
Modeling of Respiratory Diseases Evolving with Fibrosis from Organoids Derived from Human Pluripotent Stem Cells

{kind=link}
{kind=link}
Abstract
1. Introduction
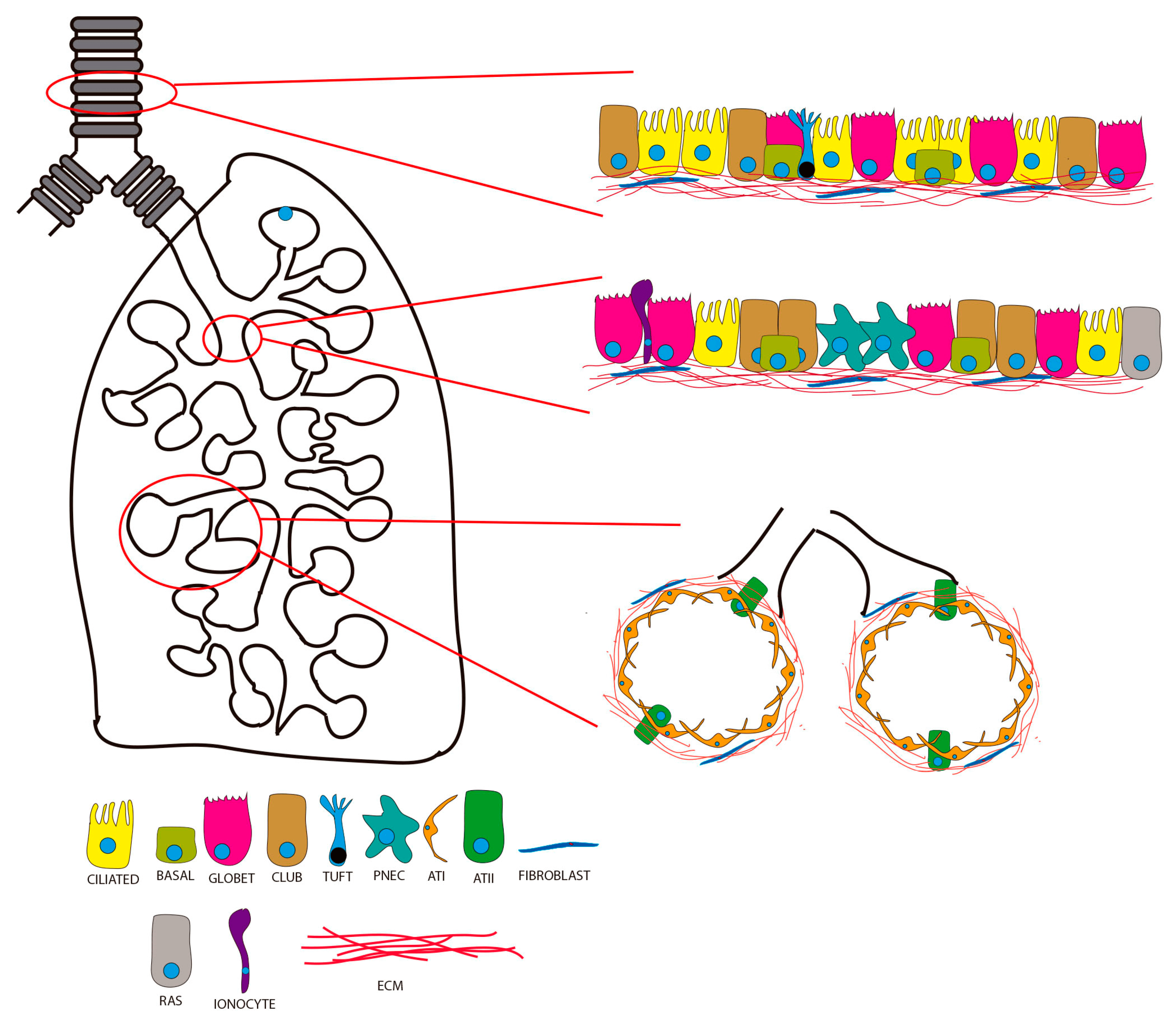
2. Lung Organoids from Human Pluripotent Stem Cells (hPSCs)
3. Idiopathic Pulmonary Fibrosis
4. Cystic Fibrosis
5. Chronic Obstructive Pulmonary Disease
6. Severe Acute Respiratory Syndrome Coronavirus Type 2 (SARS-CoV-2) and Fibrosis
- (i)
- Interaction of viral spike “S” protein with angiotensin converting enzyme (ACE2) receptor. Binding of the virus to its receptor can downregulate level of ACE2, increase levels of Ang II, and decrease level of Ang1–7, thus promoting inflammation and fibrosis.
- (ii)
- Aberrant immune response leading to so-called “cytokine storm”, resulting in increased plasma levels of IL-1β, IL-2, IL-7, and IL-10, GCSF, MIP1α, IFNy, IP-10, IL-6, IL-8, TNFα, etc. Cytokine storm accelerates disease progression and aggravates ARDS and multiple organ failure. Release of pro-inflammatory cytokines and metalloproteinases during ARDS induces damage of the epithelium, endothelium, and fibrotic remodeling.
