype relationships in primary ciliary dyskinesia. Int. J. Mol. Sci. 2021, 22, 8272. [Google Scholar] [CrossRef] [PubMed]
- van Dam, T.J.; Wheway, G.; Slaats, G.G.; SYSCILIA Study Group; Huynen, M.A.; Giles, R.H. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia 2013, 2, 7. [Google Scholar] [CrossRef]
- Vasquez, S.S.V.; van Dam, J.; Wheway, G. An updated SYSCILIA gold standard (SCGSv2) of known ciliary genes, revealing the vast progress that has been made in the cilia research field. Mol. Biol. Cell 2021, 32, br13. [Google Scholar] [CrossRef]
- Vij, S.; Rink, J.C.; Ho, H.K.; Babu, D.; Eitel, M.; Narasimhan, V.; Tiku, V.; Westbrook, J.; Schierwater, B.; Roy, S. Evolutionarily ancient association of the FoxJ1 transcription factor with the motile ciliogenic program. PLoS Genet. 2012, 8, e1003019. [Google Scholar] [CrossRef] [PubMed]
- Brody, S.L.; Yan, X.H.; Wuerffel, M.K.; Song, S.K.; Shapiro, S.D. Ciliogenesis and left-right axis defects in forkhead factor HFH-4-Null mice. Am. J. Respir. Cell. Mol. Biol. 2000, 23, 45–51. [Google Scholar] [CrossRef]
- Stubbs, J.L.; Oishi, I.; Izpisúa Belmonte, J.C.; Kintner, C. The forkhead protein Foxj1 specifies node-like cilia in xenopus and zebrafish embryos. Nat. Genet. 2008, 40, 1454–1460. [Google Scholar] [CrossRef]
- Moore, A.; Escudier, E.; Roger, G.; Tamalet, A.; Pelosse, B.; Marlin, S.; Clément, A.; Geremek, M.; Delaisi, B.; Bridoux, A.-M.; et al. RPGR Is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J. Med. Genet. 2006, 43, 326–333. [Google Scholar] [CrossRef]
- Bukowy-Bieryłło, Z.; Ziętkiewicz, E.; Loges, N.T.; Wittmer, M.; Geremek, M.; Olbrich, H.; Fliegauf, M.; Voelkel, K.; Rutkiewicz, E.; Rutland, J.; et al. RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr. Pulmonol. 2013, 48, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Murga-Zamalloa, C.A.; Chan, L.; Hitchcock, P.F.; Swaroop, A.; Khanna, H. Human retinopathy-associated ciliary protein retinitis pigmentosa GTPase regulator mediates cilia-dependent vertebrate development. Hum. Mol. Genet. 2010, 19, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Hannah, W.B.; DeBrosse, S.; Kinghorn, B.; Strausbaugh, S.; Aitken, M.L.; Rosenfeld, M.; Wolf, W.E.; Knowles, M.R.; Zariwala, M.A. The expanding phenotype of OFD1-related disorders: Hemizygous loss-of-function variants in three patients with primary ciliary dyskinesia. Mol. Genet. Genom. Med. 2019, 7, e911. [Google Scholar] [CrossRef] [PubMed]
- Bukowy-Bieryllo, Z.; Rabiasz, A.; Dabrowski, M.; Pogorzelski, A.; Wojda, A.; Dmenska, H.; Grzela, K.; Sroczynski, J.; Witt, M.; Zietkiewicz, E. Truncating mutations in exons 20 and 21 of OFD1 can cause primary ciliary dyskinesia without associated syndromic symptoms. J. Med. Genet. 2019, 56, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Bengueddach, H.; Lemullois, M.; Aubusson-Fleury, A.; Koll, F. Basal body positioning and anchoring in the multiciliated cell paramecium tetraurelia: Roles of OFD1 and VFL3. Cilia 2017, 6, 6. [Google Scholar] [CrossRef]
- Ferrante, M.I.; Zullo, A.; Barra, A.; Bimonte, S.; Messaddeq, N.; Studer, M.; Dollé, P.; Franco, B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat. Genet. 2006, 38, 112–117. [Google Scholar] [CrossRef]
- Azimzadeh, J.; Wong, M.L.; Downhour, D.M.; Sánchez Alvarado, A.; Marshall, W.F. Centrosome loss in the evolution of planarians. Science 2012, 335, 461–463. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Yin, W.-N.; Sears, P.R.; Werner, M.E.; Brotslaw, E.J.; Mitchell, B.J.; Jania, C.M.; Zeman, K.L.; Rogers, T.D.; Herring, L.E.; et al. Lack of GAS2L2 causes PCD by impairing cilia orientation and mucociliary clearance. Am. J. Hum. Genet. 2019, 104, 229–245. [Google Scholar] [CrossRef]
- Mitchison, H.M.; Schmidts, M.; Loges, N.T.; Freshour, J.; Dritsoula, A.; Hirst, R.A.; O’Callaghan, C.; Blau, H.; Al Dabbagh, M.; Olbrich, H.; et al. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012, 44, 381–389, S1-2. [Google Scholar] [CrossRef]
- Fassad, M.R.; Shoemark, A.; le Borgne, P.; Koll, F.; Patel, M.; Dixon, M.; Hayward, J.; Richardson, C.; Frost, E.; Jenkins, L.; et al. C11orf70 mutations disrupting the intraflagellar transport-dependent assembly of multiple axonemal dyneins cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2018, 102, 956–972. [Google Scholar] [CrossRef]
- Höben, I.M.; Hjeij, R.; Olbrich, H.; Dougherty, G.W.; Nöthe-Menchen, T.; Aprea, I.; Frank, D.; Pennekamp, P.; Dworniczak, B.; Wallmeier, J.; et al. Mutations in C11orf70 cause primary ciliary dyskinesia with randomization of left/right body asymmetry due to defects of outer and inner dynein arms. Am. J. Hum. Genet. 2018, 102, 973–984. [Google Scholar] [CrossRef]
- Zietkiewicz, E.; Bukowy-Bieryllo, Z.; Rabiasz, A.; Daca-Roszak, P.; Wojda, A.; Voelkel, K.; Rutkiewicz, E.; Pogorzelski, A.; Rasteiro, M.; Witt, M. CFAP300: Mutations in slavic patients with primary ciliary dyskinesia and a role in ciliary dynein arms trafficking. Am. J. Respir. Cell Mol. Biol. 2019, 61, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Ostrowski, L.E.; Loges, N.T.; Hurd, T.; Leigh, M.W.; Huang, L.; Wolf, W.E.; Carson, J.L.; Hazucha, M.J.; Yin, W.; et al. Mutations in SPAG1 cause primary ciliary dyskinesia associated with defective outer and inner dynein arms. Am. J. Hum. Genet. 2013, 93, 711–720. [Google Scholar] [CrossRef]
- Loges, N.T.; Olbrich, H.; Becker-Heck, A.; Häffner, K.; Heer, A.; Reinhard, C.; Schmidts, M.; Kispert, A.; Zariwala, M.A.; Leigh, M.W.; et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009, 85, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Duquesnoy, P.; Escudier, E.; Vincensini, L.; Freshour, J.; Bridoux, A.-M.; Coste, A.; Deschildre, A.; de Blic, J.; Legendre, M.; Montantin, G.; et al. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009, 85, 890–896. [Google Scholar] [CrossRef]
- Thi-Kim Vu, H.; Rink, J.C.; McKinney, S.A.; McClain, M.; Lakshmanaperumal, N.; Alexander, R.; Sánchez Alvarado, A. Stem cells and fluid flow drive cyst formation in an invertebrate excretory organ. eLife 2015, 4, e07405. [Google Scholar] [CrossRef]
- Van Rooijen, E.; Giles, R.H.; Voest, E.E.; van Rooijen, C.; Schulte-Merker, S.; van Eeden, F.J. LRRC50, a conserved ciliary protein implicated in polycystic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Druley, T.E.; Zariwala, M.A.; Patel, A.C.; Levinson, B.T.; Van Arendonk, L.G.; Thornton, K.C.; Giacalone, J.C.; Albee, A.J.; Wilson, K.S.; et al. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 685–693. [Google Scholar] [CrossRef]
- Diggle, C.P.; Moore, D.J.; Mali, G.; zur Lage, P.; Ait-Lounis, A.; Schmidts, M.; Shoemark, A.; Munoz, A.G.; Halachev, M.R.; Gautier, P.; et al. HEATR2 plays a conserved role in assembly of the ciliary motile apparatus. PLoS Genet. 2014, 10, e1004577. [Google Scholar] [CrossRef]
- Paff, T.; Daniels, J.M.A.; Weersink, E.J.; Lutter, R.; Vonk Noordegraaf, A.; Haarman, E.G. A randomised controlled trial on the effect of inhaled hypertonic saline on quality of life in primary ciliary dyskinesia. Eur. Respir. J. 2017, 49, 1601770. [Google Scholar] [CrossRef] [PubMed]
