


Lipids at the Nexus between Cerebrovascular Disease and Vascular Dementia: The Impact of HDL-Cholesterol and Ceramides

 ,
,  ,
, 

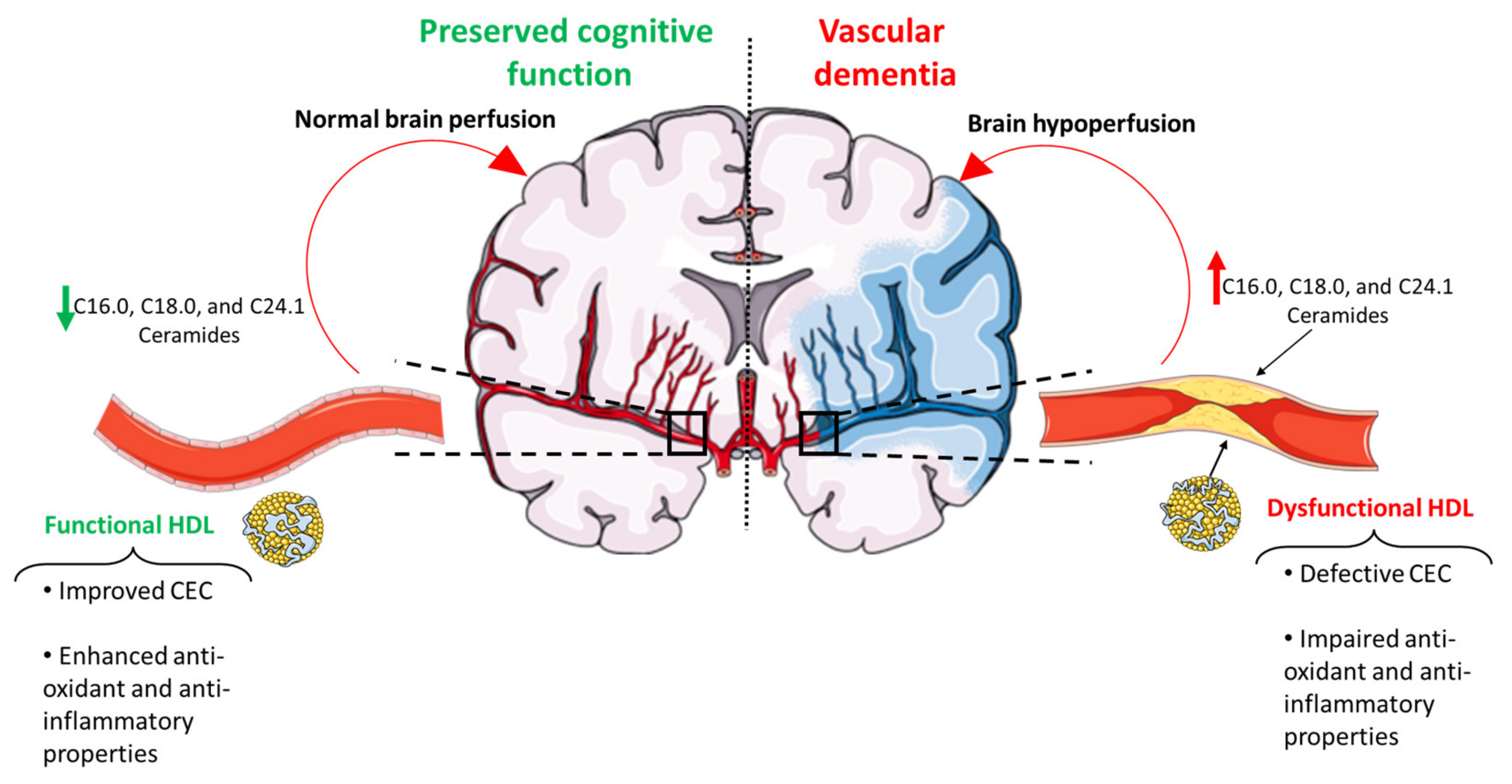{kind=link}
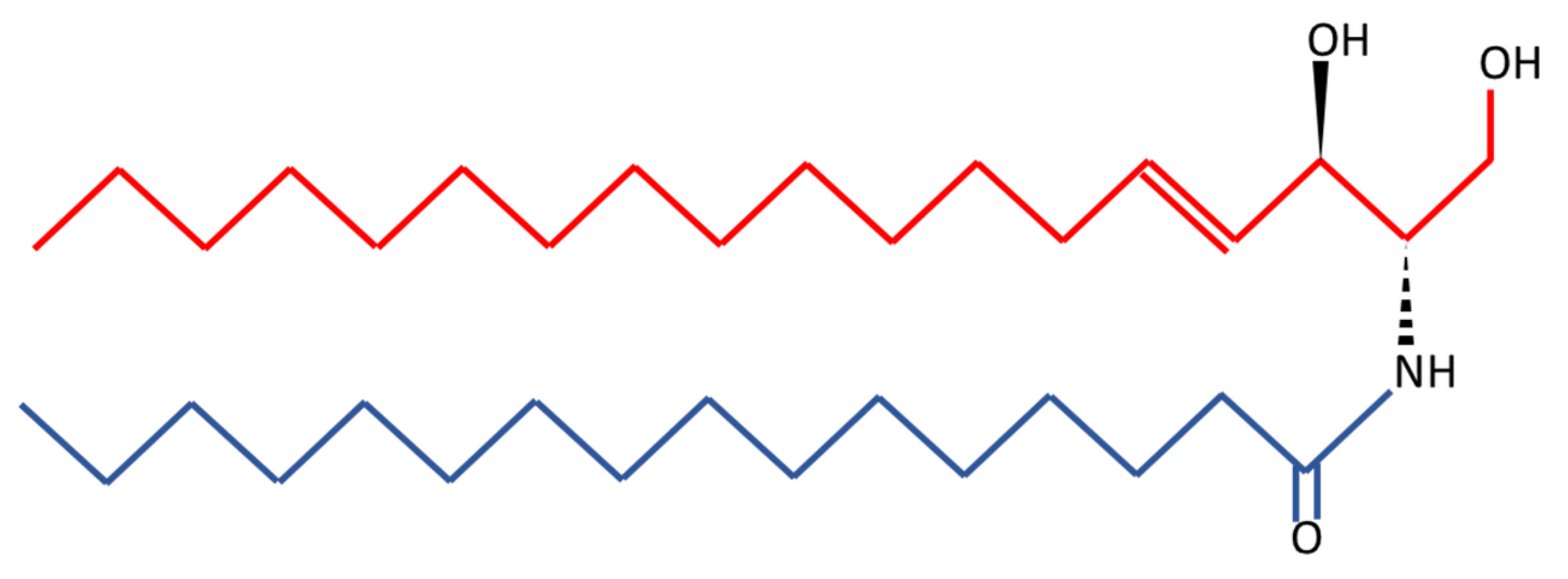{kind=link}
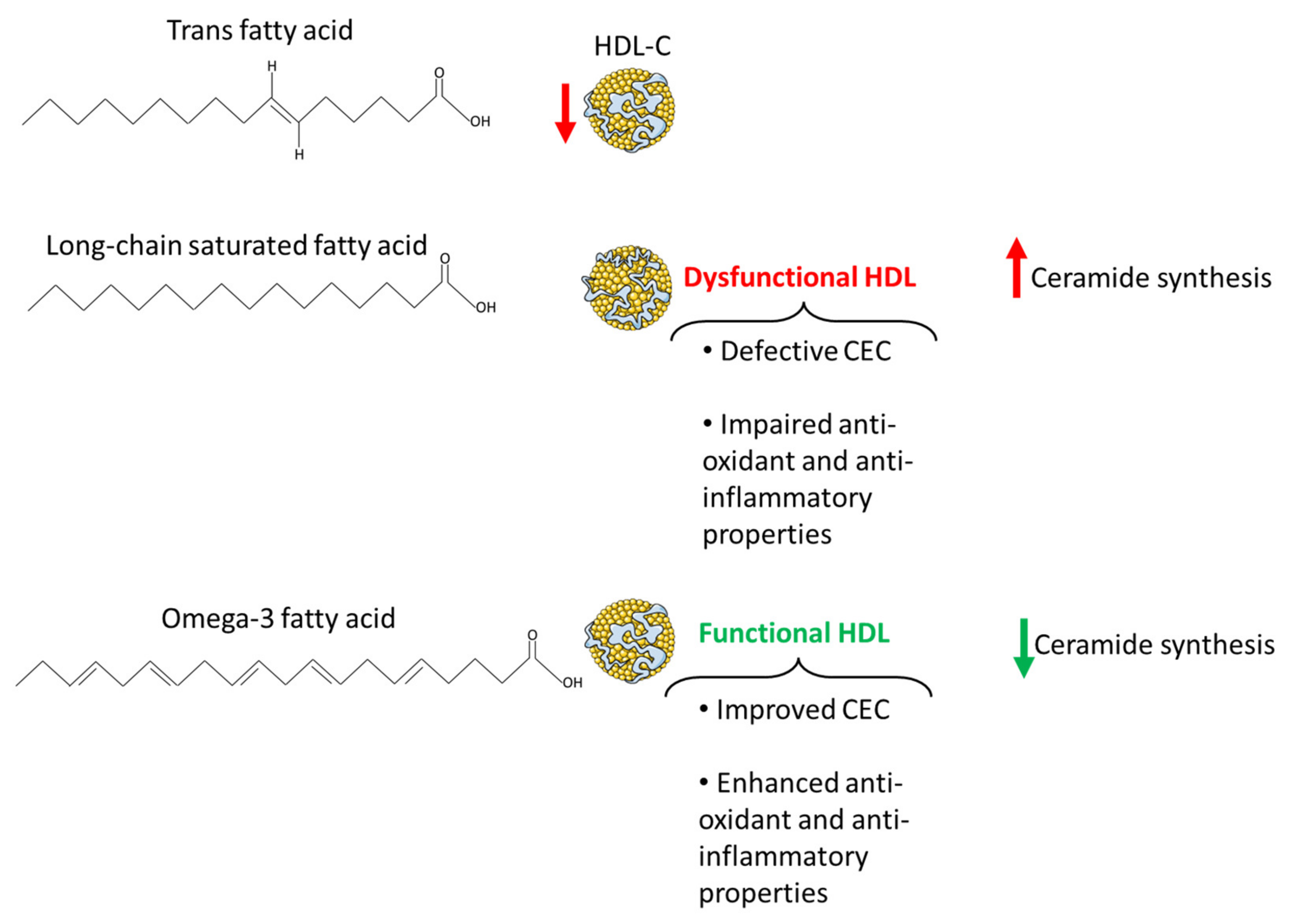{kind=link}
Abstract
1. Introduction
2. VaD
3. Dyslipidemia: A Non-Exhaustive Definition
4. Functional and Dysfunctional HDL
4.1. HDL Remodeling
4.2. Functional HDL
4.3. Evaluation of HDL Function
4.4. HDL-like Particles and Cerebrovascular Diseases
4.5. Dysfunctional HDLlp
5. Evidence from Observational Studies Linking HDL and HDLlp to VaD
Mechanisms Linking Dysfunctional HDL and HDLlp and VaD
6. Ceramides as a Novel Potential Driver of Cerebrovascular Disease
7. Dietary Lipids in the Pathogenesis of Cerebrovascular Disease
7.1. Omega-3 Fatty Acids
7.2. Long-Chain Saturated Fatty Acids
7.3. Trans Fatty Acids
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, W.V.; Fujioka, K.; Wilson, P.W.; Woodworth, K.A. Obesity: Why be concerned? Am. J. Med. 2009, 122, S4–S11. [Google Scholar] [CrossRef] [PubMed]
- Mazon, J.N.; de Mello, A.H.; Ferreira, G.K.; Rezin, G.T. The impact of obesity on neurodegenerative diseases. Life Sci. 2017, 182, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Singh-Manoux, A.; Dugravot, A.; Shipley, M.; Brunner, E.J.; Elbaz, A.; Sabia, S.; Kivimaki, M. Obesity trajectories and risk of dementia: 28 years of follow-up in the Whitehall II Study. Alzheimers Dement. 2018, 14, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Cooney, M.T.; Dudina, A.; De Bacquer, D.; Wilhelmsen, L.; Sans, S.; Menotti, A.; De Backer, G.; Jousilahti, P.; Keil, U.; Thomsen, T.; et al. HDL cholesterol protects against cardiovascular disease in both genders, at all ages and at all levels of risk. Atherosclerosis 2009, 206, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Jomard, A.; Osto, E. High Density Lipoproteins: Metabolism, Function, and Therapeutic Potential. Front. Cardiovasc. Med. 2020, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Choi, R.H.; Tatum, S.M.; Symons, J.D.; Summers, S.A.; Holland, W.L. Ceramides and other sphingolipids as drivers of cardiovascular disease. Nat. Rev. Cardiol. 2021, 18, 701–711. [Google Scholar] [CrossRef]
- Ding, M.; Rexrode, K.M. A Review of Lipidomics of Cardiovascular Disease Highlights the Importance of Isolating Lipoproteins. Metabolites 2020, 10, 163. [Google Scholar] [CrossRef]
- McGurk, K.A.; Keavney, B.D.; Nicolaou, A. Circulating ceramides as biomarkers of cardiovascular disease: Evidence from phenotypic and genomic studies. Atherosclerosis 2021, 327, 18–30. [Google Scholar] [CrossRef]
- Mishra, P.P.; Mishra, B.H.; Lyytikäinen, L.-P.; Hilvo, M.; Juonala, M.; Kähönen, M.; Hutri-Kähönen, N.; Fotiadis, D.I.; Raitakari, O.T.; Laaksonen, R. Assessment of plasma ceramides as predictor for subclinical atherosclerosis. Atheroscler. Plus 2021, 45, 25–31. [Google Scholar] [CrossRef]
- Veno, S.K.; Schmidt, E.B.; Bork, C.S. Polyunsaturated Fatty Acids and Risk of Ischemic Stroke. Nutrients 2019, 11, 1467. [Google Scholar] [CrossRef]
- de Wilde, M.C.; Farkas, E.; Gerrits, M.; Kiliaan, A.J.; Luiten, P.G. The effect of n-3 polyunsaturated fatty acid-rich diets on cognitive and cerebrovascular parameters in chronic cerebral hypoperfusion. Brain Res. 2002, 947, 166–173. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; O’Keefe, J.H. Effects of dietary fats on blood lipids: A review of direct comparison trials. Open Heart 2018, 5, e000871. [Google Scholar] [CrossRef]
- Cartolano, F.C.; Dias, G.D.; Miyamoto, S.; Damasceno, N.R.T. Omega-3 Fatty Acids Improve Functionality of High-Density Lipoprotein in Individuals With High Cardiovascular Risk: A Randomized, Parallel, Controlled and Double-Blind Clinical Trial. Front. Nutr. 2021, 8, 767535. [Google Scholar] [CrossRef]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef]
- Sergi, D.; Morris, A.C.; Kahn, D.E.; McLean, F.H.; Hay, E.A.; Kubitz, P.; MacKenzie, A.; Martinoli, M.G.; Drew, J.E.; Williams, L.M. Palmitic acid triggers inflammatory responses in N42 cultured hypothalamic cells partially via ceramide synthesis but not via TLR4. Nutr. Neurosci. 2018, 23, 321–334. [Google Scholar] [CrossRef]
- Akhter, F.; Persaud, A.; Zaokari, Y.; Zhao, Z.; Zhu, D. Vascular Dementia and Underlying Sex Differences. Front. Aging Neurosci. 2021, 13, 720715. [Google Scholar] [CrossRef]
- Collaborators, G.B.D.D.F. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Wolters, F.J.; Ikram, M.A. Epidemiology of Vascular Dementia. Arter. Thromb. Vasc. Biol. 2019, 39, 1542–1549. [Google Scholar] [CrossRef]
- Si, S.; Hou, L.; Chen, X.; Li, W.; Liu, X.; Liu, C.; Li, Y.; Yuan, T.; Li, J.; Wang, B.; et al. Exploring the Causal Roles of Circulating Remnant Lipid Profile on Cardiovascular and Cerebrovascular Diseases: Mendelian Randomization Study. J. Epidemiol. 2022, 32, 205–214. [Google Scholar] [CrossRef]