- (iii)
- Infection and damage of ATII cells. ATII cells are key mediators of the alveolar innate response and regeneration of the respiratory epithelia through its proliferation and differentiation into ATI cells. ATII can enter the senescence state and secrete a series of inflammatory mediators and metalloproteinases (SARS phenotype) that mediate tissue remodeling. For instance, TGFβ triggers proliferation and differentiation of fibroblasts into myofibroblasts, causing aberrant deposition of extracellular matrix proteins during abnormal fibrosis.
- (iv)
- Damage of endothelial cells. Endothelial cells, when injured, transform into a mesenchymal state (EndMT) with increased activity of mesenchymal protein secretion and matrix metalloproteinases, leading to accumulation of fibroblasts and myofibroblasts and induction of fibrotic remodeling in the interstitium of the lung. Transit of the virus into the lower respiratory tract the lung can be favored using mechanical ventilation. Presence of the virus in the lung may contribute to acute lung injury and secondary fibrosis.
7. Discussion
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Basil, M.C.; Cardenas-Diaz, F.L.; Kathiriya, J.J.; Morley, M.P.; Carl, J.; Brumwell, A.N.; Katzen, J.; Slovik, K.J.; Babu, A.; Zhou, S.; et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 2022, 604, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Zilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef]
- Araya, J.; Nishimura, S.L. Fibrogenic reactions in lung disease. Annu. Rev. Pathol. 2010, 5, 77–98. [Google Scholar] [CrossRef]
- Hardie, W.D.; Glasser, S.W.; Hagood, J.S. Emerging concepts in the pathogenesis of lung fibrosis. Am. J. Pathol. 2009, 175, 3–16. [Google Scholar] [CrossRef]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef]
- Clark, R.A. The commonality of cutaneous wound repair and lung injury. Chest 1991, 99 (Suppl. S3), 57S–60S. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell. Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Stancil, I.T.; Michalski, J.E.; Davis-Hall, D.; Chu, H.W.; Park, J.A.; Magin, C.M.; Yang, I.V.; Smith, B.J.; Dobrinskikh, E.; Schwartz, D.A. Pulmonary fibrosis distal airway epithelia are dynamically and structurally dysfunctional. Nat. Commun. 2021, 12, 4566. [Google Scholar] [CrossRef]
- Chen, J.; Stubbe, J. Bleomycins: Towards better therapeutics. Nat. Rev. Cancer 2005, 5, 102–112. [Google Scholar] [CrossRef]
- Denham, J.W.; Hauer-Jensen, M. The radiotherapeutic injury—A complex ‘wound’. Radiother. Oncol. 2002, 63, 129–145. [Google Scholar] [CrossRef]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free. Radic. Biol. Med. 2003, 34, 1507–1516. [Google Scholar] [CrossRef]
- Kelly, B.G.; Lok, S.S.; Hasleton, P.S.; Egan, J.J.; Stewart, J.P. A rearranged form of Epstein-Barr virus DNA is associated with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2002, 166, 510–513. [Google Scholar] [CrossRef]
- Aceves, S.S.; Broide, D.H. Airway fibrosis and angiogenesis due to eosinophil trafficking in chronic asthma. Curr. Mol. Med. 2008, 8, 350–358. [Google Scholar] [CrossRef]
- Barnes, P.J. Senescence in COPD and Its Comorbidities. Annu. Rev. Physiol. 2017, 79, 517–539. [Google Scholar] [CrossRef]
- Barnes, P.J. Small airway fibrosis in COPD. Int. J. Biochem. Cell. Biol. 2019, 116, 105598. [Google Scholar] [CrossRef]
- Gamble, J.F.; Hessel, P.A.; Nicolich, M. Relationship between silicosis and lung function. Scand. J. Work. Environ. Health 2004, 30, 5–20. [Google Scholar] [CrossRef]
- Griese, M. Pulmonary surfactant in health and human lung diseases: State of the art. Eur. Respir. J. 1999, 13, 1455–1476. [Google Scholar] [CrossRef]
- Hamblin, M.; Prosch, H.; Vasakova, M. Diagnosis, course and management of hypersensitivity pneumonitis. Eur. Respir. Rev. 2022, 31, 210169. [Google Scholar] [CrossRef] [PubMed]
- Hirota, N.; Martin, J.G. Mechanisms of airway remodeling. Chest 2013, 144, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Kligerman, S.J.; Franks, T.J.; Galvin, J.R. From the radiologic pathology archives: Organization and fibrosis as a response to lung injury in diffuse alveolar damage, organizing pneumonia, and acute fibrinous and organizing pneumonia. Radiographics 2013, 33, 1951–1975. [Google Scholar] [CrossRef] [PubMed]