- Olcese, C.; Patel, M.P.; Shoemark, A.; Kiviluoto, S.; Legendre, M.; Williams, H.J.; Vaughan, C.K.; Hayward, J.; Goldenberg, A.; Emes, R.D.; et al. X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat. Commun. 2017, 8, 14279. [Google Scholar] [CrossRef]
- Lennon, J.; zur Lage, P.; von Kriegsheim, A.; Jarman, A.P. Strongly truncated Dnaaf4 plays a conserved role in drosophila ciliary dynein assembly as part of an R2TP-Like co-chaperone complex with Dnaaf6. Front. Genet. 2022, 13, 943197. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Shinohara, K.; Botilde, Y.; Nabeshima, R.; Asai, Y.; Fukumoto, A.; Hasegawa, T.; Matsuo, M.; Takeda, H.; Shiratori, H.; et al. Pih1d3 is required for cytoplasmic preassembly of axonemal dynein in mouse sperm. J. Cell Biol. 2014, 204, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Zariwala, M.A.; Gee, H.Y.; Kurkowiak, M.; Al-Mutairi, D.A.; Leigh, M.W.; Hurd, T.W.; Hjeij, R.; Dell, S.D.; Chaki, M.; Dougherty, G.W.; et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am. J. Hum. Genet. 2013, 93, 336–345. [Google Scholar] [CrossRef]
- Moore, D.J.; Onoufriadis, A.; Shoemark, A.; Simpson, M.A.; zur Lage, P.I.; de Castro, S.C.; Bartoloni, L.; Gallone, G.; Petridi, S.; Woollard, W.J.; et al. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 93, 346–356. [Google Scholar] [CrossRef]
- Kott, E.; Duquesnoy, P.; Copin, B.; Legendre, M.; Dastot-Le Moal, F.; Montantin, G.; Jeanson, L.; Tamalet, A.; Papon, J.-F.; Siffroi, J.-P.; et al. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Kavlie, R.G.; Kernan, M.J.; Eberl, D.F. Hearing in drosophila requires TilB, a conserved protein associated with ciliary motility. Genetics 2010, 185, 177–188. [Google Scholar] [CrossRef][Green Version]
- Kishimoto, N.; Cao, Y.; Park, A.; Sun, Z. Cystic kidney gene seahorse regulates cilia-mediated processes and wnt pathways. Dev. Cell 2008, 14, 954–961. [Google Scholar] [CrossRef]
- Xue, J.C.; Goldberg, E. Identification of a novel testis-specific leucine-rich protein in humans and mice. Biol. Reprod. 2000, 62, 1278–1284. [Google Scholar] [CrossRef][Green Version]
- Austin-Tse, C.; Halbritter, J.; Zariwala, M.A.; Gilberti, R.M.; Gee, H.Y.; Hellman, N.; Pathak, N.; Liu, Y.; Panizzi, J.R.; Patel-King, R.S.; et al. Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 93, 672–686. [Google Scholar] [CrossRef]
- Omran, H.; Kobayashi, D.; Olbrich, H.; Tsukahara, T.; Loges, N.T.; Hagiwara, H.; Zhang, Q.; Leblond, G.; O’Toole, E.; Hara, C.; et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 2008, 456, 611–616. [Google Scholar] [CrossRef]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.V.; Shook, B.; Cantino, M.; Schwartz, D.; Jahnke, C.; et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.S.; Hjeij, R.; Vivante, A.; Bearce, E.A.; Dyer, L.; Wang, J.; Rawlins, L.; Kennedy, J.; Ubeyratna, N.; Fasham, J.; et al. Biallelic DAW1 variants cause a motile ciliopathy characterized by laterality defects and subtle ciliary beating abnormalities. Genet. Med. 2022, 24, 2249–2261. [Google Scholar] [CrossRef]
- Lesko, S.L.; Rouhana, L. Dynein assembly factor with WD repeat domains 1 (DAW1) is required for the function of motile cilia in the planarian schmidtea mediterranea. Dev. Growth Differ. 2020, 62, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.T.; Gao, C.; Lucker, B.F.; Cole, D.G.; Mitchell, D.R. ODA16 aids axonemal outer row dynein assembly through an interaction with the intraflagellar transport machinery. J. Cell Biol. 2008, 183, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Bouhouche, K.; Whitfield, M.; Thouvenin, G.; Coste, A.; Louis, B.; Szymanski, C.; Bequignon, E.; Papon, J.-F.; Castelli, M.; et al. TTC12 loss-of-function mutations cause primary ciliary dyskinesia and unveil distinct dynein assembly mechanisms in motile cilia versus flagella. Am. J. Hum. Genet. 2020, 106, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Pennarun, G.; Escudier, E.; Chapelin, C.; Bridoux, A.M.; Cacheux, V.; Roger, G.; Clément, A.; Goossens, M.; Amselem, S.; Duriez, B. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am. J. Hum. Genet. 1999, 65, 1508–1519. [Google Scholar] [CrossRef]
- Guichard, C.; Harricane, M.C.; Lafitte, J.J.; Godard, P.; Zaegel, M.; Tack, V.; Lalau, G.; Bouvagnet, P. Axonemal dynein intermediate-chain gene (DNAI1) mutations result in situs inversus and primary ciliary dyskinesia (kartagener syndrome). Am. J. Hum. Genet. 2001, 68, 1030–1035. [Google Scholar] [CrossRef]
- Loges, N.T.; Olbrich, H.; Fenske, L.; Mussaffi, H.; Horvath, J.; Fliegauf, M.; Kuhl, H.; Baktai, G.; Peterffy, E.; Chodhari, R.; et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008, 83, 547–558. [Google Scholar] [CrossRef]
- Rompolas, P.; Patel-King, R.S.; King, S.M. An outer arm dynein conformational switch is required for metachronal synchrony of motile cilia in planaria. Mol. Biol. Cell 2010, 21, 3669–3679. [Google Scholar] [CrossRef]
- Pennarun, G.; Chapelin, C.; Escudier, E.; Bridoux, A.M.; Dastot, F.; Cacheux, V.; Goossens, M.; Amselem, S.; Duriez, B. The human dynein intermediate chain 2 gene (DNAI2): Cloning, mapping, expression pattern, and evaluation as a candidate for primary ciliary dyskinesia. Hum. Genet. 2000, 107, 642–649. [Google Scholar] [CrossRef]
- Olbrich, H.; Häffner, K.; Kispert, A.; Völkel, A.; Volz, A.; Sasmaz, G.; Reinhardt, R.; Hennig, S.; Lehrach, H.; Konietzko, N.; et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002, 30, 143–144. [Google Scholar] [CrossRef]
- DiBella, L.M.; King, S.M. Dynein motors of the Chlamydomonas flagellum. Int. Rev. Cytol. 2001, 210, 227–268. [Google Scholar] [CrossRef]
- Duriez, B.; Duquesnoy, P.; Escudier, E.; Bridoux, A.-M.; Escalier, D.; Rayet, I.; Marcos, E.; Vojtek, A.-M.; Bercher, J.-F.; Amselem, S. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA 2007, 104, 3336–3341. [Google Scholar] [CrossRef] [PubMed]
- Padma, P.; Hozumi, A.; Ogawa, K.; Inaba, K. Molecular cloning and characterization of a thioredoxin/nucleoside diphosphate kinase related dynein intermediate chain from the ascidian, ciona intestinalis. Gene 2001, 275, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Mazor, M.; Alkrinawi, S.; Chalifa-Caspi, V.; Manor, E.; Sheffield, V.C.; Aviram, M.; Parvari, R. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet. 2011, 88, 599–607. [Google Scholar] [CrossRef][Green Version]
- Piperno, G.; Huang, B.; Luck, D.J. Two-dimensional analysis of flagellar proteins from wild-type and paralyzed mutants of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1977, 74, 1600–1604. [Google Scholar] [CrossRef]
- Baron, D.M.; Kabututu, Z.P.; Hill, K.L. Stuck in reverse: Loss of LC1 in trypanosoma brucei disrupts outer dynein arms and leads to reverse flagellar beat and backward movement. J. Cell Sci. 2007, 120, 1513–1520. [Google Scholar] [CrossRef]