- Button, E.B.; Robert, J.; Caffrey, T.M.; Fan, J.; Zhao, W.; Wellington, C.L. HDL from an Alzheimer’s disease perspective. Curr. Opin. Lipidol. 2019, 30, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Zuliani, G.; Cavalieri, M.; Galvani, M.; Volpato, S.; Cherubini, A.; Bandinelli, S.; Corsi, A.M.; Lauretani, F.; Guralnik, J.M.; Fellin, R.; et al. Relationship between low levels of high-density lipoprotein cholesterol and dementia in the elderly. The InChianti study. J. Gerontol. Biol. Sci. Med. Sci. 2010, 65, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.Y.; Johnson, C.; Kao, W.H.; Sharrett, A.R.; Arends, V.L.; Kronmal, R.; Jenny, N.S.; Jacobs, D.R., Jr.; Arnett, D.; O’Leary, D.; et al. Cholesteryl ester transfer protein genetic polymorphisms, HDL cholesterol, and subclinical cardiovascular disease in the Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2008, 200, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lou, Y.; Qiu, X.; Liu, Y.; Lu, L.; Chen, Q.; Jin, W. Association of cholesteryl ester transfer protein (CETP) gene polymorphism, high density lipoprotein cholesterol and risk of coronary artery disease: A meta-analysis using a Mendelian randomization approach. BMC Med. Genet. 2014, 15, 118. [Google Scholar] [CrossRef]
- Phillips, M.C. New insights into the determination of HDL structure by apolipoproteins: Thematic review series: High density lipoprotein structure, function, and metabolism. J. Lipid Res. 2013, 54, 2034–2048. [Google Scholar] [CrossRef]
- Kontush, A.; Chantepie, S.; Chapman, M.J. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1881–1888. [Google Scholar] [CrossRef]
- Kontush, A. HDL particle number and size as predictors of cardiovascular disease. Front. Pharmacol. 2015, 6, 218. [Google Scholar] [CrossRef]
- Soedamah-Muthu, S.S.; Chang, Y.F.; Otvos, J.; Evans, R.W.; Orchard, T.J.; Pittsburgh Epidemiology of Diabetes Complications Study. Lipoprotein subclass measurements by nuclear magnetic resonance spectroscopy improve the prediction of coronary artery disease in Type 1 diabetes. A prospective report from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetologia 2003, 46, 674–682. [Google Scholar] [CrossRef]
- Dias, I.H.; Polidori, M.C.; Li, L.; Weber, D.; Stahl, W.; Nelles, G.; Grune, T.; Griffiths, H.R. Plasma levels of HDL and carotenoids are lower in dementia patients with vascular comorbidities. J. Alzheimers Dis. 2014, 40, 399–408. [Google Scholar] [CrossRef]
- Adorni, M.P.; Ronda, N.; Bernini, F.; Zimetti, F. High Density Lipoprotein Cholesterol Efflux Capacity and Atherosclerosis in Cardiovascular Disease: Pathophysiological Aspects and Pharmacological Perspectives. Cells 2021, 10, 574. [Google Scholar] [CrossRef]
- Cervellati, C.; Vigna, G.B.; Trentini, A.; Sanz, J.M.; Zimetti, F.; Dalla Nora, E.; Morieri, M.L.; Zuliani, G.; Passaro, A. Paraoxonase-1 activities in individuals with different HDL circulating levels: Implication in reverse cholesterol transport and early vascular damage. Atherosclerosis 2019, 285, 64–70. [Google Scholar] [CrossRef]
- Kobayashi, A.; Takanezawa, Y.; Hirata, T.; Shimizu, Y.; Misasa, K.; Kioka, N.; Arai, H.; Ueda, K.; Matsuo, M. Efflux of sphingomyelin, cholesterol, and phosphatidylcholine by ABCG1. J. Lipid Res. 2006, 47, 1791–1802. [Google Scholar] [CrossRef]
- Mangaraj, M.; Nanda, R.; Panda, S. Apolipoprotein A-I: A Molecule of Diverse Function. Indian J. Clin. Biochem. 2016, 31, 253–259. [Google Scholar] [CrossRef]
- Aviram, M.; Rosenblat, M.; Billecke, S.; Erogul, J.; Sorenson, R.; Bisgaier, C.L.; Newton, R.S.; La Du, B. Human serum paraoxonase (PON 1) is inactivated by oxidized low density lipoprotein and preserved by antioxidants. Free Radic. Biol. Med. 1999, 26, 892–904. [Google Scholar] [CrossRef]
- van der Steeg, W.A.; Holme, I.; Boekholdt, S.M.; Larsen, M.L.; Lindahl, C.; Stroes, E.S.; Tikkanen, M.J.; Wareham, N.J.; Faergeman, O.; Olsson, A.G.; et al. High-density lipoprotein cholesterol, high-density lipoprotein particle size, and apolipoprotein A-I: Significance for cardiovascular risk: The IDEAL and EPIC-Norfolk studies. J. Am. Coll. Cardiol. 2008, 51, 634–642. [Google Scholar] [CrossRef]
- Qi, Y.; Fan, J.; Liu, J.; Wang, W.; Wang, M.; Sun, J.; Liu, J.; Xie, W.; Zhao, F.; Li, Y.; et al. Cholesterol-overloaded HDL particles are independently associated with progression of carotid atherosclerosis in a cardiovascular disease-free population: A community-based cohort study. J. Am. Coll. Cardiol. 2015, 65, 355–363. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef]
- Tolle, M.; Huang, T.; Schuchardt, M.; Jankowski, V.; Prufer, N.; Jankowski, J.; Tietge, U.J.; Zidek, W.; van der Giet, M. High-density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-inflammatory-serum amyloid A. Cardiovasc. Res. 2012, 94, 154–162. [Google Scholar] [CrossRef]
- Marsche, G.; Stadler, J.T.; Kargl, J.; Holzer, M. Understanding Myeloperoxidase-Induced Damage to HDL Structure and Function in the Vessel Wall: Implications for HDL-Based Therapies. Antioxidants 2022, 11, 556. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, Z.; Riwanto, M.; Gao, S.; Levison, B.S.; Gu, X.; Fu, X.; Wagner, M.A.; Besler, C.; Gerstenecker, G.; et al. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J. Clin. Investig. 2013, 123, 3815–3828. [Google Scholar] [CrossRef]