- Michalik, M.; Wojcik-Pszczola, K.; Paw, M.; Wnuk, D.; Koczurkiewicz, P.; Sanak, M.; Pekala, E.; Madeja, Z. Fibroblast-to-myofibroblast transition in bronchial asthma. Cell. Mol. Life Sci. 2018, 75, 3943–3961. [Google Scholar] [CrossRef]
- Nobauer-Huhmann, I.M.; Eibenberger, K.; Schaefer-Prokop, C.; Steltzer, H.; Schlick, W.; Strasser, K.; Fridrich, P.; Herold, C.J. Changes in lung parenchyma after acute respiratory distress syndrome (ARDS): Assessment with high-resolution computed tomography. Eur. Radiol. 2001, 11, 2436–2443. [Google Scholar] [CrossRef]
- Patterson, K.C.; Strek, M.E. Pulmonary fibrosis in sarcoidosis. Clinical features and outcomes. Ann. Am. Thorac. Soc. 2013, 10, 362–370. [Google Scholar] [CrossRef]
- Philip, A.G. Chronic lung disease of prematurity: A short history. Semin. Fetal Neonatal Med. 2009, 14, 333–338. [Google Scholar] [CrossRef]
- Powers, K. Acute respiratory distress syndrome. JAAPA 2022, 35, 29–33. [Google Scholar] [CrossRef]
- Sahni, M.; Mowes, A.K. Bronchopulmonary Dysplasi; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell. Signal. 2020, 66, 109482. [Google Scholar] [CrossRef]
- Selman, M.; Thannickal, V.J.; Pardo, A.; Zisman, D.A.; Martinez, F.J.; Lynch, J.P., 3rd. Idiopathic pulmonary fibrosis: Pathogenesis and therapeutic approaches. Drugs 2004, 64, 405–430. [Google Scholar] [CrossRef]
- Werlein, C.; Ackermann, M.; Hoffmann, T.L.; Laenger, F.; Jonigk, D. Fibrotic remodeling of the lung following lung and stem-cell transplantation. Pathologe 2021, 42, 17–24. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Calvert, B.A.; Ryan Firth, A.L. Application of iPSC to Modelling of Respiratory Diseases. Adv. Exp. Med. Biol. 2020, 1237, 1–16. [Google Scholar] [CrossRef]
- Carpenedo, R.L.; Julian, L.M.; Stanford, W.L. Human pluripotent stem cells. Methods 2016, 101, 1–3. [Google Scholar] [CrossRef]
- Mora, C.; Serzanti, M.; Consiglio, A.; Memo, M.; Dell’Era, P. Clinical potentials of human pluripotent stem cells. Cell. Biol. Toxicol. 2017, 33, 351–360. [Google Scholar] [CrossRef]
- Prentice, D.A. Adult Stem Cells. Circ. Res. 2019, 124, 837–839. [Google Scholar] [CrossRef]
- Cheng, X.; Ying, L.; Lu, L.; Galvao, A.M.; Mills, J.A.; Lin, H.C.; Kotton, D.N.; Shen, S.S.; Nostro, M.C.; Choi, J.K.; et al. Self-renewing endodermal progenitor lines generated from human pluripotent stem cells. Cell Stem Cell 2012, 10, 371–384. [Google Scholar] [CrossRef]
- Dailey, L.; Ambrosetti, D.; Mansukhani, A.; Basilico, C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor. Rev. 2005, 16, 233–247. [Google Scholar] [CrossRef]
- Green, M.D.; Chen, A.; Nostro, M.C.; d’Souza, S.L.; Schaniel, C.; Lemischka, I.R.; Gouon-Evans, V.; Keller, G.; Snoeck, H.W. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 267–272. [Google Scholar] [CrossRef]
- Kadzik, R.S.; Morrisey, E.E. Directing lung endoderm differentiation in pluripotent stem cells. Cell Stem Cell 2012, 10, 355–361. [Google Scholar] [CrossRef]
- Kubo, A.; Shinozaki, K.; Shannon, J.M.; Kouskoff, V.; Kennedy, M.; Woo, S.; Fehling, H.J.; Keller, G. Development of definitive endoderm from embryonic stem cells in culture. Development 2004, 131, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Longmire, T.A.; Ikonomou, L.; Hawkins, F.; Christodoulou, C.; Cao, Y.; Jean, J.C.; Kwok, L.W.; Mou, H.; Rajagopal, J.; Shen, S.S.; et al. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell 2012, 10, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Rankin, S.A.; Zorn, A.M. Gene regulatory networks governing lung specification. J. Cell. Biochem. 2014, 115, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Gao, X.; Xue, Y.; Randell, S.H.; Kong, Y.Y.; Hogan, B.L. Notch-dependent differentiation of adult airway basal stem cells. Cell Stem Cell 2011, 8, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.P.; Bear, C.E.; Chin, S.; Pasceri, P.; Thompson, T.O.; Huan, L.J.; Ratjen, F.; Ellis, J.; Rossant, J. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat. Biotechnol. 2012, 30, 876–882. [Google Scholar] [CrossRef]