- Schwabe, G.C.; Hoffmann, K.; Loges, N.T.; Birker, D.; Rossier, C.; de Santi, M.M.; Olbrich, H.; Fliegauf, M.; Failly, M.; Liebers, U.; et al. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum. Mutat. 2008, 29, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, G.W.; Loges, N.T.; Klinkenbusch, J.A.; Olbrich, H.; Pennekamp, P.; Menchen, T.; Raidt, J.; Wallmeier, J.; Werner, C.; Westermann, C.; et al. DNAH11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes. Am. J. Respir. Cell. Mol. Biol. 2016, 55, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Loges, N.T.; Antony, D.; Maver, A.; Deardorff, M.A.; Güleç, E.Y.; Gezdirici, A.; Nöthe-Menchen, T.; Höben, I.M.; Jelten, L.; Frank, D.; et al. Recessive DNAH9 loss-of-function mutations cause laterality defects and subtle respiratory ciliary-beating defects. Am. J. Hum. Genet. 2018, 103, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Fassad, M.R.; Shoemark, A.; Legendre, M.; Hirst, R.A.; Koll, F.; le Borgne, P.; Louis, B.; Daudvohra, F.; Patel, M.P.; Thomas, L.; et al. Mutations in outer dynein arm heavy chain DNAH9 cause motile cilia defects and situs inversus. Am. J. Hum. Genet. 2018, 103, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Leigh, M.W.; Ostrowski, L.E.; Huang, L.; Carson, J.L.; Hazucha, M.J.; Yin, W.; Berg, J.S.; Davis, S.D.; Dell, S.D.; et al. Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 92, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Paff, T.; Antony, D.; Shoemark, A.; Micha, D.; Kuyt, B.; Schmidts, M.; Petridi, S.; Dankert-Roelse, J.E.; Haarman, E.G.; et al. Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 92, 88–98. [Google Scholar] [CrossRef]
- Hjeij, R.; Lindstrand, A.; Francis, R.; Zariwala, M.A.; Liu, X.; Li, Y.; Damerla, R.; Dougherty, G.W.; Abouhamed, M.; Olbrich, H.; et al. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet. 2013, 93, 357–367. [Google Scholar] [CrossRef]
- Wallmeier, J.; Shiratori, H.; Dougherty, G.W.; Edelbusch, C.; Hjeij, R.; Loges, N.T.; Menchen, T.; Olbrich, H.; Pennekamp, P.; Raidt, J.; et al. TTC25 Deficiency results in defects of the outer dynein arm docking machinery and primary ciliary dyskinesia with left-right body asymmetry randomization. Am. J. Hum. Genet. 2016, 99, 460–469. [Google Scholar] [CrossRef]
- Xu, Y.; Cao, J.; Huang, S.; Feng, D.; Zhang, W.; Zhu, X.; Yan, X. Characterization of tetratricopeptide repeat-containing proteins critical for cilia formation and function. PLoS ONE 2015, 10, e0124378. [Google Scholar] [CrossRef]
- Hjeij, R.; Onoufriadis, A.; Watson, C.M.; Slagle, C.E.; Klena, N.T.; Dougherty, G.W.; Kurkowiak, M.; Loges, N.T.; Diggle, C.P.; Morante, N.F.C.; et al. CCDC151 mutations cause primary ciliary dyskinesia by disruption of the outer dynein arm docking complex formation. Am. J. Hum. Genet. 2014, 95, 257–274. [Google Scholar] [CrossRef]
- Alsaadi, M.M.; Erzurumluoglu, A.M.; Rodriguez, S.; Guthrie, P.A.I.; Gaunt, T.R.; Omar, H.Z.; Mubarak, M.; Alharbi, K.K.; Al-Rikabi, A.C.; Day, I.N.M. Nonsense mutation in coiled-coil domain containing 151 gene (CCDC151) causes primary ciliary dyskinesia. Hum. Mutat. 2014, 35, 1446–1448. [Google Scholar] [CrossRef]
- Dean, A.B.; Mitchell, D.R. Chlamydomonas ODA10 is a conserved axonemal protein that plays a unique role in outer dynein arm assembly. Mol. Biol. Cell 2013, 24, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Panizzi, J.R.; Becker-Heck, A.; Castleman, V.H.; Al-Mutairi, D.A.; Liu, Y.; Loges, N.T.; Pathak, N.; Austin-Tse, C.; Sheridan, E.; Schmidts, M.; et al. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet. 2012, 44, 714–719. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Patel-King, R.S. The oligomeric outer dynein arm assembly factor CCDC103 is tightly integrated within the ciliary axoneme and exhibits periodic binding to microtubules. J. Biol. Chem. 2015, 290, 7388–7401. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, S.; Watson, C.M.; Kernohan, K.D.; Lemos, M.; Hutchinson, S.; Poulter, J.A.; Crinnion, L.A.; Berry, I.; Simmonds, J.; Vasudevan, P.; et al. Biallelic mutations in LRRC56, encoding a protein associated with intraflagellar transport, cause mucociliary clearance and laterality defects. Am. J. Hum. Genet. 2018, 103, 727–739. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Horani, A.; Stoyanova, M.; Charng, W.-L.; Bottier, M.; Sears, P.R.; Yin, W.-N.; Daniels, L.A.; Bowen, H.; Conrad, D.F.; et al. Mutation of CFAP57, a protein required for the asymmetric targeting of a subset of inner dynein arms in Chlamydomonas, causes primary ciliary dyskinesia. PLoS Genet. 2020, 16, e1008691. [Google Scholar] [CrossRef]
- Lin, J.; Le, T.V.; Augspurger, K.; Tritschler, D.; Bower, R.; Fu, G.; Perrone, C.; O’Toole, E.T.; Mills, K.V.; Dymek, E.; et al. FAP57/WDR65 targets assembly of a subset of inner arm dyneins and connects to regulatory hubs in cilia. Mol. Biol. Cell 2019, 30, 2659–2680. [Google Scholar] [CrossRef]
- Merveille, A.-C.; Davis, E.E.; Becker-Heck, A.; Legendre, M.; Amirav, I.; Bataille, G.; Belmont, J.; Beydon, N.; Billen, F.; Clément, A.; et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011, 43, 72–78. [Google Scholar] [CrossRef]
- Becker-Heck, A.; Zohn, I.E.; Okabe, N.; Pollock, A.; Lenhart, K.B.; Sullivan-Brown, J.; McSheene, J.; Loges, N.T.; Olbrich, H.; Haeffner, K.; et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011, 43, 79–84. [Google Scholar] [CrossRef]
- Olbrich, H.; Cremers, C.; Loges, N.T.; Werner, C.; Nielsen, K.G.; Marthin, J.K.; Philipsen, M.; Wallmeier, J.; Pennekamp, P.; Menchen, T.; et al. Loss-of-function GAS8 mutations cause primary ciliary dyskinesia and disrupt the nexin-dynein regulatory complex. Am. J. Hum. Genet. 2015, 97, 546–554. [Google Scholar] [CrossRef]
- Rupp, G.; Porter, M.E. A subunit of the dynein regulatory complex in chlamydomonas is a homologue of a growth arrest-specific gene product. J. Cell Biol. 2003, 162, 47–57. [Google Scholar] [CrossRef]
- Hutchings, N.R.; Donelson, J.E.; Hill, K.L. Trypanin is a cytoskeletal linker protein and is required for cell motility in African trypanosomes. J. Cell Biol. 2002, 156, 867–877. [Google Scholar] [CrossRef]
- Colantonio, J.R.; Vermot, J.; Wu, D.; Langenbacher, A.D.; Fraser, S.; Chen, J.-N.; Hill, K.L. The dynein regulatory complex is required for ciliary motility and otolith biogenesis in the inner ear. Nature 2009, 457, 10. [Google Scholar] [CrossRef] [PubMed]
- Wirschell, M.; Olbrich, H.; Werner, C.; Tritschler, D.; Bower, R.; Sale, W.S.; Loges, N.T.; Pennekamp, P.; Lindberg, S.; Stenram, U.; et al. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat. Genet. 2013, 45, 262–268. [Google Scholar] [CrossRef]
- Horani, A.; Brody, S.L.; Ferkol, T.W.; Shoseyov, D.; Wasserman, M.G.; Ta-shma, A.; Wilson, K.S.; Bayly, P.V.; Amirav, I.; Cohen-Cymberknoh, M.; et al. CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia. PLoS ONE 2013, 8, e72299. [Google Scholar] [CrossRef] [PubMed]
- Bower, R.; Tritschler, D.; VanderWaal, K.; Perrone, C.A.; Mueller, J.; Fox, L.; Sale, W.S.; Porter, M.E. The N-DRC forms a conserved biochemical complex that maintains outer doublet alignment and limits microtubule sliding in motile axonemes. Mol. Biol. Cell 2013, 24, 1134–1152. [Google Scholar] [CrossRef]