- Shao, B.; Cavigiolio, G.; Brot, N.; Oda, M.N.; Heinecke, J.W. Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-I. Proc. Natl. Acad. Sci. USA 2008, 105, 12224–12229. [Google Scholar] [CrossRef]
- Sorci-Thomas, M.G.; Bhat, S.; Thomas, M.J. Activation of lecithin:cholesterol acyltransferase by HDL ApoA-I central helices. Clin. Lipidol. 2009, 4, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A.; Genest, J. High density lipoproteins: Measurement techniques and potential biomarkers of cardiovascular risk. BBA Clin. 2015, 3, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Tang, Q.; Cheng, C.; Xu, S. Low serum lipid levels, use of statin and cerebral microbleeds: A systematic review and meta-analysis. J. Clin. Neurosci. 2021, 94, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Tang, M.X.; Luchsinger, J.; Mayeux, R. Relation of plasma lipids to Alzheimer disease and vascular dementia. Arch. Neurol. 2004, 61, 705–714. [Google Scholar] [CrossRef]
- Barter, P.; Gotto, A.M.; LaRosa, J.C.; Maroni, J.; Szarek, M.; Grundy, S.M.; Kastelein, J.J.; Bittner, V.; Fruchart, J.C.; Treating to New Targets Investigators. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N. Engl. J. Med. 2007, 357, 1301–1310. [Google Scholar] [CrossRef]
- Woodward, M.; Barzi, F.; Feigin, V.; Gu, D.; Huxley, R.; Nakamura, K.; Patel, A.; Ho, S.; Jamrozik, K.; Asia Pacific Cohort Studies, C. Associations between high-density lipoprotein cholesterol and both stroke and coronary heart disease in the Asia Pacific region. Eur. Heart J. 2007, 28, 2653–2660. [Google Scholar] [CrossRef]
- Michikawa, M. Cholesterol paradox: Is high total or low HDL cholesterol level a risk for Alzheimer’s disease? J. Neurosci. Res. 2003, 72, 141–146. [Google Scholar] [CrossRef]
- Willey, J.; Gonzalez-Castellon, M. Cholesterol level and stroke: A complex relationship. JAMA Intern. Med. 2013, 173, 1765–1766. [Google Scholar] [CrossRef]
- Anstey, K.J.; Ashby-Mitchell, K.; Peters, R. Updating the Evidence on the Association between Serum Cholesterol and Risk of Late-Life Dementia: Review and Meta-Analysis. J. Alzheimers Dis. 2017, 56, 215–228. [Google Scholar] [CrossRef]
- Bots, M.L.; Elwood, P.C.; Nikitin, Y.; Salonen, J.T.; Freire de Concalves, A.; Inzitari, D.; Sivenius, J.; Benetou, V.; Tuomilehto, J.; Koudstaal, P.J.; et al. Total and HDL cholesterol and risk of stroke. EUROSTROKE: A collaborative study among research centres in Europe. J. Epidemiol. Community Health 2002, 56 (Suppl. S1), i19–i24. [Google Scholar] [CrossRef]
- Duong, M.T.; Nasrallah, I.M.; Wolk, D.A.; Chang, C.C.Y.; Chang, T.Y. Cholesterol, Atherosclerosis, and APOE in Vascular Contributions to Cognitive Impairment and Dementia (VCID): Potential Mechanisms and Therapy. Front. Aging Neurosci. 2021, 13, 647990. [Google Scholar] [CrossRef]
- Fung, K.Y.; Wang, C.; Nyegaard, S.; Heit, B.; Fairn, G.D.; Lee, W.L. SR-BI Mediated Transcytosis of HDL in Brain Microvascular Endothelial Cells Is Independent of Caveolin, Clathrin, and PDZK1. Front. Physiol. 2017, 8, 841. [Google Scholar] [CrossRef]
- Rhea, E.M.; Banks, W.A. Interactions of Lipids, Lipoproteins, and Apolipoproteins with the Blood-Brain Barrier. Pharm. Res. 2021, 38, 1469–1475. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5644–5651. [Google Scholar] [CrossRef]
- Koch, S.; Donarski, N.; Goetze, K.; Kreckel, M.; Stuerenburg, H.J.; Buhmann, C.; Beisiegel, U. Characterization of four lipoprotein classes in human cerebrospinal fluid. J. Lipid Res. 2001, 42, 1143–1151. [Google Scholar] [CrossRef]
- Zimetti, F.; Adorni, M.P.; Marsillach, J.; Marchi, C.; Trentini, A.; Valacchi, G.; Cervellati, C. Connection between the Altered HDL Antioxidant and Anti-Inflammatory Properties and the Risk to Develop Alzheimer’s Disease: A Narrative Review. Oxid. Med. Cell Longev. 2021, 2021, 6695796. [Google Scholar] [CrossRef]
- Van Valkenburgh, J.; Meuret, C.; Martinez, A.E.; Kodancha, V.; Solomon, V.; Chen, K.; Yassine, H.N. Understanding the Exchange of Systemic HDL Particles Into the Brain and Vascular Cells Has Diagnostic and Therapeutic Implications for Neurodegenerative Diseases. Front. Physiol. 2021, 12, 700847. [Google Scholar] [CrossRef]
- Salazar, J.G.; Marsillach, J.; Reverte, I.; Mackness, B.; Mackness, M.; Joven, J.; Camps, J.; Colomina, M.T. Paraoxonase-1 and -3 Protein Expression in the Brain of the Tg2576 Mouse Model of Alzheimer’s Disease. Antioxidants 2021, 10, 339. [Google Scholar] [CrossRef]
- Vitali, C.; Wellington, C.L.; Calabresi, L. HDL and cholesterol handling in the brain. Cardiovasc. Res. 2014, 103, 405–413. [Google Scholar] [CrossRef]
- Feringa, F.M.; van der Kant, R. Cholesterol and Alzheimer’s Disease; From Risk Genes to Pathological Effects. Front. Aging Neurosci. 2021, 13, 690372. [Google Scholar] [CrossRef] [PubMed]
- Marsillach, J.; Adorni, M.P.; Zimetti, F.; Papotti, B.; Zuliani, G.; Cervellati, C. HDL Proteome and Alzheimer’s Disease: Evidence of a Link. Antioxidants 2020, 9, 1224. [Google Scholar] [CrossRef] [PubMed]
