
Recent Progress in the Identification of Early Transition Biomarkers from Relapsing-Remitting to Progressive Multiple Sclerosis

 , , , ,
, , , ,
Abstract
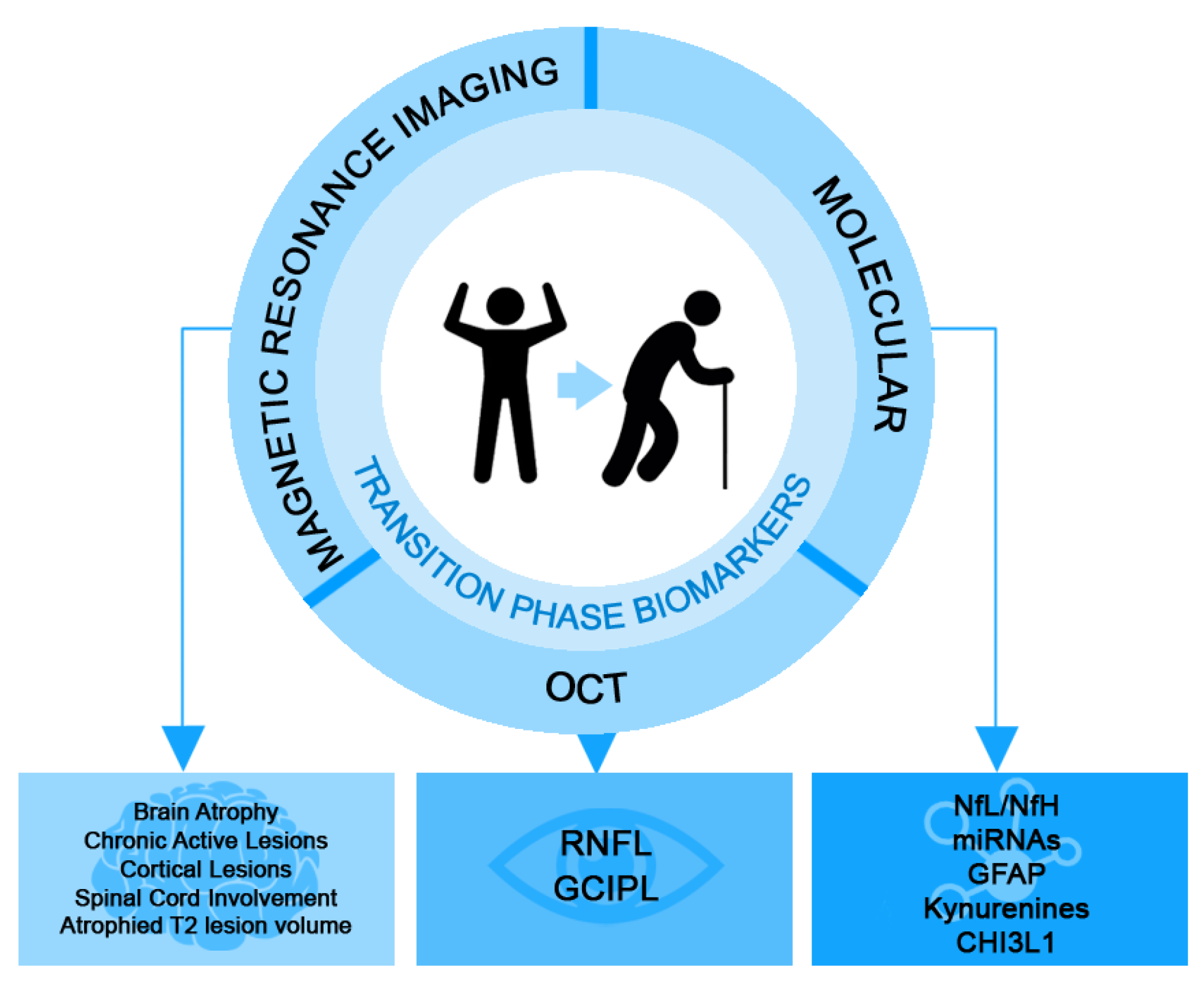
1. Introduction
2. Molecular Biomarkers
2.1. Neurofilaments
2.2. Circulating microRNAs

{kind=link}
| Study | Reported Study Population | Evaluated miRNAs | Selected Reported Results |
|---|---|---|---|
| Regev et al., 2016 [69] | Discovery phase: 7 RRMS, 9 SPMS, 10 PPMS and 20 HCs Validation phase: 29 RRMS, 19 SPMS, 10 PPMS and 30 HCs | 652 miRNAs were measured in the discovery phase and 40 in the validation phase (serum) | miR-27a-3p and miR-376b-3p were the only miRNAs showing significantly different expressions between RRMS and SPMS. After multiple comparations, only miR-27a-3p remained significant with an AUC of 0.78. |
| Gandhi et al., 2013 [62] | Discovery phase: 10 RRMS, 9 SPMS and 9 HCs Validation phase: 50 RRMS, 51 SPMS and 32 HCs | 368 miRNAs were measured in the discovery phase and 19 miRNAs in the validation phase (plasma) | hsa-miR-92a-1, let-7 family, hsa-miR-454 and miR-145 had different expressions in RRMS vs. SPMS patients. |
| Haghikia et al., 2012 [60] | Discovery phase: 10 MS and 10 with OND Validation phase: 17 RRMS, 30 SPMS, 6 PPMS, 20 patients with OND | 760 CSF miRNAs were measured in the discovery phase and the validation phase: miR-132, miR-181c, miR-494, miR-633 and miR-922 (CSF) | Only miR-181c and miR-633 differentiated RRMS patients from SPMS patients with a specificity of 82% and a sensitivity of 69%. |
| Kramer et al., 2019 [61] | 81 RRMS, 106 SPMS, 12 PPMS and 218 patients with OND | miR-181c, miR-633 and miR-922 (CSF) | miR-181c levels were significantly different between RRMS and SPMS patients (p = 0.036) |
| Regev et al., 2018 [71] | Discovery phase: 7 RRMS, 9 SPMS and 20 HCs Validation phase: 29 RRMS, 19 SPMS and 30 HCs Reproducibility phase: 24 RRMS, 18 SPM and 30 HCs Transportability phase: 91 RRMS, 33 SPMS, and 58 HCs | Discovery phase: 652 miRNAs Validation phase: 192 miRNAs Reproducibility phase: 73 miRNAs Transportability phase: 73 miRNAs (serum) | hsa-miR-337-3p is under-expressed in SPMS compared with RRMS. |
| Sharaf-Eldin et al., 2017 [72] | 18 RRMS, 19 SPMS, 20 patients with OND and 23 HCs | miR-145, miR-223 and miR-326 (serum) | miR-326 had a significantly lower expression in SPMS patients compared with RRMS patients (p = 0.017). |
| Khedr et al., 2022 [74] | 50 MS patients and 50 HCs | miRNA-22 (serum) | miRNA-22 had a significantly higher expression in SPMS patients compared with RRMS patients (p = 0.042). |
| Ibrahim et al., 2020 [75] | 39 RRMS, 35 SPMS and 10 HCs | miR-300 and miR-450b-5p (serum) | miR-300 and miR-450b-5p expression were significantly lower in SPMS patients, regardless of the EDSS score, compared with RRMS patients with EDSS ≤5. |