- Kott, E.; Legendre, M.; Copin, B.; Papon, J.-F.; Dastot-Le Moal, F.; Montantin, G.; Duquesnoy, P.; Piterboth, W.; Amram, D.; Bassinet, L.; et al. Loss-of-function mutations in RSPH1 cause primary ciliary dyskinesia with central-complex and radial-spoke defects. Am. J. Hum. Genet. 2013, 93, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Ostrowski, L.E.; Leigh, M.W.; Sears, P.R.; Davis, S.D.; Wolf, W.E.; Hazucha, M.J.; Carson, J.L.; Olivier, K.N.; Sagel, S.D.; et al. Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am. J. Respir. Crit. Care Med. 2014, 189, 707–717. [Google Scholar] [CrossRef]
- Yin, W.; Livraghi-Butrico, A.; Sears, P.R.; Rogers, T.D.; Burns, K.A.; Grubb, B.R.; Ostrowski, L.E. Mice with a deletion of Rsph1 exhibit a low level of mucociliary clearance and develop a primary ciliary dyskinesia phenotype. Am. J. Respir. Cell Mol. Biol. 2019, 61, 312–321. [Google Scholar] [CrossRef]
- Castleman, V.H.; Romio, L.; Chodhari, R.; Hirst, R.A.; de Castro, S.C.P.; Parker, K.A.; Ybot-Gonzalez, P.; Emes, R.D.; Wilson, S.W.; Wallis, C.; et al. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009, 84, 197–209. [Google Scholar] [CrossRef]
- Yoke, H.; Ueno, H.; Narita, A.; Sakai, T.; Horiuchi, K.; Shingyoji, C.; Hamada, H.; Shinohara, K. Rsph4a is essential for the triplet radial spoke head assembly of the mouse motile cilia. PLoS Genet. 2020, 16, e1008664. [Google Scholar] [CrossRef]
- Jeanson, L.; Copin, B.; Papon, J.-F.; Dastot-Le Moal, F.; Duquesnoy, P.; Montantin, G.; Cadranel, J.; Corvol, H.; Coste, A.; Désir, J.; et al. RSPH3 mutations cause primary ciliary dyskinesia with central-complex defects and a near absence of radial spokes. Am. J. Hum. Genet. 2015, 97, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, A.R.; Diener, D.R.; Rosenbaum, J.L.; Sale, W.S. Flagellar radial spoke protein 3 is an a-kinase anchoring protein (Akap). J. Cell Biol. 2001, 153, 443–448. [Google Scholar] [CrossRef] [PubMed]
- El Khouri, E.; Thomas, L.; Jeanson, L.; Bequignon, E.; Vallette, B.; Duquesnoy, P.; Montantin, G.; Copin, B.; Dastot-Le Moal, F.; Blanchon, S.; et al. Mutations in DNAJB13, encoding an HSP40 family member, cause primary ciliary dyskinesia and male infertility. Am. J. Hum. Genet. 2016, 99, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Yuan, L. A heat-shock protein 40, DNAJB13, is an axoneme-associated component in mouse spermatozoa. Mol. Reprod. Dev. 2008, 75, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Owen, H.A.; Yang, P. Dimeric heat shock protein 40 binds radial spokes for generating coupled power strokes and recovery strokes of 9 + 2 flagella. J. Cell Biol. 2008, 180, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.H.; Huh, H.J.; Jeong, I.; Lee, N.Y.; Koh, W.-J.; Park, H.-C.; Ki, C.-S. A nonsense variant in NME5 causes human primary ciliary dyskinesia with radial spoke defects. Clin. Genet. 2020, 98, 64–68. [Google Scholar] [CrossRef]
- Anderegg, L.; Im Hof Gut, M.; Hetzel, U.; Howerth, E.W.; Leuthard, F.; Kyöstilä, K.; Lohi, H.; Pettitt, L.; Mellersh, C.; Minor, K.M.; et al. NME5 frameshift variant in alaskan malamutes with primary ciliary dyskinesia. PLoS Genet. 2019, 15, e1008378. [Google Scholar] [CrossRef]
- Olbrich, H.; Schmidts, M.; Werner, C.; Onoufriadis, A.; Loges, N.T.; Raidt, J.; Banki, N.F.; Shoemark, A.; Burgoyne, T.; Al Turki, S.; et al. Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012, 91, 672–684. [Google Scholar] [CrossRef]
- Cindrić, S.; Dougherty, G.W.; Olbrich, H.; Hjeij, R.; Loges, N.T.; Amirav, I.; Philipsen, M.C.; Marthin, J.K.; Nielsen, K.G.; Sutharsan, S.; et al. SPEF2- and HYDIN-mutant cilia lack the central pair-associated protein SPEF2, aiding primary ciliary dyskinesia diagnostics. Am. J. Respir. Cell Mol. Biol. 2020, 62, 382–396. [Google Scholar] [CrossRef]
- Broadhead, R.; Dawe, H.R.; Farr, H.; Griffiths, S.; Hart, S.R.; Portman, N.; Shaw, M.K.; Ginger, M.L.; Gaskell, S.J.; McKean, P.G.; et al. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature 2006, 440, 224–227. [Google Scholar] [CrossRef]
- Lechtreck, K.-F.; Delmotte, P.; Robinson, M.L.; Sanderson, M.J.; Witman, G.B. Mutations in hydin impair ciliary motility in mice. J. Cell Biol. 2008, 180, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Lechtreck, K.-F.; Witman, G.B. Chlamydomonas reinhardtii hydin is a central pair protein required for flagellar motility. J. Cell Biol. 2007, 176, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Edelbusch, C.; Cindrić, S.; Dougherty, G.W.; Loges, N.T.; Olbrich, H.; Rivlin, J.; Wallmeier, J.; Pennekamp, P.; Amirav, I.; Omran, H. Mutation of serine/threonine protein kinase 36 (STK36) causes primary ciliary dyskinesia with a central pair defect. Hum. Mutat. 2017, 38, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.W.; Nguyen, C.T.; Chen, M.-H.; Yang, J.-H.; Gacayan, R.; Huang, J.; Chen, J.-N.; Chuang, P.-T. Fused has evolved divergent roles in vertebrate hedgehog signaling and motile ciliogenesis. Nature 2009, 459, 98–102. [Google Scholar] [CrossRef]
- Rink, J.C.; Gurley, K.A.; Elliott, S.A.; Sánchez Alvarado, A. Planarian Hh signaling regulates regeneration polarity and links Hh pathway evolution to cilia. Science 2009, 326, 1406–1410. [Google Scholar] [CrossRef]
- Sironen, A.; Kotaja, N.; Mulhern, H.; Wyatt, T.A.; Sisson, J.H.; Pavlik, J.A.; Miiluniemi, M.; Fleming, M.D.; Lee, L. Loss of SPEF2 function in mice results in spermatogenesis defects and primary ciliary dyskinesia. Biol. Reprod. 2011, 85, 690–701. [Google Scholar] [CrossRef]
- Biebach, L.; Cindrić, S.; Koenig, J.; Aprea, I.; Dougherty, G.W.; Raidt, J.; Bracht, D.; Ruppel, R.; Schreiber, J.; Hjeij, R.; et al. Recessive mutations in CFAP74 cause primary ciliary dyskinesia with normal ciliary ultrastructure. Am. J. Respir. Cell Mol. Biol. 2022, 67, 409–413. [Google Scholar] [CrossRef]
- DiPetrillo, C.G.; Smith, E.F. Pcdp1 is a central apparatus protein that binds Ca2+-calmodulin and regulates ciliary motility. J. Cell Biol. 2010, 189, 601–612. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Shapiro, A.; Sears, P.R.; Charng, W.-L.; Conrad, D.F.; Leigh, M.W.; Knowles, M.R.; Ostrowski, L.E.; Zariwala, M.A. Identification of genetic variants in CFAP221 as a cause of primary ciliary dyskinesia. J. Hum. Genet. 2020, 65, 175–180. [Google Scholar] [CrossRef]
- Lee, L.; Campagna, D.R.; Pinkus, J.L.; Mulhern, H.; Wyatt, T.A.; Sisson, J.H.; Pavlik, J.A.; Pinkus, G.S.; Fleming, M.D. Primary ciliary dyskinesia in mice lacking the novel ciliary protein Pcdp1. Mol. Cell Biol. 2008, 28, 949–957. [Google Scholar] [CrossRef]
- Dougherty, G.W.; Mizuno, K.; Nöthe-Menchen, T.; Ikawa, Y.; Boldt, K.; Ta-Shma, A.; Aprea, I.; Minegishi, K.; Pang, Y.-P.; Pennekamp, P.; et al. CFAP45 deficiency causes situs abnormalities and asthenospermia by disrupting an axonemal adenine nucleotide homeostasis module. Nat. Commun. 2020, 11, 5520. [Google Scholar] [CrossRef] [PubMed]