- Borras, C.; Mercer, A.; Sirisi, S.; Alcolea, D.; Escola-Gil, J.C.; Blanco-Vaca, F.; Tondo, M. HDL-like-Mediated Cell Cholesterol Trafficking in the Central Nervous System and Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2022, 23, 9356. [Google Scholar] [CrossRef] [PubMed]
- Marchi, C.; Adorni, M.P.; Caffarra, P.; Ronda, N.; Spallazzi, M.; Barocco, F.; Galimberti, D.; Bernini, F.; Zimetti, F. ABCA1- and ABCG1-mediated cholesterol efflux capacity of cerebrospinal fluid is impaired in Alzheimer’s disease. J. Lipid Res. 2019, 60, 1449–1456. [Google Scholar] [CrossRef]
- Mahley, R.W. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1305–1315. [Google Scholar] [CrossRef]
- Eberle, D.; Kim, R.Y.; Luk, F.S.; de Mochel, N.S.; Gaudreault, N.; Olivas, V.R.; Kumar, N.; Posada, J.M.; Birkeland, A.C.; Rapp, J.H.; et al. Apolipoprotein E4 domain interaction accelerates diet-induced atherosclerosis in hypomorphic Arg-61 apoe mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1116–1123. [Google Scholar] [CrossRef]
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef]
- Yang, Y.; Cao, Z.; Tian, L.; Garvey, W.T.; Cheng, G. VPO1 mediates ApoE oxidation and impairs the clearance of plasma lipids. PLoS ONE 2013, 8, e57571. [Google Scholar] [CrossRef]
- Pecorelli, A.; Cervellati, C.; Cortelazzo, A.; Cervellati, F.; Sticozzi, C.; Mirasole, C.; Guerranti, R.; Trentini, A.; Zolla, L.; Savelli, V.; et al. Proteomic analysis of 4-hydroxynonenal and nitrotyrosine modified proteins in RTT fibroblasts. Int. J. Biochem. Cell Biol. 2016, 81, 236–245. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef]
- Duell, P.B.; Oram, J.F.; Bierman, E.L. Nonenzymatic glycosylation of HDL and impaired HDL-receptor-mediated cholesterol efflux. Diabetes 1991, 40, 377–384. [Google Scholar] [CrossRef]
- Romani, A.; Trentini, A.; Flier, W.M.V.; Bellini, T.; Zuliani, G.; Cervellati, C.; Teunissen, C.E. Arylesterase Activity of Paraoxonase-1 in Serum and Cerebrospinal Fluid of Patients with Alzheimer’s Disease and Vascular Dementia. Antioxidants 2020, 9, 456. [Google Scholar] [CrossRef]
- Stoye, N.M.; Jung, P.; Guilherme, M.D.S.; Lotz, J.; Fellgiebel, A.; Endres, K. Apolipoprotein A1 in Cerebrospinal Fluid Is Insufficient to Distinguish Alzheimer’s Disease from Other Dementias in a Naturalistic, Clinical Setting. J. Alzheimers Dis. Rep. 2020, 4, 15–19. [Google Scholar] [CrossRef]
- Zuin, M.; Cervellati, C.; Trentini, A.; Passaro, A.; Rosta, V.; Zimetti, F.; Zuliani, G. Association between Serum Concentrations of Apolipoprotein A-I (ApoA-I) and Alzheimer’s Disease: Systematic Review and Meta-Analysis. Diagnostics 2021, 11, 984. [Google Scholar] [CrossRef]
- Faiz, F.; Hu, M.; Hooper, A.J.; Tomlinson, B.; van Bockxmeer, F.M. Molecular characterization of a Chinese woman homozygous for the familial hypercholesterolemia LDLR c. 1474G> A (p. Asp492Asn) mutation. Clin. Lipidol. 2014, 9, 163–170. [Google Scholar] [CrossRef]
- Miida, T.; Yamada, T.; Seino, U.; Ito, M.; Fueki, Y.; Takahashi, A.; Kosuge, K.; Soda, S.; Hanyu, O.; Obayashi, K.; et al. Serum amyloid A (SAA)-induced remodeling of CSF-HDL. Biochim. Biophys. Acta 2006, 1761, 424–433. [Google Scholar] [CrossRef]
- Ortiz-Munoz, G.; Couret, D.; Lapergue, B.; Bruckert, E.; Meseguer, E.; Amarenco, P.; Meilhac, O. Dysfunctional HDL in acute stroke. Atherosclerosis 2016, 253, 75–80. [Google Scholar] [CrossRef]
- Bednarz-Misa, I.; Berdowska, I.; Zboch, M.; Misiak, B.; Zielinski, B.; Placzkowska, S.; Fleszar, M.; Wisniewski, J.; Gamian, A.; Krzystek-Korpacka, M. Paraoxonase 1 decline and lipid peroxidation rise reflect a degree of brain atrophy and vascular impairment in dementia. Adv. Clin. Exp. Med. 2020, 29, 71–78. [Google Scholar] [CrossRef]
- Cervellati, C.; Romani, A.; Bergamini, C.M.; Bosi, C.; Sanz, J.M.; Passaro, A.; Zuliani, G. PON-1 and ferroxidase activities in older patients with mild cognitive impairment, late onset Alzheimer’s disease or vascular dementia. Clin. Chem. Lab. Med. 2015, 53, 1049–1056. [Google Scholar] [CrossRef]
- Paragh, G.; Balla, P.; Katona, E.; Seres, I.; Egerhazi, A.; Degrell, I. Serum paraoxonase activity changes in patients with Alzheimer’s disease and vascular dementia. Eur. Arch. Psychiatry Clin. Neurosci. 2002, 252, 63–67. [Google Scholar] [CrossRef]
- Lp, P.L.A.S.C.; Thompson, A.; Gao, P.; Orfei, L.; Watson, S.; Di Angelantonio, E.; Kaptoge, S.; Ballantyne, C.; Cannon, C.P.; Criqui, M.; et al. Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: Collaborative analysis of 32 prospective studies. Lancet 2010, 375, 1536–1544. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lu, Y.B.; Huang, X.L.; Lao, Y.F.; Zhang, L.; Yang, J.; Shi, M.; Ma, H.L.; Pan, Y.W.; Zhang, Y.N. Myeloperoxidase: A new target for the treatment of stroke? Neural. Regen. Res. 2022, 17, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Cervellati, C.; Valacchi, G.; Tisato, V.; Zuliani, G.; Marsillach, J. Evaluating the link between Paraoxonase-1 levels and Alzheimer’s disease development. Minerva. Med. 2019, 110, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Cervellati, C.; Trentini, A.; Romani, A.; Bellini, T.; Bosi, C.; Ortolani, B.; Zurlo, A.; Passaro, A.; Seripa, D.; Zuliani, G. Serum paraoxonase and arylesterase activities of paraoxonase-1 (PON-1), mild cognitive impairment, and 2-year conversion to dementia: A pilot study. J. Neurochem. 2015, 135, 395–401. [Google Scholar] [CrossRef]