- Wong, A.P.; Rossant, J. Generation of Lung Epithelium from Pluripotent Stem Cells. Curr. Pathobiol. Rep. 2013, 1, 137–145. [Google Scholar] [CrossRef]
- Dye, B.R.; Hill, D.R.; Ferguson, M.A.; Tsai, Y.H.; Nagy, M.S.; Dyal, R.; Wells, J.M.; Mayhew, C.N.; Nattiv, R.; Klein, O.D.; et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife 2015, 4, e05098. [Google Scholar] [CrossRef]
- Firth, A.L.; Dargitz, C.T.; Qualls, S.J.; Menon, T.; Wright, R.; Singer, O.; Gage, F.H.; Khanna, A.; Verma, I.M. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E1723–E1730. [Google Scholar] [CrossRef]
- Huang, S.X.; Green, M.D.; de Carvalho, A.T.; Mumau, M.; Chen, Y.W.; D’Souza, S.L.; Snoeck, H.W. The in vitro generation of lung and airway progenitor cells from human pluripotent stem cells. Nat. Protoc. 2015, 10, 413–425. [Google Scholar] [CrossRef]
- Huang, S.X.; Islam, M.N.; O’Neill, J.; Hu, Z.; Yang, Y.G.; Chen, Y.W.; Mumau, M.; Green, M.D.; Vunjak-Novakovic, G.; Bhattacharya, J.; et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 84–91. [Google Scholar] [CrossRef]
- Mou, H.; Vinarsky, V.; Tata, P.R.; Brazauskas, K.; Choi, S.H.; Crooke, A.K.; Zhang, B.; Solomon, G.M.; Turner, B.; Bihler, H.; et al. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell Stem Cell 2016, 19, 217–231. [Google Scholar] [CrossRef]
- Chen, Y.W.; Huang, S.X.; de Carvalho, A.; Ho, S.H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell. Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef]
- Magro-Lopez, E.; Guijarro, T.; Martinez, I.; Martin-Vicente, M.; Liste, I.; Zambrano, A. A Two-Dimensional Human Minilung System (Model) for Respiratory Syncytial Virus Infections. Viruses 2017, 9, 379. [Google Scholar] [CrossRef]
- Magro-Lopez, E.; Palmer, C.; Manso, J.; Liste, I.; Zambrano, A. Effects of lung and airway epithelial maturation cocktail on the structure of lung bud organoids. Stem Cell. Res. Ther. 2018, 9, 186. [Google Scholar] [CrossRef]
- Gotoh, S.; Ito, I.; Nagasaki, T.; Yamamoto, Y.; Konishi, S.; Korogi, Y.; Matsumoto, H.; Muro, S.; Hirai, T.; Funato, M.; et al. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell. Rep. 2014, 3, 394–403. [Google Scholar] [CrossRef]
- Porotto, M.; Ferren, M.; Chen, Y.W.; Siu, Y.; Makhsous, N.; Rima, B.; Briese, T.; Greninger, A.L.; Snoeck, H.W.; Moscona, A. Authentic Modeling of Human Respiratory Virus Infection in Human Pluripotent Stem Cell-Derived Lung Organoids. mBio 2019, 10, 3. [Google Scholar] [CrossRef]
- Lim, H.K.; Huang, S.X.L.; Chen, J.; Kerner, G.; Gilliaux, O.; Bastard, P.; Dobbs, K.; Hernandez, N.; Goudin, N.; Hasek, M.L.; et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J. Exp. Med. 2019, 216, 2038–2056. [Google Scholar] [CrossRef]
- Sempere, J.; Rossi, S.A.; Chamorro-Herrero, I.; Gonzalez-Camacho, F.; de Lucas, M.P.; Rojas-Cabaneros, J.M.; Taborda, C.P.; Zaragoza, O.; Yuste, J.; Zambrano, A. Minilungs from Human Embryonic Stem Cells to Study the Interaction of Streptococcus pneumoniae with the Respiratory Tract. Microbiol. Spectr. 2022, 10, e0045322. [Google Scholar] [CrossRef]
- McCauley, K.B.; Hawkins, F.; Serra, M.; Thomas, D.C.; Jacob, A.; Kotton, D.N. Efficient Derivation of Functional Human Airway Epithelium from Pluripotent Stem Cells via Temporal Regulation of Wnt Signaling. Cell Stem Cell 2017, 20, 844–857.e6. [Google Scholar] [CrossRef]
- Strikoudis, A.; Cieslak, A.; Loffredo, L.; Chen, Y.W.; Patel, N.; Saqi, A.; Lederer, D.J.; Snoeck, H.W. Modeling of Fibrotic Lung Disease Using 3D Organoids Derived from Human Pluripotent Stem Cells. Cell. Rep. 2019, 27, 3709–3723.e5. [Google Scholar] [CrossRef]
- Jacob, A.; Morley, M.; Hawkins, F.; McCauley, K.B.; Jean, J.C.; Heins, H.; Na, C.L.; Weaver, T.E.; Vedaie, M.; Hurley, K.; et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 2017, 21, 472–488.e10. [Google Scholar] [CrossRef] [PubMed]