- Ta-Shma, A.; Perles, Z.; Yaacov, B.; Werner, M.; Frumkin, A.; Rein, A.J.J.T.; Elpeleg, O. A human laterality disorder associated with a homozygous WDR16 deletion. Eur. J. Hum. Genet. 2015, 23, 1262–1265. [Google Scholar] [CrossRef] [PubMed]
- Wallmeier, J.; Bracht, D.; Alsaif, H.S.; Dougherty, G.W.; Olbrich, H.; Cindric, S.; Dzietko, M.; Heyer, C.; Teig, N.; Thiels, C.; et al. Mutations in TP73 cause impaired mucociliary clearance and lissencephaly. Am. J. Hum. Genet. 2021, 108, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Nemajerova, A.; Kramer, D.; Siller, S.S.; Herr, C.; Shomroni, O.; Pena, T.; Gallinas Suazo, C.; Glaser, K.; Wildung, M.; Steffen, H.; et al. TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes. Dev. 2016, 30, 1300–1312. [Google Scholar] [CrossRef]
- Al Mutairi, F.; Alkhalaf, R.; Alkhorayyef, A.; Alroqi, F.; Yusra, A.; Umair, M.; Nouf, F.; Khan, A.; Meshael, A.; Hamad, A.; et al. Homozygous truncating NEK10 mutation, associated with primary ciliary dyskinesia: A case report. BMC Pulm. Med. 2020, 20, 141. [Google Scholar] [CrossRef]
- Porpora, M.; Sauchella, S.; Rinaldi, L.; Delle Donne, R.; Sepe, M.; Torres-Quesada, O.; Intartaglia, D.; Garbi, C.; Insabato, L.; Santoriello, M.; et al. Counterregulation of CAMP-directed kinase activities controls ciliogenesis. Nat. Commun. 2018, 9, 1224. [Google Scholar] [CrossRef]
- Ryan, R.; Failler, M.; Reilly, M.L.; Garfa-Traore, M.; Delous, M.; Filhol, E.; Reboul, T.; Bole-Feysot, C.; Nitschké, P.; Baudouin, V.; et al. Functional characterization of tektin-1 in motile cilia and evidence for TEKT1 as a new candidate gene for motile ciliopathies. Hum. Mol. Genet. 2018, 27, 266–282. [Google Scholar] [CrossRef]
- Devlin, L.A.; Coles, J.; Jackson, C.L.; Barroso-Gil, M.; Green, B.; Walker, W.T.; Thomas, N.S.; Thompson, J.; Rock, S.A.; Neatu, R.; et al. Biallelic variants in CEP164 cause a motile ciliopathy-like syndrome. Clin. Genet. 2022, 103, 330–334. [Google Scholar] [CrossRef]
- Reed, M.; Takemaru, K.-I.; Ying, G.; Frederick, J.M.; Baehr, W. Deletion of CEP164 in mouse photoreceptors post-ciliogenesis interrupts ciliary intraflagellar transport (IFT). PLoS Genet. 2022, 18, e1010154. [Google Scholar] [CrossRef]
- Beckers, A.; Adis, C.; Schuster-Gossler, K.; Tveriakhina, L.; Ott, T.; Fuhl, F.; Hegermann, J.; Boldt, K.; Serth, K.; Rachev, E.; et al. The FOXJ1 target Cfap206 is required for sperm motility, mucociliary clearance of the airways and brain development. Development 2020, 147, dev188052. [Google Scholar] [CrossRef]
- Vasudevan, K.K.; Song, K.; Alford, L.M.; Sale, W.S.; Dymek, E.E.; Smith, E.F.; Hennessey, T.; Joachimiak, E.; Urbanska, P.; Wloga, D.; et al. FAP206 is a microtubule-docking adapter for ciliary radial spoke 2 and dynein c. Mol. Biol. Cell 2015, 26, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Tritschler, D.; Song, K.; Barber, C.F.; Cobb, J.S.; Porter, M.E.; Nicastro, D. Building blocks of the nexin-dynein regulatory complex in Chlamydomonas flagella. J. Biol. Chem. 2011, 286, 29175–29191. [Google Scholar] [CrossRef] [PubMed]
- Ta-Shma, A.; Hjeij, R.; Perles, Z.; Dougherty, G.W.; Abu Zahira, I.; Letteboer, S.J.F.; Antony, D.; Darwish, A.; Mans, D.A.; Spittler, S.; et al. Homozygous loss-of-function mutations in MNS1 cause laterality defects and likely male infertility. PLoS Genet. 2018, 14, e1007602. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, F.; Leu, N.A.; Wang, P.J. MNS1 is essential for spermiogenesis and motile ciliary functions in mice. PLoS Genet. 2012, 8, e1002516. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Tu, C.-F.; Yang, D.-H.; Ding, S.-Z.; Lei, C.; Wang, R.-C.; Liu, L.; Kang, X.; Shen, X.-Q.; Yang, Y.-F.; et al. Bi-Allelic BRWD1 variants cause male infertility with asthenoteratozoospermia and likely primary ciliary dyskinesia. Hum. Genet. 2021, 140, 761–773. [Google Scholar] [CrossRef]
- Rachev, E.; Schuster-Gossler, K.; Fuhl, F.; Ott, T.; Tveriakhina, L.; Beckers, A.; Hegermann, J.; Boldt, K.; Mai, M.; Kremmer, E.; et al. CFAP43 modulates ciliary beating in mouse and xenopus. Dev. Biol. 2020, 459, 109–125. [Google Scholar] [CrossRef]
- Narasimhan, V.; Hjeij, R.; Vij, S.; Loges, N.T.; Wallmeier, J.; Koerner-Rettberg, C.; Werner, C.; Thamilselvam, S.K.; Boey, A.; Choksi, S.P.; et al. Mutations in CCDC11, which encodes a coiled-coil containing ciliary protein, causes situs inversus due to dysmotility of monocilia in the left-right organizer. Hum. Mutat. 2015, 36, 307–318. [Google Scholar] [CrossRef]
- Silva, E.; Betleja, E.; John, E.; Spear, P.; Moresco, J.J.; Zhang, S.; Yates, J.R.; Mitchell, B.J.; Mahjoub, M.R. Ccdc11 is a novel centriolar satellite protein essential for ciliogenesis and establishment of left–right asymmetry. Mol. Biol. Cell 2016, 27, 48–63. [Google Scholar] [CrossRef]
- Shapiro, A.J.; Leigh, M.W. Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: Genetic defects with normal and non-diagnostic ciliary ultrastructure. Ultrastruct. Pathol. 2017, 41, 373–385. [Google Scholar] [CrossRef]
- Amirav, I.; Mussaffi, H.; Roth, Y.; Schmidts, M.; Omran, H.; Werner, C. Israeli PCD consortium investigators a reach-out system for video microscopy analysis of ciliary motions aiding PCD diagnosis. BMC Res. Notes 2015, 8, 71. [Google Scholar] [CrossRef]
- Knowles, M.R.; Leigh, M.W.; Carson, J.L.; Davis, S.D.; Dell, S.D.; Ferkol, T.W.; Olivier, K.N.; Sagel, S.D.; Rosenfeld, M.; Burns, K.A.; et al. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax 2012, 67, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, R.; Hogg, C. Primary ciliary dyskinesia: A major player in a bigger game. Breathe 2020, 16, 200047. [Google Scholar] [CrossRef] [PubMed]
- Budny, B.; Chen, W.; Omran, H.; Fliegauf, M.; Tzschach, A.; Wisniewska, M.; Jensen, L.R.; Raynaud, M.; Shoichet, S.A.; Badura, M.; et al. A novel x-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum. Genet. 2006, 120, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Yiallouros, P.K.; Kouis, P.; Pirpa, P.; Michailidou, K.; Loizidou, M.A.; Potamiti, L.; Kalyva, M.; Koutras, G.; Kyriacou, K.; Hadjisavvas, A. Wide phenotypic variability in RSPH9-associated primary ciliary dyskinesia: Review of a case-series from cyprus. J. Thorac. Dis. 2019, 11, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Sha, Y.; Li, Y.; Mei, L.; Lin, S.; Huang, X.; Lu, J.; Ding, L.; Kong, S.; Lu, Z. Loss-of-function mutations in SPEF2 cause multiple morphological abnormalities of the sperm flagella (MMAF). J. Med. Genet. 2019, 56, 678–684. [Google Scholar] [CrossRef]
- Ostrowski, L.E.; Yin, W.; Smith, A.J.; Sears, P.R.; Bustamante-Marin, X.M.; Dang, H.; Hildebrandt, F.; Daniels, L.A.; Capps, N.A.; Sullivan, K.M.; et al. Expression of a truncated form of ODAD1 associated with an unusually mild primary ciliary dyskinesia phenotype. Int. J. Mol. Sci. 2022, 23, 1753. [Google Scholar] [CrossRef]
- Preston, C.G.; Wright, M.W.; Madhavrao, R.; Harrison, S.M.; Goldstein, J.L.; Luo, X.; Wand, H.; Wulf, B.; Cheung, G.; Mandell, M.E.; et al. ClinGen variant curation interface: A variant classification platform for the application of evidence criteria from ACMG/AMP guidelines. Genome Med. 2022, 14, 6. [Google Scholar] [CrossRef]