- Dahabreh, I.J.; Kitsios, G.D.; Kent, D.M.; Trikalinos, T.A. Paraoxonase 1 polymorphisms and ischemic stroke risk: A systematic review and meta-analysis. Genet. Med. 2010, 12, 606–615. [Google Scholar] [CrossRef]
- Kotur-Stevuljevic, J.; Bogavac-Stanojevic, N.; Jelic-Ivanovic, Z.; Stefanovic, A.; Gojkovic, T.; Joksic, J.; Sopic, M.; Gulan, B.; Janac, J.; Milosevic, S. Oxidative stress and paraoxonase 1 status in acute ischemic stroke patients. Atherosclerosis 2015, 241, 192–198. [Google Scholar] [CrossRef]
- Lapergue, B.; Moreno, J.A.; Dang, B.Q.; Coutard, M.; Delbosc, S.; Raphaeli, G.; Auge, N.; Klein, I.; Mazighi, M.; Michel, J.B.; et al. Protective effect of high-density lipoprotein-based therapy in a model of embolic stroke. Stroke 2010, 41, 1536–1542. [Google Scholar] [CrossRef]
- Kang, L.; Yu, H.; Yang, X.; Zhu, Y.; Bai, X.; Wang, R.; Cao, Y.; Xu, H.; Luo, H.; Lu, L.; et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat. Commun. 2020, 11, 2488. [Google Scholar] [CrossRef]
- Mannheim, D.; Herrmann, J.; Versari, D.; Gossl, M.; Meyer, F.B.; McConnell, J.P.; Lerman, L.O.; Lerman, A. Enhanced expression of Lp-PLA2 and lysophosphatidylcholine in symptomatic carotid atherosclerotic plaques. Stroke 2008, 39, 1448–1455. [Google Scholar] [CrossRef]
- de la Torre, J.C. Alzheimer disease as a vascular disorder: Nosological evidence. Stroke 2002, 33, 1152–1162. [Google Scholar] [CrossRef]
- Zuliani, G.; Trentini, A.; Rosta, V.; Guerrini, R.; Pacifico, S.; Bonazzi, S.; Guiotto, A.; Passaro, A.; Seripa, D.; Valacchi, G.; et al. Increased blood BACE1 activity as a potential common pathogenic factor of vascular dementia and late onset Alzheimer’s disease. Sci. Rep. 2020, 10, 14980. [Google Scholar] [CrossRef]
- Klohs, J. An Integrated View on Vascular Dysfunction in Alzheimer’s Disease. Neurodegener. Dis. 2019, 19, 109–127. [Google Scholar] [CrossRef]
- Jakel, L.; De Kort, A.M.; Klijn, C.J.M.; Schreuder, F.; Verbeek, M.M. Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimers Dement. 2022, 18, 10–28. [Google Scholar] [CrossRef]
- Mulder, M.; Terwel, D. Possible link between lipid metabolism and cerebral amyloid angiopathy in Alzheimer’s disease: A role for high-density lipoproteins? Haemostasis 1998, 28, 174–194. [Google Scholar] [CrossRef]
- Bonaterra-Pastra, A.; Fernandez-de-Retana, S.; Rivas-Urbina, A.; Puig, N.; Benitez, S.; Pancorbo, O.; Rodriguez-Luna, D.; Pujadas, F.; Freijo, M.D.M.; Tur, S.; et al. Comparison of Plasma Lipoprotein Composition and Function in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Biomedicines 2021, 9, 72. [Google Scholar] [CrossRef]
- Bennett, S.; Grant, M.M.; Aldred, S. Oxidative stress in vascular dementia and Alzheimer’s disease: A common pathology. J. Alzheimers Dis. 2009, 17, 245–257. [Google Scholar] [CrossRef]
- Pratico, D.; Lee, V.M.Y.; Trojanowski, J.Q.; Rokach, J.; Fitzgerald, G.A. Increased F2-isoprostanes in Alzheimer’s disease: Evidence for enhanced lipid peroxidation in vivo. FASEB J. 1998, 12, 1777–1783. [Google Scholar] [CrossRef]
- Cervellati, C.; Wood, P.L.; Romani, A.; Valacchi, G.; Squerzanti, M.; Sanz, J.M.; Ortolani, B.; Zuliani, G. Oxidative challenge in Alzheimer’s disease: State of knowledge and future needs. J. Investig. Med. 2016, 64, 21–32. [Google Scholar] [CrossRef]
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef]
- De Silva, T.M.; Miller, A.A. Cerebral Small Vessel Disease: Targeting Oxidative Stress as a Novel Therapeutic Strategy? Front. Pharmacol. 2016, 7, 61. [Google Scholar] [CrossRef]
- Hottman, D.A.; Chernick, D.; Cheng, S.; Wang, Z.; Li, L. HDL and cognition in neurodegenerative disorders. Neurobiol. Dis. 2014, 72, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Boon, J.; Hoy, A.J.; Stark, R.; Brown, R.D.; Meex, R.C.; Henstridge, D.C.; Schenk, S.; Meikle, P.J.; Horowitz, J.F.; Kingwell, B.A.; et al. Ceramides contained in LDL are elevated in type 2 diabetes and promote inflammation and skeletal muscle insulin resistance. Diabetes 2013, 62, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Zietzer, A.; Dusing, P.; Reese, L.; Nickenig, G.; Jansen, F. Ceramide Metabolism in Cardiovascular Disease: A Network With High Therapeutic Potential. Arter. Thromb. Vasc. Biol. 2022, 42, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, E.; Blachnio-Zabielska, A. The Role of Ceramides in Insulin Resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Galadari, A.; Thayyullathil, F. Role of ceramide in diabetes mellitus: Evidence and mechanisms. Lipids Health Dis. 2013, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef]
- Ruvolo, P.P. Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol. Res. 2003, 47, 383–392. [Google Scholar] [CrossRef]
- Czubowicz, K.; Jesko, H.; Wencel, P.; Lukiw, W.J.; Strosznajder, R.P. The Role of Ceramide and Sphingosine-1-Phosphate in Alzheimer’s Disease and Other Neurodegenerative Disorders. Mol. Neurobiol. 2019, 56, 5436–5455. [Google Scholar] [CrossRef]