- Korogi, Y.; Gotoh, S.; Ikeo, S.; Yamamoto, Y.; Sone, N.; Tamai, K.; Konishi, S.; Nagasaki, T.; Matsumoto, H.; Ito, I.; et al. In Vitro Disease Modeling of Hermansky-Pudlak Syndrome Type 2 Using Human Induced Pluripotent Stem Cell-Derived Alveolar Organoids. Stem Cell. Rep. 2019, 13, 235. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Poran, A.; Unni, A.M.; Huang, S.X.; Elemento, O.; Snoeck, H.W.; Varmus, H. Generation of pulmonary neuroendocrine cells and SCLC-like tumors from human embryonic stem cells. J. Exp. Med. 2019, 216, 674–687. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Itano, J.; Tanimoto, Y.; Kimura, G.; Hamada, N.; Tanaka, H.; Ninomiya, S.; Kosaki, K.; Miyahara, N.; Maeda, Y.; Kiura, K. Interstitial Pneumonia Secondary to Hermansky-Pudlak Syndrome Type 4 Treated with Different Antifibrotic Agents. Int. Med. 2021, 60, 783–788. [Google Scholar] [CrossRef]
- Lawson, W.E.; Grant, S.W.; Ambrosini, V.; Womble, K.E.; Dawson, E.P.; Lane, K.B.; Markin, C.; Renzoni, E.; Lympany, P.; Thomas, A.Q.; et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 2004, 59, 977–980. [Google Scholar] [CrossRef]
- Nogee, L.M.; Dunbar, A.E., 3rd; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 2001, 344, 573–579. [Google Scholar] [CrossRef]
- Nureki, S.I.; Tomer, Y.; Venosa, A.; Katzen, J.; Russo, S.J.; Jamil, S.; Barrett, M.; Nguyen, V.; Kopp, M.; Mulugeta, S.; et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Investig. 2018, 128, 4008–4024. [Google Scholar] [CrossRef]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef]
- Noble, P.W.; Homer, R.J. Idiopathic pulmonary fibrosis: New insights into pathogenesis. Clin. Chest Med. 2004, 25, 749–758, vii. [Google Scholar] [CrossRef]
- Raghu, G.; Chang, J. Idiopathic pulmonary fibrosis: Current trends in management. Clin. Chest Med. 2004, 25, 621–636, v. [Google Scholar] [CrossRef]
- Selman, M.; King, T.E.; Pardo, A.; American Thoracic Society; European Respiratory Society; American College of Chest Physicians. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Int. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef]
- Chilosi, M.; Carloni, A.; Rossi, A.; Poletti, V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl. Res. 2013, 162, 156–173. [Google Scholar] [CrossRef]
- Kuwano, K.; Araya, J.; Hara, H.; Minagawa, S.; Takasaka, N.; Ito, S.; Kobayashi, K.; Nakayama, K. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir. Investig. 2016, 54, 397–406. [Google Scholar] [CrossRef]
- Kuwano, K.; Kunitake, R.; Kawasaki, M.; Nomoto, Y.; Hagimoto, N.; Nakanishi, Y.; Hara, N. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1996, 154, 477–483. [Google Scholar] [CrossRef]
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. (Lausanne) 2017, 4, 154. [Google Scholar] [CrossRef]
- Kropski, J.A.; Blackwell, T.S.; Loyd, J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1717–1727. [Google Scholar] [CrossRef]
- Kropski, J.A.; Loyd, J.E. Telomeres revisited: RTEL1 variants in pulmonary fibrosis. Eur. Respir. J. 2015, 46, 312–314. [Google Scholar] [CrossRef]
- Aoshiba, K.; Tsuji, T.; Nagai, A. Bleomycin induces cellular senescence in alveolar epithelial cells. Eur. Respir. J. 2003, 22, 436–443. [Google Scholar] [CrossRef]
- Chua, F.; Gauldie, J.; Laurent, G.J. Pulmonary fibrosis: Searching for model answers. Am. J. Respir. Cell. Mol. Biol. 2005, 33, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, T.; Magro-Lopez, E.; Manso, J.; Garcia-Martinez, R.; Fernandez-Acenero, M.J.; Liste, I.; Zambrano, A. Detrimental pro-senescence effects of vitamin D on lung fibrosis. Mol. Med. 2018, 24, 64. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.X.; Chen, Y.H.; Xu, S.; Qin, H.Y.; Zhang, C.; Zhao, H.; Xu, D.X. Calcitriol inhibits bleomycin-induced early pulmonary inflammatory response and epithelial-mesenchymal transition in mice. Toxicol. Lett. 2016, 240, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, X.; Fang, X.; Liang, A.; Yu, Z.; Gu, P.; Zeng, Y.; He, J.; Zhu, H.; Li, S.; et al. Preventive effects of vitamin D treatment on bleomycin-induced pulmonary fibrosis. Sci. Rep. 2015, 5, 17638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.M.; Gu, P.; Yi, X.H.; Fang, X.; Zeng, Y.; Zhang, S.X.; Zhu, X.Y.; Zhang, Y.D.; Gu, J.; Qiu, W.Z.; et al. Effects of 1, 25 (OH)2D3 on bleomycin-induced pulmonary fibrosis in mice. Zhonghua Jie He He Hu Xi Za Zhi 2013, 36, 814–820. [Google Scholar]