- Bukowy-Bieryłło, Z. Long-term differentiating primary human airway epithelial cell cultures: How far are we? Cell Commun. Signal 2021, 19, 63. [Google Scholar] [CrossRef]
- Poprzeczko, M.; Bicka, M.; Farahat, H.; Bazan, R.; Osinka, A.; Fabczak, H.; Joachimiak, E.; Wloga, D. Rare human diseases: Model organisms in deciphering the molecular basis of primary ciliary dyskinesia. Cells 2019, 8, 1614. [Google Scholar] [CrossRef]
- Niziolek, M.; Bicka, M.; Osinka, A.; Samsel, Z.; Sekretarska, J.; Poprzeczko, M.; Bazan, R.; Fabczak, H.; Joachimiak, E.; Wloga, D. PCD genes-from patients to model organisms and back to humans. Int. J. Mol. Sci. 2022, 23, 1749. [Google Scholar] [CrossRef]
- Newmark, P.A.; Sánchez Alvarado, A. Not your father’s planarian: A classic model enters the era of functional genomics. Nat. Rev. Genet. 2002, 3, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.H. Experimental studies of the regeneration of planaria maculata. Arch. Entwickelungsmechanik Org. 1898, 7, 364–397. [Google Scholar] [CrossRef]
- Vila-Farré, M.; C Rink, J. The ecology of freshwater planarians. Methods Mol. Biol. 2018, 1774, 173–205. [Google Scholar] [CrossRef]
- Reddien, P.W.; Sánchez Alvarado, A. Fundamentals of planarian regeneration. Annu. Rev. Cell Dev. Biol. 2004, 20, 725–757. [Google Scholar] [CrossRef] [PubMed]
- Reddien, P.W. The cellular and molecular basis for planarian regeneration. Cell 2018, 175, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Oviedo, N.J.; Nicolas, C.L.; Adams, D.S.; Levin, M. Planarians: A versatile and powerful model system for molecular studies of regeneration, adult stem cell regulation, aging, and behavior. CSH Protoc. 2008, 2008, pdb.emo101. [Google Scholar] [CrossRef]
- Deochand, N.; Costello, M.S.; Deochand, M.E. Behavioral research with planaria. Perspect. Behav. Sci. 2018, 41, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Basquin, C.; Orfila, A.-M.; Azimzadeh, J. The planarian Schmidtea mediterranea as a model for studying motile cilia and multiciliated cells. Methods Cell Biol. 2015, 127, 243–262. [Google Scholar] [CrossRef]
- Saló, E.; Pineda, D.; Marsal, M.; Gonzalez, J.; Gremigni, V.; Batistoni, R. Genetic network of the eye in Platyhelminthes: Expression and functional analysis of some players during planarian regeneration. Gene 2002, 287, 67–74. [Google Scholar] [CrossRef]
- LoCascio, S.A.; Lapan, S.W.; Reddien, P.W. Eye absence does not regulate planarian stem cells during eye regeneration. Dev. Cell 2017, 40, 381–391.e3. [Google Scholar] [CrossRef]
- Önal, P.; Grün, D.; Adamidi, C.; Rybak, A.; Solana, J.; Mastrobuoni, G.; Wang, Y.; Rahn, H.-P.; Chen, W.; Kempa, S.; et al. Gene expression of pluripotency determinants is conserved between mammalian and planarian stem cells. EMBO J. 2012, 31, 2755–2769. [Google Scholar] [CrossRef] [PubMed]
- Rompolas, P.; Azimzadeh, J.; Marshall, W.F.; King, S.M. Analysis of ciliary assembly and function in planaria. Methods Enzym. 2013, 525, 245–264. [Google Scholar] [CrossRef]
- Merryman, M.S.; Alvarado, A.S.; Jenkin, J.C. Culturing planarians in the laboratory. Methods Mol. Biol. 2018, 1774, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Alvarado, A.; Newmark, P.A. Double-stranded RNA specifically disrupts gene expression during planarian regeneration. Proc. Natl. Acad. Sci. USA 1999, 96, 5049–5054. [Google Scholar] [CrossRef]
- Reddien, P.W.; Bermange, A.L.; Murfitt, K.J.; Jennings, J.R.; Sánchez Alvarado, A. Identification of genes needed for regeneration, stem cell function, and tissue homeostasis by systematic gene perturbation in planaria. Dev. Cell 2005, 8, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Rouhana, L.; Weiss, J.A.; Forsthoefel, D.J.; Lee, H.; King, R.S.; Inoue, T.; Shibata, N.; Agata, K.; Newmark, P.A. RNA interference by feeding in vitro-synthesized double-stranded rna to planarians: Methodology and dynamics. Dev. Dyn. 2013, 242, 718–730. [Google Scholar] [CrossRef]
- Fincher, C.T.; Wurtzel, O.; de Hoog, T.; Kravarik, K.M.; Reddien, P.W. Cell Type transcriptome atlas for the planarian Schmidtea mediterranea. Science 2018, 360, eaaq1736. [Google Scholar] [CrossRef]
- Plass, M.; Solana, J.; Wolf, F.A.; Ayoub, S.; Misios, A.; Glažar, P.; Obermayer, B.; Theis, F.J.; Kocks, C.; Rajewsky, N. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science 2018, 360, eaaq1723. [Google Scholar] [CrossRef]
- Grohme, M.A.; Schloissnig, S.; Rozanski, A.; Pippel, M.; Young, G.R.; Winkler, S.; Brandl, H.; Henry, I.; Dahl, A.; Powell, S.; et al. The genome of Schmidtea mediterranea and the evolution of core cellular mechanisms. Nature 2018, 554, 56–61. [Google Scholar] [CrossRef]
- Benham-Pyle, B.W.; Brewster, C.E.; Kent, A.M.; Mann, F.G.; Chen, S.; Scott, A.R.; Box, A.C.; Sánchez Alvarado, A. Identification of rare post-mitotic cell states induced by injury and required for whole-body regeneration in Schmidtea mediterranea. Nat. Cell Biol. 2021, 23, 939–952. [Google Scholar] [CrossRef]
- Robb, S.M.C.; Ross, E.; Sánchez Alvarado, A. SmedGD: The Schmidtea mediterranea genome database. Nucleic Acids Res. 2008, 36, D599–D606. [Google Scholar] [CrossRef]
- Robb, S.M.C.; Gotting, K.; Ross, E.; Sánchez Alvarado, A. SmedGD 2.0: The Schmidtea mediterranea genome database. Genesis 2015, 53, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Rozanski, A.; Moon, H.; Brandl, H.; Martín-Durán, J.M.; Grohme, M.A.; Hüttner, K.; Bartscherer, K.; Henry, I.; Rink, J.C. PlanMine 3.0-improvements to a mineable resource of flatworm biology and biodiversity. Nucleic Acids Res. 2019, 47, D812–D820. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Lara, S.; Abril, J.F. PlanNET: Homology-based predicted interactome for multiple planarian transcriptomes. Bioinformatics 2018, 34, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Rompolas, P.; Patel-King, R.S.; King, S.M. Schmidtea mediterranea: A model system for analysis of motile cilia. Methods Cell Biol. 2009, 93, 81–98. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Patel-King, R.S. Planaria as a model system for the analysis of ciliary assembly and motility. Methods Mol. Biol. 2016, 1454, 245–254. [Google Scholar] [CrossRef]
- Azimzadeh, J.; Basquin, C. Basal bodies across eukaryotes series: Basal bodies in the freshwater planarian Schmidtea mediterranea. Cilia 2016, 5, 15. [Google Scholar] [CrossRef]
- Li, Y.; Guo, F.; Jing, Q.; Zhu, X.; Yan, X. Characterisation of centriole biogenesis during multiciliation in planarians. Biol. Cell 2020, 112, 398–408. [Google Scholar] [CrossRef]
- Harrath, A.H.; Gammoudi, M.; Mansour, L.; Ahmed, M.; Sirotkin, A.V.; Al Omar, S.Y.; Ibrahim, K.E.; Alwasel, S.H. Investigation of the ultrastructure of dendrocoelum constrictum (Platyhelminthes, Tricladida) spermatogenesis and mature spermatozoa. C. R. Biol. 2014, 337, 513–520. [Google Scholar] [CrossRef]