- Chaurasia, B.; Summers, S.A. Ceramides in Metabolism: Key Lipotoxic Players. Annu. Rev. Physiol. 2021, 83, 303–330. [Google Scholar] [CrossRef]
- Chaurasia, B.; Talbot, C.L.; Summers, S.A. Adipocyte Ceramides-The Nexus of Inflammation and Metabolic Disease. Front. Immunol. 2020, 11, 576347. [Google Scholar] [CrossRef]
- Shu, H.; Peng, Y.; Hang, W.; Li, N.; Zhou, N.; Wang, D.W. Emerging Roles of Ceramide in Cardiovascular Diseases. Aging Dis. 2022, 13, 232–245. [Google Scholar] [CrossRef]
- Vasile, V.C.; Meeusen, J.W.; Medina Inojosa, J.R.; Donato, L.J.; Scott, C.G.; Hyun, M.S.; Vinciguerra, M.; Rodeheffer, R.R.; Lopez-Jimenez, F.; Jaffe, A.S. Ceramide Scores Predict Cardiovascular Risk in the Community. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1558–1569. [Google Scholar] [CrossRef]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J. Alzheimers Dis. 2012, 29, 537–547. [Google Scholar] [CrossRef]
- de Wit, N.M.; den Hoedt, S.; Martinez-Martinez, P.; Rozemuller, A.J.; Mulder, M.T.; de Vries, H.E. Astrocytic ceramide as possible indicator of neuroinflammation. J. Neuroinflamm. 2019, 16, 48. [Google Scholar] [CrossRef]
- Mun, K.T.; Hinman, J.D. Inflammation and the Link to Vascular Brain Health: Timing Is Brain. Stroke 2022, 53, 427–436. [Google Scholar] [CrossRef]
- Gonzalez, S.; Huerta, J.M.; Fernandez, S.; Patterson, A.M.; Lasheras, C. The relationship between dietary lipids and cognitive performance in an elderly population. Int. J. Food Sci. Nutr. 2010, 61, 217–225. [Google Scholar] [CrossRef]
- Morris, M.C.; Tangney, C.C. Dietary fat composition and dementia risk. Neurobiol. Aging 2014, 35 (Suppl. S2), S59–S64. [Google Scholar] [CrossRef]
- Huang, Y.; Deng, Y.; Zhang, P.; Lin, J.; Guo, D.; Yang, L.; Liu, D.; Xu, B.; Huang, C.; Zhang, H. Associations of fish oil supplementation with incident dementia: Evidence from the UK Biobank cohort study. Front. Neurosci. 2022, 16, 910977. [Google Scholar] [CrossRef]
- Thomas, A.; Baillet, M.; Proust-Lima, C.; Feart, C.; Foubert-Samier, A.; Helmer, C.; Catheline, G.; Samieri, C. Blood polyunsaturated omega-3 fatty acids, brain atrophy, cognitive decline, and dementia risk. Alzheimers Dement. 2020, 17, 407–416. [Google Scholar] [CrossRef]
- Ma, H.; Zhou, T.; Li, X.; Heianza, Y.; Qi, L. Use of fish oil supplements is differently related to incidence of all-cause and vascular dementia among people with the distinct APOE epsilon4 dosage. Clin. Nutr. 2022, 41, 731–736. [Google Scholar] [CrossRef]
- Daiello, L.A.; Gongvatana, A.; Dunsiger, S.; Cohen, R.A.; Ott, B.R.; Alzheimer’s Disease Neuroimaging Initiative. Association of fish oil supplement use with preservation of brain volume and cognitive function. Alzheimers Dement. 2015, 11, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Preston Mason, R. New Insights into Mechanisms of Action for Omega-3 Fatty Acids in Atherothrombotic Cardiovascular Disease. Curr. Atheroscler. Rep. 2019, 21, 2. [Google Scholar] [CrossRef] [PubMed]
- Skulas-Ray, A.C.; Wilson, P.W.F.; Harris, W.S.; Brinton, E.A.; Kris-Etherton, P.M.; Richter, C.K.; Jacobson, T.A.; Engler, M.B.; Miller, M.; Robinson, J.G.; et al. Omega-3 Fatty Acids for the Management of Hypertriglyceridemia: A Science Advisory From the American Heart Association. Circulation 2019, 140, e673–e691. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Ren, H.; Yao, X.; Shi, Z.; Liang, F.; Kang, J.X.; Wan, J.B.; Pei, Z.; Su, K.P.; Su, H. Enriched Brain Omega-3 Polyunsaturated Fatty Acids Confer Neuroprotection against Microinfarction. EBioMedicine 2018, 32, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Stevens, S.; Gorman, D.; Pan, A.; Warnakula, S.; Chowdhury, S.; Ward, H.; Johnson, L.; Crowe, F.; Hu, F.B.; et al. Association between fish consumption, long chain omega 3 fatty acids, and risk of cerebrovascular disease: Systematic review and meta-analysis. BMJ 2012, 345, e6698. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef]
- Gustavsson, A.M.; van Westen, D.; Stomrud, E.; Engstrom, G.; Nagga, K.; Hansson, O. Midlife Atherosclerosis and Development of Alzheimer or Vascular Dementia. Ann. Neurol. 2020, 87, 52–62. [Google Scholar] [CrossRef]
- Khan, S.U.; Lone, A.N.; Khan, M.S.; Virani, S.S.; Blumenthal, R.S.; Nasir, K.; Miller, M.; Michos, E.D.; Ballantyne, C.M.; Boden, W.E.; et al. Effect of omega-3 fatty acids on cardiovascular outcomes: A systematic review and meta-analysis. EClinicalMedicine 2021, 38, 100997. [Google Scholar] [CrossRef]
- Griffin, M.D.; Sanders, T.A.; Davies, I.G.; Morgan, L.M.; Millward, D.J.; Lewis, F.; Slaughter, S.; Cooper, J.A.; Miller, G.J.; Griffin, B.A. Effects of altering the ratio of dietary n-6 to n-3 fatty acids on insulin sensitivity, lipoprotein size, and postprandial lipemia in men and postmenopausal women aged 45-70 y: The OPTILIP Study. Am. J. Clin. Nutr. 2006, 84, 1290–1298. [Google Scholar] [CrossRef]
- Wilkinson, P.; Leach, C.; Ah-Sing, E.E.; Hussain, N.; Miller, G.J.; Millward, D.J.; Griffin, B.A. Influence of alpha-linolenic acid and fish-oil on markers of cardiovascular risk in subjects with an atherogenic lipoprotein phenotype. Atherosclerosis 2005, 181, 115–124. [Google Scholar] [CrossRef]