- Araya, J.; Kuwano, K. Cellular senescence-an aging hallmark in chronic obstructive pulmonary disease pathogenesis. Respir. Investig. 2022, 60, 33–44. [Google Scholar] [CrossRef]
- Magro-Lopez, E.; Chamorro-Herrero, I.; Zambrano, A. Effects of Hypocalcemic Vitamin D Analogs in the Expression of DNA Damage Induced in Minilungs from hESCs: Implications for Lung Fibrosis. Int. J. Mol. Sci. 2022, 23, 4921. [Google Scholar] [CrossRef]
- Wilkinson, D.C.; Alva-Ornelas, J.A.; Sucre, J.M.; Vijayaraj, P.; Durra, A.; Richardson, W.; Jonas, S.J.; Paul, M.K.; Karumbayaram, S.; Dunn, B.; et al. Development of a Three-Dimensional Bioengineering Technology to Generate Lung Tissue for Personalized Disease Modeling. Stem Cells Transl. Med. 2017, 6, 622–633. [Google Scholar] [CrossRef]
- Schruf, E.; Schroeder, V.; Le, H.Q.; Schonberger, T.; Raedel, D.; Stewart, E.L.; Fundel-Clemens, K.; Bluhmki, T.; Weigle, S.; Schuler, M.; et al. Recapitulating idiopathic pulmonary fibrosis related alveolar epithelial dysfunction in a human iPSC-derived air-liquid interface model. FASEB J. 2020, 34, 7825–7846. [Google Scholar] [CrossRef]
- Suezawa, T.; Kanagaki, S.; Moriguchi, K.; Masui, A.; Nakao, K.; Toyomoto, M.; Tamai, K.; Mikawa, R.; Hirai, T.; Murakami, K.; et al. Disease modeling of pulmonary fibrosis using human pluripotent stem cell-derived alveolar organoids. Stem Cell. Rep. 2021, 16, 2973–2987. [Google Scholar] [CrossRef]
- Scotet, V.; Dugueperoux, I.; Saliou, P.; Rault, G.; Roussey, M.; Audrezet, M.P.; Ferec, C. Evidence for decline in the incidence of cystic fibrosis: A 35-year observational study in Brittany, France. Orphanet J. Rare Dis. 2012, 7, 14. [Google Scholar] [CrossRef]
- Spoonhower, K.A.; Davis, P.B. Epidemiology of Cystic Fibrosis. Clin. Chest Med. 2016, 37, 1–8. [Google Scholar] [CrossRef]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Kartner, N.; Hanrahan, J.W.; Jensen, T.J.; Naismith, A.L.; Sun, S.Z.; Ackerley, C.A.; Reyes, E.F.; Tsui, L.C.; Rommens, J.M.; Bear, C.E.; et al. Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produces a regulated anion conductance. Cell 1991, 64, 681–691. [Google Scholar] [CrossRef]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef]
- Knowles, M.R.; Stutts, M.J.; Spock, A.; Fischer, N.; Gatzy, J.T.; Boucher, R.C. Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science 1983, 221, 1067–1070. [Google Scholar] [CrossRef]
- Quinton, P.M. Chloride impermeability in cystic fibrosis. Nature 1983, 301, 421–422. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C. Airway surface dehydration in cystic fibrosis: Pathogenesis and therapy. Annu. Rev. Med. 2007, 58, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Garland, A.L.; Walton, W.G.; Coakley, R.D.; Tan, C.D.; Gilmore, R.C.; Hobbs, C.A.; Tripathy, A.; Clunes, L.A.; Bencharit, S.; Stutts, M.J.; et al. Molecular basis for pH-dependent mucosal dehydration in cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2013, 110, 15973–15978. [Google Scholar] [CrossRef] [PubMed]
- Abou Alaiwa, M.H.; Beer, A.M.; Pezzulo, A.A.; Launspach, J.L.; Horan, R.A.; Stoltz, D.A.; Starner, T.D.; Welsh, M.J.; Zabner, J. Neonates with cystic fibrosis have a reduced nasal liquid pH; a small pilot study. J. Cyst. Fibros. 2014, 13, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef]
- Song, Y.; Salinas, D.; Nielson, D.W.; Verkman, A.S. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am. J. Physiol. Cell. Physiol. 2006, 290, C741–C749. [Google Scholar] [CrossRef]
- Malhotra, S.; Hayes, D., Jr.; Wozniak, D.J. Cystic Fibrosis and Pseudomonas aeruginosa: The Host-Microbe Interface. Clin. Microbiol. Rev. 2019, 32, 3. [Google Scholar] [CrossRef]
- Malhotra, S.; Hayes, D., Jr.; Wozniak, D.J. Mucoid Pseudomonas aeruginosa and regional inflammation in the cystic fibrosis lung. J. Cyst. Fibros. 2019, 18, 796–803. [Google Scholar] [CrossRef]