- Christman, D.A.; Curry, H.N.; Rouhana, L. Heterotrimeric kinesin II is required for flagellar assembly and elongation of nuclear morphology during spermiogenesis in Schmidtea mediterranea. Dev. Biol. 2021, 477, 191–204. [Google Scholar] [CrossRef]
- Rink, J.C.; Vu, H.T.-K.; Sánchez Alvarado, A. The maintenance and regeneration of the planarian excretory system are regulated by EGFR signaling. Development 2011, 138, 3769–3780. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. WNT signaling pathway and stem cell signaling network. Clin. Cancer Res. 2007, 13, 4042–4045. [Google Scholar] [CrossRef]
- Peiris, T.H.; Ramirez, D.; Barghouth, P.G.; Oviedo, N.J. The akt signaling pathway is required for tissue maintenance and regeneration in planarians. BMC Dev. Biol. 2016, 16, 7. [Google Scholar] [CrossRef]
- Petersen, C.P.; Reddien, P.W. Polarized notum activation at wounds inhibits wnt function to promote planarian head regeneration. Science 2011, 332, 852–855. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, E.M. Wnt signaling in axial patterning and regeneration: Lessons from planaria. Sci. Signal. 2010, 3, pe21. [Google Scholar] [CrossRef] [PubMed]
- Glazer, A.M.; Wilkinson, A.W.; Backer, C.B.; Lapan, S.W.; Gutzman, J.H.; Cheeseman, I.M.; Reddien, P.W. The Zn finger protein iguana impacts hedgehog signaling by promoting ciliogenesis. Dev. Biol. 2010, 337, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Currie, K.W.; Pearson, B.J. Transcription factors Lhx1/5-1 and Pitx are required for the maintenance and regeneration of serotonergic neurons in planarians. Development 2013, 140, 3577–3588. [Google Scholar] [CrossRef]
- Jerber, J.; Baas, D.; Soulavie, F.; Chhin, B.; Cortier, E.; Vesque, C.; Thomas, J.; Durand, B. The coiled-coil domain containing protein CCDC151 is required for the function of IFT-dependent motile cilia in animals. Hum. Mol. Genet. 2014, 23, 563–577. [Google Scholar] [CrossRef]
- Ahmed, N.T.; Mitchell, D.R. ODA16p, a Chlamydomonas flagellar protein needed for dynein assembly. Mol. Biol. Cell 2005, 16, 5004–5012. [Google Scholar] [CrossRef]
- Gao, C.; Wang, G.; Amack, J.D.; Mitchell, D.R. Oda16/Wdr69 is essential for axonemal dynein assembly and ciliary motility during zebrafish embryogenesis. Dev. Dyn. 2010, 239, 2190–2197. [Google Scholar] [CrossRef]
- Solomon, G.M.; Francis, R.; Chu, K.K.; Birket, S.E.; Gabriel, G.; Trombley, J.E.; Lemke, K.L.; Klena, N.; Turner, B.; Tearney, G.J.; et al. Assessment of ciliary phenotype in primary ciliary dyskinesia by micro-optical coherence tomography. JCI Insight 2017, 2, e91702. [Google Scholar] [CrossRef] [PubMed]
- Taschner, M.; Mourão, A.; Awasthi, M.; Basquin, J.; Lorentzen, E. Structural basis of outer dynein arm intraflagellar transport by the transport adaptor protein ODA16 and the intraflagellar transport protein IFT46. J. Biol. Chem. 2017, 292, 7462–7473. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Witman, G.B. The N-terminus of IFT46 mediates intraflagellar transport of outer arm dynein and its cargo-adaptor ODA16. Mol. Biol. Cell 2017, 28, 2420–2433. [Google Scholar] [CrossRef] [PubMed]
- Patel-King, R.S.; King, S.M. An outer arm dynein light chain acts in a conformational switch for flagellar motility. J. Cell Biol. 2009, 186, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Horváth, J.; Fliegauf, M.; Olbrich, H.; Kispert, A.; King, S.M.; Mitchison, H.; Zariwala, M.A.; Knowles, M.R.; Sudbrak, R.; Fekete, G.; et al. Identification and analysis of axonemal dynein light chain 1 in primary ciliary dyskinesia patients. Am. J. Respir. Cell Mol. Biol. 2005, 33, 41–47. [Google Scholar] [CrossRef]
- Takada, S.; Wilkerson, C.G.; Wakabayashi, K.; Kamiya, R.; Witman, G.B. The outer dynein arm-docking complex: Composition and characterization of a subunit (Oda1) necessary for outer arm assembly. Mol. Biol. Cell 2002, 13, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.M.; Inaba, K.; Pazour, G.J.; Takada, S.; Wakabayashi, K.; Wilkerson, C.G.; Kamiya, R.; Witman, G.B. DC3, the 21-KDa subunit of the outer dynein arm-docking complex (ODA-DC), is a novel EF-hand protein important for assembly of both the outer arm and the ODA-DC. Mol. Biol. Cell 2003, 14, 3650–3663. [Google Scholar] [CrossRef]
- Takada, S.; Kamiya, R. Functional reconstitution of chlamydomonas outer dynein arms from alpha-beta and gamma subunits: Requirement of a third factor. J. Cell Biol. 1994, 126, 737–745. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Shoemark, A.; Munye, M.M.; James, C.T.; Schmidts, M.; Patel, M.; Rosser, E.M.; Bacchelli, C.; Beales, P.L.; Scambler, P.J.; et al. Combined exome and whole-genome sequencing identifies mutations in ARMC4 as a cause of primary ciliary dyskinesia with defects in the outer dynein arm. J. Med. Genet. 2014, 51, 61–67. [Google Scholar] [CrossRef]
- Young, S.A.M.; Miyata, H.; Satouh, Y.; Kato, H.; Nozawa, K.; Isotani, A.; Aitken, R.J.; Baker, M.A.; Ikawa, M. CRISPR/Cas9-mediated rapid generation of multiple mouse lines identified Ccdc63 as essential for spermiogenesis. Int. J. Mol. Sci. 2015, 16, 24732–24750. [Google Scholar] [CrossRef]
- Kyuji, A.; Patel-King, R.S.; Hisabori, T.; King, S.M.; Wakabayashi, K.-I. Cilia loss and dynein assembly defects in planaria lacking an outer dynein arm-docking complex subunit. Zoolog. Sci. 2020, 37, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Patel-King, R.S.; King, S.M. A prefoldin-associated WD-repeat protein (WDR92) is required for the correct architectural assembly of motile cilia. Mol. Biol. Cell 2016, 27, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- Zur Lage, P.; Stefanopoulou, P.; Styczynska-Soczka, K.; Quinn, N.; Mali, G.; von Kriegsheim, A.; Mill, P.; Jarman, A.P. Ciliary dynein motor preassembly is regulated by Wdr92 in association with HSP90 co-chaperone, R2TP. J. Cell Biol. 2018, 217, 2583–2598. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, L.; Pan, J. Chlamydomonas WDR92 in association with R2TP-like complex and multiple DNAAFs to regulate ciliary dynein preassembly. J. Mol. Cell Biol. 2019, 11, 770–780. [Google Scholar] [CrossRef]
- Patel-King, R.S.; Sakato-Antoku, M.; Yankova, M.; King, S.M. WDR92 Is required for axonemal dynein heavy chain stability in cytoplasm. Mol. Biol. Cell 2019, 30, 1834–1845. [Google Scholar] [CrossRef]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef]
- Moyer, J.H.; Lee-Tischler, M.J.; Kwon, H.Y.; Schrick, J.J.; Avner, E.D.; Sweeney, W.E.; Godfrey, V.L.; Cacheiro, N.L.; Wilkinson, J.E.; Woychik, R.P. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 1994, 264, 1329–1333. [Google Scholar] [CrossRef]
- Patel-King, R.S.; Gilberti, R.M.; Hom, E.F.Y.; King, S.M. WD60/FAP163 Is a dynein intermediate chain required for retrograde intraflagellar transport in cilia. Mol. Biol. Cell 2013, 24, 2668–2677. [Google Scholar] [CrossRef]
- Scimone, M.L.; Srivastava, M.; Bell, G.W.; Reddien, P.W. A regulatory program for excretory system regeneration in planarians. Development 2011, 138, 4387–4398. [Google Scholar] [CrossRef]
- Marra, A.N.; Li, Y.; Wingert, R.A. Antennas of organ morphogenesis: The roles of cilia in vertebrate kidney development. Genesis 2016, 54, 457–469. [Google Scholar] [CrossRef]