- Yanai, H.; Masui, Y.; Katsuyama, H.; Adachi, H.; Kawaguchi, A.; Hakoshima, M.; Waragai, Y.; Harigae, T.; Sako, A. An Improvement of Cardiovascular Risk Factors by Omega-3 Polyunsaturated Fatty Acids. J. Clin. Med. Res. 2018, 10, 281–289. [Google Scholar] [CrossRef]
- Ghorbanihaghjo, A.; Kolahi, S.; Seifirad, S.; Rashtchizadeh, N.; Argani, H.; Hajialilo, M.; Khabazi, A.; Alizadeh, S.; Bahreini, E. Effect of fish oil supplements on serum paraoxonase activity in female patients with rheumatoid arthritis: A double-blind randomized controlled trial. Arch. Iran. Med. 2012, 15, 549–552. [Google Scholar]
- Golzari, M.H.; Hosseini, S.; Koohdani, F.; Saboor Yaraghi, A.A.; Javanbakht, M.H.; Mohammadzadeh-Honarvar, N.; Djalali, M. The Effect of Eicosapentaenoic Acid on the Serum Levels and Enzymatic Activity of Paraoxonase 1 in the Patients with Type 2 Diabetes Mellitus. Acta Med. Iran. 2017, 55, 486–495. [Google Scholar]
- Tanaka, N.; Ishida, T.; Nagao, M.; Mori, T.; Monguchi, T.; Sasaki, M.; Mori, K.; Kondo, K.; Nakajima, H.; Honjo, T.; et al. Administration of high dose eicosapentaenoic acid enhances anti-inflammatory properties of high-density lipoprotein in Japanese patients with dyslipidemia. Atherosclerosis 2014, 237, 577–583. [Google Scholar] [CrossRef]
- Lankinen, M.; Schwab, U.; Erkkila, A.; Seppanen-Laakso, T.; Hannila, M.L.; Mussalo, H.; Lehto, S.; Uusitupa, M.; Gylling, H.; Oresic, M. Fatty fish intake decreases lipids related to inflammation and insulin signaling—A lipidomics approach. PLoS ONE 2009, 4, e5258. [Google Scholar] [CrossRef]
- Ottestad, I.; Hassani, S.; Borge, G.I.; Kohler, A.; Vogt, G.; Hyotylainen, T.; Oresic, M.; Bronner, K.W.; Holven, K.B.; Ulven, S.M.; et al. Fish oil supplementation alters the plasma lipidomic profile and increases long-chain PUFAs of phospholipids and triglycerides in healthy subjects. PLoS ONE 2012, 7, e42550. [Google Scholar] [CrossRef]
- Pinel, A.; Rigaudiere, J.P.; Laillet, B.; Pouyet, C.; Malpuech-Brugere, C.; Prip-Buus, C.; Morio, B.; Capel, F. N-3PUFA differentially modulate palmitate-induced lipotoxicity through alterations of its metabolism in C2C12 muscle cells. Biochim. Biophys. Acta 2016, 1861, 12–20. [Google Scholar] [CrossRef]
- Jin, J.; Lu, Z.; Li, Y.; Cowart, L.A.; Lopes-Virella, M.F.; Huang, Y. Docosahexaenoic acid antagonizes the boosting effect of palmitic acid on LPS inflammatory signaling by inhibiting gene transcription and ceramide synthesis. PLoS ONE 2018, 13, e0193343. [Google Scholar] [CrossRef]
- Kasbi-Chadli, F.; Ferchaud-Roucher, V.; Krempf, M.; Ouguerram, K. Direct and maternal n-3 long-chain polyunsaturated fatty acid supplementation improved triglyceridemia and glycemia through the regulation of hepatic and muscle sphingolipid synthesis in offspring hamsters fed a high-fat diet. Eur. J. Nutr. 2016, 55, 589–599. [Google Scholar] [CrossRef]
- Dong, Y.Q.; Zhang, X.Z.; Sun, L.L.; Zhang, S.Y.; Liu, B.; Liu, H.Y.; Wang, X.; Jiang, C.T. Omega-3 PUFA ameliorates hyperhomocysteinemia-induced hepatic steatosis in mice by inhibiting hepatic ceramide synthesis. Acta Pharmacol. Sin. 2017, 38, 1601–1610. [Google Scholar] [CrossRef]
- Chacinska, M.; Zabielski, P.; Ksiazek, M.; Szalaj, P.; Jarzabek, K.; Kojta, I.; Chabowski, A.; Blachnio-Zabielska, A.U. The Impact of OMEGA-3 Fatty Acids Supplementation on Insulin Resistance and Content of Adipocytokines and Biologically Active Lipids in Adipose Tissue of High-Fat Diet Fed Rats. Nutrients 2019, 11, 835. [Google Scholar] [CrossRef] [PubMed]
- Barnard, N.D.; Bunner, A.E.; Agarwal, U. Saturated and trans fats and dementia: A systematic review. Neurobiol. Aging 2014, 35 (Suppl. S2), S65–S73. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, B.; Kundu, P.; Rooney, W.D.; Raber, J. The Effect of High Fat Diet on Cerebrovascular Health and Pathology: A Species Comparative Review. Molecules 2021, 26, 3406. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.R.; Haley-Zitlin, V.; Rosenberger, D.S.; Granholm, A.C. Damaging effects of a high-fat diet to the brain and cognition: A review of proposed mechanisms. Nutr. Neurosci. 2014, 17, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. The pathology and pathophysiology of vascular dementia. Neuropharmacology 2018, 134, 226–239. [Google Scholar] [CrossRef]
- Astrup, A.; Magkos, F.; Bier, D.M.; Brenna, J.T.; de Oliveira Otto, M.C.; Hill, J.O.; King, J.C.; Mente, A.; Ordovas, J.M.; Volek, J.S.; et al. Saturated Fats and Health: A Reassessment and Proposal for Food-Based Recommendations: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 844–857. [Google Scholar] [CrossRef]
- Mensink, R.P.; Zock, P.L.; Kester, A.D.; Katan, M.B. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: A meta-analysis of 60 controlled trials. Am. J. Clin. Nutr. 2003, 77, 1146–1155. [Google Scholar] [CrossRef]