- Hilliard, T.N.; Regamey, N.; Shute, J.K.; Nicholson, A.G.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling in children with cystic fibrosis. Thorax 2007, 62, 1074–1080. [Google Scholar] [CrossRef]
- Huaux, F.; Noel, S.; Dhooghe, B.; Panin, N.; Lo Re, S.; Lison, D.; Wallemacq, P.; Marbaix, E.; Scholte, B.J.; Lebecque, P.; et al. Dysregulated proinflammatory and fibrogenic phenotype of fibroblasts in cystic fibrosis. PLoS ONE 2013, 8, e64341. [Google Scholar] [CrossRef]
- Geurts, M.H.; de Poel, E.; Amatngalim, G.D.; Oka, R.; Meijers, F.M.; Kruisselbrink, E.; van Mourik, P.; Berkers, G.; de Winter-de Groot, K.M.; Michel, S.; et al. CRISPR-Based Adenine Editors Correct Nonsense Mutations in a Cystic Fibrosis Organoid Biobank. Cell Stem Cell 2020, 26, 503–510.e7. [Google Scholar] [CrossRef]
- Decramer, M.; Janssens, W.; Miravitlles, M. Chronic obstructive pulmonary disease. Lancet 2012, 379, 1341–1351. [Google Scholar] [CrossRef]
- Papi, A.; Bellettato, C.M.; Braccioni, F.; Romagnoli, M.; Casolari, P.; Caramori, G.; Fabbri, L.M.; Johnston, S.L. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am. J. Respir. Crit. Care Med. 2006, 173, 1114–1121. [Google Scholar] [CrossRef]
- Alsumrain, M.; De Giacomi, F.; Nasim, F.; Koo, C.W.; Bartholmai, B.J.; Levin, D.L.; Moua, T. Combined pulmonary fibrosis and emphysema as a clinicoradiologic entity: Characterization of presenting lung fibrosis and implications for survival. Respir. Med. 2019, 146, 106–112. [Google Scholar] [CrossRef]
- Hogg, J.C.; Pare, P.D.; Hackett, T.L. The Contribution of Small Airway Obstruction to the Pathogenesis of Chronic Obstructive Pulmonary Disease. Physiol. Rev. 2017, 97, 529–552. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Hassibi, S.; Fenwick, P.S.; Donnelly, L.E.; Ito, K.; Barnes, P.J. MicroRNA-570 is a novel regulator of cellular senescence and inflammaging. FASEB J. 2019, 33, 1605–1616. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative stress dependent microRNA-34a activation via PI3Kalpha reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell. Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]
- De Rose, V.; Molloy, K.; Gohy, S.; Pilette, C.; Greene, C.M. Airway Epithelium Dysfunction in Cystic Fibrosis and COPD. Mediators Inflamm. 2018, 2018, 1309746. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.; Fieldes, M.; Bourguignon, C.; Mianne, J.; Petit, A.; Jory, M.; Cazevieille, C.; Boukhaddaoui, H.; Garnett, J.P.; Hirtz, C.; et al. Differentiation of Human Induced Pluripotent Stem Cells from Patients with Severe COPD into Functional Airway Epithelium. Cells 2022, 11, 2422. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.; Fieldes, M.; Mianne, J.; Bourguignon, C.; Nasri, A.; Vachier, I.; Assou, S.; Bourdin, A.; De Vos, J. Generation of four severe early-onset chronic obstructive pulmonary disease (COPD) patient-derived induced pluripotent stem cell lines from peripheral blood mononuclear cells. Stem Cell Res. 2021, 56, 102550. [Google Scholar] [CrossRef]
- Basma, H.; Gunji, Y.; Iwasawa, S.; Nelson, A.; Farid, M.; Ikari, J.; Liu, X.; Wang, X.; Michalski, J.; Smith, L.; et al. Reprogramming of COPD lung fibroblasts through formation of induced pluripotent stem cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L552–L565. [Google Scholar] [CrossRef]
- Nasser, M.; Larrieu, S.; Si-Mohamed, S.; Ahmad, K.; Boussel, L.; Brevet, M.; Chalabreysse, L.; Fabre, C.; Marque, S.; Revel, D.; et al. Progressive fibrosing interstitial lung disease: A clinical cohort (the PROGRESS study). Eur. Respir. J. 2021, 57, 2002718. [Google Scholar] [CrossRef]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm. Med. 2020, 2020, 6175964. [Google Scholar] [CrossRef]
- Spagnolo, P.; Balestro, E.; Aliberti, S.; Cocconcelli, E.; Biondini, D.; Casa, G.D.; Sverzellati, N.; Maher, T.M. Pulmonary fibrosis secondary to COVID-19: A call to arms? Lancet Respir. Med. 2020, 8, 750–752. [Google Scholar] [CrossRef]