| Ebrahimkhani et al., 2017 [73] | Discovery phase: 14 RRMS, 7 SPMS, 4 PPMS and 11 HCs Validation phase: 14 RRMS, 9 SPMS, 2 PPMS and 11 HCs | 168 miRNAs from exosomes (serum) | Nine miRNAs were identified with a different expression between RRMS and progressive MS patients: miR-15b-5p, miR-23a-3p, miR-223-3p, miR-374a-5p, miR-30b-5p, miR-433-3p, miR-485-3p, miR-342-3p, miR-432-5p). A combination of miR-223-3p, miR-485-3p and miR-30b-5p showed a 95% accuracy rate in distinguishing progressive forms of MS from RRMS. |
| Magner et al., 2016 [76] | 59 RRMS, 15 SPMS and 25 HCs | 64 miRNAs miR-103, miR-191, miR−423 (whole blood EDTA) | hsa-miR-874, hsa-miR-99a-5p, hsa-miR-141-3p, hsa-miR-125b-5p, hsa-miR-342-3p had different expressions in SPMS patients who responded poorly to IFNβ1a compared with RRMS patients who responded poorly (p < 0.01). |
2.3. Glial Fibrillary Acid Protein
2.4. Kynurenines
2.5. Chitinase-3-like-1 and Chemokine C-X-C Motif Ligand 13
2.6. Other Potential Serum and CSF Biomarkers
3. Optical Coherence Tomography Biomarkers
| Study | Reported Study Population | Selected Evaluated Parameters | Selected Reported Results |
|---|---|---|---|
| Yurtogullari et al., 2022 [123] | 66 RRMS, 31 SPMS | RFNL GCIPL | RNFL and GCIPL were thinner in the SPMS group compared with the RRMS group in patients who had no previous history of ON. |
| Pisa et al., 2020 [124] | 26 NMOSD 29 RRMS, 20 SPMS and 3 PPMS | RNFL GCIPL | RRMS patients meeting NEDA-3 criteria had no significant RNFL thinning during the follow-up (13 patients; –0.271 µm/year; 95% CI = –0.892 to 0.349 µm/year; p = 0.392). The opposite is valid for PMS patients with NEDA-3 criteria, which had a significant RNFL loss during the study (11 patients; –0.556 µm/year; 95% CI = –0.985 to −0.127 µm/year; p = 0.011). Regarding the evaluation of GCIPL, in stable RRMS patients no significant thinning of this layer was observed, but in the case of progressive MS patients, with both active and stable forms of the disease, a significant thinning of GCIPL was observed. |
| Pulicken et al., 2007 [114] | 135 RRMS, 12 PPMS and 16 SPMS | RNFL MV | The average RNFL thickness was reduced in both ON and non-ON MS patients compared with controls. The PPMS and SPMS groups had a lower baseline RNFL thickness than the RRMS group. A statistically significant reduced mean MV was noted in the SPMS patients compared with RRMS and SPMS patients compared with controls. |
| Gelfand et al., 2012 [113] | 45 CIS, 403 RRMS, 60 SPMS and 33 PPMS | RNFL MV | In patients that had more advanced stages of the disease, the RNFL thinning was progressively increased based on the MS type (CIS = 98.2 ± 8.4 µm, RRMS = 92.9 ± 13 µm, SPMS = 85.5 ± 14.3 µm, PPMS = 80.5 ± 15.4 µm). |