- Reddien, P.W.; Newmark, P.A.; Sánchez Alvarado, A. Gene nomenclature guidelines for the planarian Schmidtea mediterranea. Dev. Dyn. 2008, 237, 3099–3101. [Google Scholar] [CrossRef] [PubMed]
- Nowotarski, S.H.; Davies, E.L.; Robb, S.M.C.; Ross, E.J.; Matentzoglu, N.; Doddihal, V.; Mir, M.; McClain, M.; Sánchez Alvarado, A. Planarian anatomy ontology: A resource to connect data within and across experimental platforms. Development 2021, 148, dev196097. [Google Scholar] [CrossRef] [PubMed]
- Brandl, H.; Moon, H.; Vila-Farré, M.; Liu, S.-Y.; Henry, I.; Rink, J.C. PlanMine--a mineable resource of planarian biology and biodiversity. Nucleic Acids Res. 2016, 44, D764–D773. [Google Scholar] [CrossRef] [PubMed]
- van Wolfswinkel, J.C.; Wagner, D.E.; Reddien, P.W. Single-cell analysis reveals functionally distinct classes within the planarian stem cell compartment. Cell Stem Cell 2014, 15, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.C.; Cheng, L.-C.; TK Vu, H.; Lange, J.J.; McKinney, S.A.; Seidel, C.W.; Sánchez Alvarado, A. Egr-5 is a post-mitotic regulator of planarian epidermal differentiation. eLife 2015, 4, e10501. [Google Scholar] [CrossRef]
- Wurtzel, O.; Oderberg, I.M.; Reddien, P.W. Planarian epidermal stem cells respond to positional cues to promote cell type diversity. Dev. Cell 2017, 40, 491–504.e5. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene (Alias) | Role in Motile Cilia Biogenesis or Part of Axonemal Structure | Defects (Mode of Detection) | Confirmation from Selected Animal Models | Seminal References |
|---|---|---|---|---|
| Cytoplasmic proteins | ||||
| CCNO MCIDAS | Centriole amplification „ | Sparse cilia (light microscope, TEM, IF against AcAT) | Xenopus Xenopus | [30] [29,30] |
| FOXJ11; autos. dominant RPGR; X-linked OFD1; X-linked GAS2L2 | BB stabiliz./orientation BB docking BB docking BB stabilization | BB mislocalized and more (TEM–except for GAS2L2; IF against BB elements) | Xenopus, Drerio, mouse, Smed Drerio Paramecium, Smed, mouse Mouse, Xenopus | [31,48,49,50] [51,52,53] [54,55,56,57,58] [59] |
| DNAAF3 (c19orf51) CFAP300 (c11orf70) SPAG1 (DNAAF13) DNAAF1 (LRRC50) DNAAF5 (HEATR2) DNAAF6 (PIH1D3); X-linked DNAAF7 (ZMYND10) DNAAF11 (LRRC6) CFAP298 (c21orf59) DNAAF2 (KTU) DNAAF4 (DYX1C1) DAW1 (WDR69,ODA16) | ODA/IDA preassembly „ „ „ „ „ „ „ „ Distal ODA preassembly “ “ | Absent/shortened DA (TEM; IF against ODA or IDA elements) | Chlamy, Drerio Chlamy, Paramecium, Smed Drerio Chlamy, Tryp, Drerio, Smed Drosi, mouse Drerio, Drosi, mouse Drerio, Drosi, Xenopus, mouse Drosi, Drerio, mouse Drerio, Chlamy, Smed medaka, Chlamy, mouse Drerio, mouse Drerio, Smed, mouse, Chlamy | [60] [61,62,63] [64] [65,66,67,68] [69,70] [71,72,73,74] [75,76] [75,77,78,79,80] [81] [82] [83] [84,85,86] |
| TTC12 | IDA assembly (in sperm IDA and ODA) | Some IDA types absent (TEM; IF against IDA elements, e.g., DNALI1) | Paramecium | [87] |
| Elements of axonemal ultrastructure | ||||
| DNAI1 DNAI2 DNAH52 TXNDC3 (NME8; DNAI8) DNAL1 DNAH113 DNAH93 | ODA “ “ “ “ Proximal ODA Distal ODA | Absent/shortened ODA (TEM–except for DNAH11; IF against ODA elements, e.g., DNAH5, DNAI2) | Chlamy Chlamy, Smed Chlamy Ciona Chlamy, Smed, Tryp Chlamy, mouse Chlamy, Paramecium, mouse | [88,89] [90,91,92] [93,94] [95,96] [91,97,98,99] [94,100,101] [94,102,103] |
| CCDC114 (ODAD1) ARMC4 (ODAD2) TTC25 (ODAD4) CCDC151 (ODAD3) CCDC1034 LRRC56 (DNAAF12) | ODA targeting/docking „ „ „ Distal ODA targeting/docking “ | Absent/shortened ODA (TEM; IF against ODA elements) | Chlamy Drerio, mouse Xenopus, mouse, Drerio Chlamy, Smed, Drerio, mouse Chlamy, Drerio Tryp | [104,105] [106] [107,108] [109,110,111] [112,113] [114] |
| CFAP57(WDR65) | IDA assembly | No TEM defect (IF) | Chlamy | [115,116] |
| CCDC39 (CFAP59) CCDC40 (CFAP172) | AR “ | Mislocalized MTs, absent IDA (TEM; IF against AR elements or GAS8) | Drerio, mouse, dog Drerio, mouse | [117] [118] |
| GAS8 (GAS11; DRC4) DRC1 (CCDC164) CCDC65 (DRC2; CFAP250) | NDR complex „ „ | MT mislocalized or no visible defect (IF against GAS8) | Chlamy, Tryp, Drerio, mouse Chlamy Chlamy, Drerio | [119,120,121,122] [123] [81,124,125] |
| RSPH1 RSPH4A RSPH9 RSPH3 DNAJB13 (RSPH16A) NME5 (RSPH23) | RS head „ „ RS stalk „ RS neck | Central pair and MTs mislocalized (TEM except for DNAJB13; IF against RS’ elements) | mouse mouse Chlamy, Drerio, mouse Chlamy Chlamy, mouse Drerio, dog | [126,127,128] [129,130] [129] [131,132] [133,134,135] [136,137] |
| HYDIN STK36 (FUSED) SPEF2 CFAP74 | CP complex “ “ “ | CP complex defects (no visible defect in TEM; IF against SPEF2, STK36; not for CFAP74) | Chlamy, mouse, Tryp Smed, Drerio, mouse mouse Chlamy | [138,139,140,141,142] [143,144,145] [139,146] [147,148] |
| CFAP221 (PCDP1) | “ | No TEM defect | mouse, Chlamy | [148,149,150] |
| Gene (Alias) | Role in Motile Cilia Biogenesis or Part of Axonemal Structure | Relevance for PCD | Confirmation from Selected Animal Models | Seminal References |
|---|---|---|---|---|
| CCDC19 (CFAP45) | Inner lumen protein | Only mild respiratory symptoms | Ch. reinhardtii, S. mediterranea, mouse | [24,151] |
| WDR16 (CFAP52) | Inner lumen protein | Only mild respiratory symptoms | Ch. reinhardtii, S. mediterranea | [24,151,152] |
| TP73; lissencephaly | Ciliogenesis | Strong PCD candidate | mouse | [153,154] |
| NEK10 | Centrosome; kinase | PCD candidate | medaka | [155,156] |
| CCDC113 (CCDC96) | NDR complex | PCD candidate | T. thermophila | [22] |
| TEKT1 | Centrosome, BB, axoneme | PCD candidate | D. rerio | [157] |
| CEP164 | Centriole; BB docking | PCD candidate | mouse | [158,159] |
| CFAP206 | BB and axoneme | PCD candidate | T. thermophila, Ch. reinhardtii, X. laevis, mouse | [160,161,162] |
| MNS1 | ODA docking | Laterality defect, male infertility | mouse | [163,164] |
| BRWD1 | Axoneme | Male infertility | - | [165] |
| CFAP43 | Axoneme | Male infertility | X. laevis, mouse | [166] |
| CCDC11 (CFAP53) | Ciliogenesis | Laterality defects | X. laevis | [167,168] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabiasz, A.; Ziętkiewicz, E. Schmidtea mediterranea as a Model Organism to Study the Molecular Background of Human Motile Ciliopathies. Int. J. Mol. Sci. 2023, 24, 4472. https://doi.org/10.3390/ijms24054472
Rabiasz A, Ziętkiewicz E. Schmidtea mediterranea as a Model Organism to Study the Molecular Background of Human Motile Ciliopathies. International Journal of Molecular Sciences. 2023; 24(5):4472. https://doi.org/10.3390/ijms24054472
Chicago/Turabian StyleRabiasz, Alicja, and Ewa Ziętkiewicz. 2023. "Schmidtea mediterranea as a Model Organism to Study the Molecular Background of Human Motile Ciliopathies" International Journal of Molecular Sciences 24, no. 5: 4472. https://doi.org/10.3390/ijms24054472
APA StyleRabiasz, A., & Ziętkiewicz, E. (2023). Schmidtea mediterranea as a Model Organism to Study the Molecular Background of Human Motile Ciliopathies. International Journal of Molecular Sciences, 24(5), 4472. https://doi.org/10.3390/ijms24054472