- Tian, S.; Xiong, Y.; Liu, H.; Niu, L.; Guo, J.; Liao, M.; Xiao, S.Y. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod. Pathol. 2020, 33, 1007–1014. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Aveyard, P.; Gao, M.; Lindson, N.; Hartmann-Boyce, J.; Watkinson, P.; Young, D.; Coupland, C.A.C.; Tan, P.S.; Clift, A.K.; Harrison, D.; et al. Association between pre-existing respiratory disease and its treatment, and severe COVID-19: A population cohort study. Lancet Respir. Med. 2021, 9, 909–923. [Google Scholar] [CrossRef]
- Drake, T.M.; Docherty, A.B.; Harrison, E.M.; Quint, J.K.; Adamali, H.; Agnew, S.; Babu, S.; Barber, C.M.; Barratt, S.; Bendstrup, E.; et al. Outcome of Hospitalization for COVID-19 in Patients with Interstitial Lung Disease. An International Multicenter Study. Am. J. Respir. Crit. Care Med. 2020, 202, 1656–1665. [Google Scholar] [CrossRef]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef]
- Collard, H.R.; Yow, E.; Richeldi, L.; Anstrom, K.J.; Glazer, C.; IPFnet investigators. Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir. Res. 2013, 14, 73. [Google Scholar] [CrossRef]
- Shen, H.; Zhang, N.; Liu, Y.; Yang, X.; He, Y.; Li, Q.; Shen, X.; Zhu, Y.; Yang, Y. The Interaction Between Pulmonary Fibrosis and COVID-19 and the Application of Related Anti-Fibrotic Drugs. Front. Pharmacol. 2021, 12, 805535. [Google Scholar] [CrossRef]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffre, F.; et al. A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 2020, 27, 125–136. [Google Scholar] [CrossRef]
- Han, Y.; Duan, X.; Yang, L.; Nilsson-Payant, B.E.; Wang, P.; Duan, F.; Tang, X.; Yaron, T.M.; Zhang, T.; Uhl, S.; et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 2021, 589, 270–275. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef]
- Bellusci, S.; Furuta, Y.; Rush, M.G.; Henderson, R.; Winnier, G.; Hogan, B.L. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development 1997, 124, 53–63. [Google Scholar] [CrossRef]
- Bellusci, S.; Grindley, J.; Emoto, H.; Itoh, N.; Hogan, B.L. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 1997, 124, 4867–4878. [Google Scholar] [CrossRef] [PubMed]
- Dye, B.R.; Dedhia, P.H.; Miller, A.J.; Nagy, M.S.; White, E.S.; Shea, L.D.; Spence, J.R. A bioengineered niche promotes in vivo engraftment and maturation of pluripotent stem cell derived human lung organoids. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Konishi, S.; Gotoh, S.; Tateishi, K.; Yamamoto, Y.; Korogi, Y.; Nagasaki, T.; Matsumoto, H.; Muro, S.; Hirai, T.; Ito, I.; et al. Directed Induction of Functional Multi-ciliated Cells in Proximal Airway Epithelial Spheroids from Human Pluripotent Stem Cells. Stem Cell Rep. 2016, 6, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Okubo, T.; Hogan, B.L. Hyperactive Wnt signaling changes the developmental potential of embryonic lung endoderm. J. Biol. 2004, 3, 11. [Google Scholar] [CrossRef]
- Rawlins, E.L.; Clark, C.P.; Xue, Y.; Hogan, B.L. The Id2+ distal tip lung epithelium contains individual multipotent embryonic progenitor cells. Development 2009, 136, 3741–3745. [Google Scholar] [CrossRef]
- Rawlins, E.L.; Okubo, T.; Xue, Y.; Brass, D.M.; Auten, R.L.; Hasegawa, H.; Wang, F.; Hogan, B.L. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 2009, 4, 525–534. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamorro-Herrero, I.; Zambrano, A. Modeling of Respiratory Diseases Evolving with Fibrosis from Organoids Derived from Human Pluripotent Stem Cells. Int. J. Mol. Sci. 2023, 24, 4413. https://doi.org/10.3390/ijms24054413
Chamorro-Herrero I, Zambrano A. Modeling of Respiratory Diseases Evolving with Fibrosis from Organoids Derived from Human Pluripotent Stem Cells. International Journal of Molecular Sciences. 2023; 24(5):4413. https://doi.org/10.3390/ijms24054413
Chicago/Turabian StyleChamorro-Herrero, Irene, and Alberto Zambrano. 2023. "Modeling of Respiratory Diseases Evolving with Fibrosis from Organoids Derived from Human Pluripotent Stem Cells" International Journal of Molecular Sciences 24, no. 5: 4413. https://doi.org/10.3390/ijms24054413
APA StyleChamorro-Herrero, I., & Zambrano, A. (2023). Modeling of Respiratory Diseases Evolving with Fibrosis from Organoids Derived from Human Pluripotent Stem Cells. International Journal of Molecular Sciences, 24(5), 4413. https://doi.org/10.3390/ijms24054413