| Oberwahrenbrock et al., 2012 [115] | 308 RRMS, 65 SPMS and 41 PPMS | RNFL MV | The thickness of RNFL was reduced in SPMS eyes compared with RRMS eyes (p = 0.007), while total MV was lower in SPMS and PPMS eyes compared with RRMS eyes (all p < 0.05). |
| Balk et al., 2014. [125] | 140 RRMS, 61 SPMS, 29 PPMS and 59 BMS | RNFL GCC | RNFL and GCC thickness were significantly lower in SPMS patients with no prior history of ON than in RRMS patients. |
| Jankowska-Lech et al., 2019 [126] | 26 RRMS 3 PPMS, 7 SPMS and 12 PRMS | RNFL | The RNFL thickness in the temporal quadrant in RRMS patients (98.82 µm ± 12.35) was higher compared with PMS patients (86.47 µm ± 11.76), p = 0.025. |
| Estiasari et al., 2021 [127] | 25 RRMS, 7 SPMS, 22 HC | RNFL GCIPL | SPMS patients have a reduced GCIPL and RNFL thickness, except for the nasal quadrant, compared with RRMS patients. |
| Jakimovski et al., 2021 [128] | 109 RRMS, 35 PMS | RNFL MV | There was no difference in macular volume found. During follow-up, progressive MS patients with disability progression had decreased RNFL thickness compared with stable, progressive MS patients (69.53 μm vs. 79.9 μm, p = 0.007). A reduction in average RNFL was associated with progressive evolution, increased age and ON history. |
| Uzunköprü C et al., 2021 [42] | 30 RRMS, 16 SPMS and 29 HCs | RNFL | SPMS patients had lower RNFL thickness in each eye as compared with the RRMS patients (p < 0.001). |
| Cellerino M et al., 2021 [129] | 101 RRMS and 79 PMS | RNFL GCIPL | Reduced RNFL and GCIPL thickness were found in PMS patients rather than RRMS patients (all p < 0.05). Using multivariate models in the RRMS group, a correlation between GCIPL and follow-up disability was proved (0.04 increase in the EDSS for each 1 μm decrease in the baseline GCIPL, p = 0.02). |
| Sotirchos et al., 2020 [130] | 178 RRMS, 126 SPMS, 60 PPMS and 66 HCs | RNFL GCIPL | All MS patients, regardless of their subtype and ON history, compared with HCs, had reduced GCIPL and RNFL thickness. When comparing MS subtypes, the SPMS and PPMS patients had lower GCIPL and RNFL thickness compared with RRMS patients. In the subgroup with no previous history of ON, the GCIPL and RNFL were thinner in SPMS and PPMS patients relative to RRMS patients. |
| Eslami et al., 2020 [131] | 14 CIS, 92 RRMS and 14 SPMS | RNFL VM | RNFL thickness did not significantly differ between MS subtypes. The SPMS patients had the lowest total MV thickness (p = 0.04). |