- Zock, P.L.; Katan, M.B. Hydrogenation alternatives: Effects of trans fatty acids and stearic acid versus linoleic acid on serum lipids and lipoproteins in humans. J. Lipid Res. 1992, 33, 399–410. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; Lucan, S.C.; O’Keefe, J.H. The Evidence for Saturated Fat and for Sugar Related to Coronary Heart Disease. Prog. Cardiovasc. Dis. 2016, 58, 464–472. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Lundman, P.; Harmer, J.A.; Cutri, B.; Griffiths, K.A.; Rye, K.A.; Barter, P.J.; Celermajer, D.S. Consumption of saturated fat impairs the anti-inflammatory properties of high-density lipoproteins and endothelial function. J. Am. Coll. Cardiol. 2006, 48, 715–720. [Google Scholar] [CrossRef]
- Brassard, D.; Arsenault, B.J.; Boyer, M.; Bernic, D.; Tessier-Grenier, M.; Talbot, D.; Tremblay, A.; Levy, E.; Asztalos, B.; Jones, P.J.H.; et al. Saturated Fats from Butter but Not from Cheese Increase HDL-Mediated Cholesterol Efflux Capacity from J774 Macrophages in Men and Women with Abdominal Obesity. J. Nutr. 2018, 148, 573–580. [Google Scholar] [CrossRef]
- van der Westhuyzen, D.R.; de Beer, F.C.; Webb, N.R. HDL cholesterol transport during inflammation. Curr. Opin. Lipidol. 2007, 18, 147–151. [Google Scholar] [CrossRef]
- Ronsein, G.E.; Vaisar, T. Inflammation, remodeling, and other factors affecting HDL cholesterol efflux. Curr. Opin. Lipidol. 2017, 28, 52–59. [Google Scholar] [CrossRef]
- Berg, J.; Seyedsadjadi, N.; Grant, R. Saturated Fatty Acid Intake Is Associated With Increased Inflammation, Conversion of Kynurenine to Tryptophan, and Delta-9 Desaturase Activity in Healthy Humans. Int. J. Tryptophan. Res. 2020, 13, 1178646920981946. [Google Scholar] [CrossRef]
- Gupta, S.; Knight, A.G.; Keller, J.N.; Bruce-Keller, A.J. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J. Neurochem. 2012, 120, 1060–1071. [Google Scholar] [CrossRef]
- Kim, F.; Pham, M.; Luttrell, I.; Bannerman, D.D.; Tupper, J.; Thaler, J.; Hawn, T.R.; Raines, E.W.; Schwartz, M.W. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ. Res. 2007, 100, 1589–1596. [Google Scholar] [CrossRef]
- Sergi, D.; Luscombe-Marsh, N.; Naumovski, N.; Abeywardena, M.; O’Callaghan, N. Palmitic Acid, but Not Lauric Acid, Induces Metabolic Inflammation, Mitochondrial Fragmentation, and a Drop in Mitochondrial Membrane Potential in Human Primary Myotubes. Front. Nutr. 2021, 8, 663838. [Google Scholar] [CrossRef]
- Sergi, D.; Luscombe-Marsh, N.; Heilbronn, L.K.; Birch-Machin, M.; Naumovski, N.; Lionetti, L.; Proud, C.G.; Abeywardena, M.Y.; O’Callaghan, N. The Inhibition of Metabolic Inflammation by EPA Is Associated with Enhanced Mitochondrial Fusion and Insulin Signaling in Human Primary Myotubes. J. Nutr. 2021, 151, 810–819. [Google Scholar] [CrossRef]
- Rosqvist, F.; Kullberg, J.; Stahlman, M.; Cedernaes, J.; Heurling, K.; Johansson, H.E.; Iggman, D.; Wilking, H.; Larsson, A.; Eriksson, O.; et al. Overeating Saturated Fat Promotes Fatty Liver and Ceramides Compared with Polyunsaturated Fat: A Randomized Trial. J. Clin. Endocrinol. Metab. 2019, 104, 6207–6219. [Google Scholar] [CrossRef]
- Dickson, R.C.; Lester, R.L.; Nagiec, M.M. Serine palmitoyltransferase. Methods Enzymol. 2000, 311, 3–9. [Google Scholar] [CrossRef]
- Oteng, A.B.; Kersten, S. Mechanisms of Action of trans Fatty Acids. Adv. Nutr. 2020, 11, 697–708. [Google Scholar] [CrossRef] [PubMed]
- de Roos, N.M.; Bots, M.L.; Katan, M.B. Replacement of dietary saturated fatty acids by trans fatty acids lowers serum HDL cholesterol and impairs endothelial function in healthy men and women. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Brandt, E.J.; Myerson, R.; Perraillon, M.C.; Polonsky, T.S. Hospital Admissions for Myocardial Infarction and Stroke Before and After the Trans-Fatty Acid Restrictions in New York. JAMA Cardiol. 2017, 2, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Aggarwal, N.; Schneider, J.; Wilson, R.S. Dietary fats and the risk of incident Alzheimer disease. Arch. Neurol. 2003, 60, 194–200. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sergi, D.; Zauli, E.; Tisato, V.; Secchiero, P.; Zauli, G.; Cervellati, C. Lipids at the Nexus between Cerebrovascular Disease and Vascular Dementia: The Impact of HDL-Cholesterol and Ceramides. Int. J. Mol. Sci. 2023, 24, 4403. https://doi.org/10.3390/ijms24054403
Sergi D, Zauli E, Tisato V, Secchiero P, Zauli G, Cervellati C. Lipids at the Nexus between Cerebrovascular Disease and Vascular Dementia: The Impact of HDL-Cholesterol and Ceramides. International Journal of Molecular Sciences. 2023; 24(5):4403. https://doi.org/10.3390/ijms24054403
Chicago/Turabian StyleSergi, Domenico, Enrico Zauli, Veronica Tisato, Paola Secchiero, Giorgio Zauli, and Carlo Cervellati. 2023. "Lipids at the Nexus between Cerebrovascular Disease and Vascular Dementia: The Impact of HDL-Cholesterol and Ceramides" International Journal of Molecular Sciences 24, no. 5: 4403. https://doi.org/10.3390/ijms24054403
APA StyleSergi, D., Zauli, E., Tisato, V., Secchiero, P., Zauli, G., & Cervellati, C. (2023). Lipids at the Nexus between Cerebrovascular Disease and Vascular Dementia: The Impact of HDL-Cholesterol and Ceramides. International Journal of Molecular Sciences, 24(5), 4403. https://doi.org/10.3390/ijms24054403