| Behbehani et al., 2017 [111] | 84 RRMS, 27 SPMS, 2 PPMS and 38 HCs | RNFL GCIPL | Statistically significant differences were found in PMS compared with RRMS patients regarding RNFL (p = 0.02) and GCIPL (p = 0.006), with lower values in the first group. |
| Study | Reported Study Population | Selected Reported Results |
|---|---|---|
| Berek et al., 2022 [132] | 93 MS | Low baseline values of RNFL and GCIPL are associated with disability progression after 6 years. GCIPL thinning is associated with EDSS worsening. |
| Balıkçı et al., 2021 [133] | 7 CIS, 51 RRMS, 21 SPMS, 4 PP and 57 HCs | EDSS negatively correlated with mean RNFL (p < 0.001) and GCC measurements (p < 0.001). |
| Piedrabuena et al., 2022 [134] | 52 RRMS | A negative correlation between EDSS and RNFL was found. No other significant differences were found in the assessments for the 2-year follow-up. |
| Barreiro-González et al., 2022 [135] | 64 MS | Patients’ EDSS values correlated with age (r = 0.543, p = 0.001), spinal cord volume (r = −0.301, p = 0.034) and GCL (GCL, r = −0.354, p = 0.012). |
| Bsteh et al., 2019 [136] | 151 RRMS | In patients with RNFL thickness ≤88 µm, there was three times increased risk of EDSS progression (p < 0.001) and 2.7 times increased risk of cognitive decline in the next 3 years (p < 0.001). |
| Schurz et al., 2021 [137] | 53 RRMS and 7 SPMS | When comparing to the stable group, the patients that had disability worsening had a reduced thickness of RNFL (83.4 µm vs. 97.7 µm, p = 0.001) and GCIPL (69.3 µm vs. 78.2 µm, p < 0.001), p = 0.672). Patients with GCIPL thickness <77 µm at baseline were associated with four times increased risk of disability worsening during the follow-up. Patients with RNFL ≤88μm had a weaker significant association with increased risk of disability worsening (HR 3.1; 95% CI = 1.4–7.0; p = 0.019. |
| Lambe et al., 2021 [121] | 106 RRMS and 26 SPMS | In patients with an average baseline GCIPL <70 µm, there was an increased four times risk of EDSS worsening association (OR: 3.97, 95% CI = 1.24–12.70; p = 0.02). An independent association was found between a lower baseline GCIPL thickness and long-term disability worsening. |
| Cilingir et al., 2021 [138] | 137 RRMS | Both intermediate (94–100 µm) and reduced (<94 µm) RNFL thickness are associated with an increase in EDSS progression in the first 2 years. |
| Bitirgen et al., 2020 [139] | 31 RRMS | A significant reduction in RNFL thickness (p = 0.004) was associated with an increase in EDSS over 2 years. |
| Cilingir et al., 2020 [140] | 134 RRMS | A significant correlation was found between EDSS progression and RNFL in both early-stage and late-stage RRMS patient groups (r = −0.471, p < 0.001, and r = −0.567, p < 0.001). |
| Bsteh et al., 2021 [141] | 183 RRMS | An increased risk of disability progression was found in patients with baseline GCIPL thickness <77 μm (HR: 2.7, 95% CI = 1.5–4.7, p < 0.001). A strong predictor for clinical progression was the annual loss of macular GCIPL, with a cut-off value ≥1 µm, which accurately identified clinically progressive patients (87% sensitivity at 90% specificity, OR: 18.3, 95% CI = 8.8–50.3). |
| Vidal-Jordana et al., 2020 [142] | 109 RRMS | Patients with a reduced RNFL thickness had associated decreased BPF, SC area values, a higher T2 lesion volume and a higher lesion volume within the optic radiations (all p < 0.05). Reduced GCIPL volumes were correlated with decreased BPF values and SC area values. Patients with RNFL thickness ≤86 µm had 4.7–6.7 times increased chance of reaching an EDSS ≥3.0. Also, patients with GCIPL volume ≤1.77 mm3 had a 9.7–11.9 times increased chance of reaching EDSS ≥ 3.0. |
| Koraysha et al., 2019 [143] | 49 RRMS | RRMS patients with an EDSS >2, compared with those with an EDSS ≤ 2.0, had lower RNFL thickness (p = 0.01). |
| Cordano et al., 2018 [144] | 228 RRMS, 29 SPMS, 32 CIS and 10 PPMS | A multivariate linear regression analysis was used to analyse the association between baseline RNFL and EDSS, adjusted by age and sex. It was found that every 1 µm decrease in the thickness of RNFL was associated with a 0.024 increase in EDSS (95% CI = 0.011–0.037; p < 0.001). Similar results were found in sensitivity analysis during a >5-year follow-up (0.02 increase in the EDSS, 95% CI = 0.03–0.01, p = 0.001). |
| Garcia-Martin et al., 2017 [145] | 100 RRMS and 50 HC | Correlation between high EDSS scores and temporal and superior RNFL thickness reduction. |
| Albrecht et al., 2012 [146] | 42 RRMS, 41 SPMS and 12 PPMS | Statistically significant correlations were found between mean RNFL and EDSS (r = −0.36, r = −0.35 and r = 0.33; all p < 0.05). The correlations remained significant even after excluding any previous ON history (r = −0.36, r = −0.35 and r = 0.33; all p < 0.05). EDSS had negative correlations with RNFL. |
| Bsteh et al., 2020 [147] | 171 RRMS | There was no difference in RNFL and GCIPL thickness related to relapses or relapse-free during the follow-up. Multivariate regression analysis found an association between PIRA and the reduction of GCIPL and RNFL thickness. |
| Bsteh et al., 2019 [148] | 141 RRMS | When comparing RRMS patients that progressed during the study with those that remained clinically stable, they found an increase in RNFL thinning in the progressing group (0.5 μm, p < 0.001). A cut-off value of RNFL >1.5 um was used to distinguish between stable and progressing RRMS (specificity 90%, sensitivity 76.1%). The exact cut-off value was associated with a 15-fold increase in the risk of clinically progressing MS (p < 0.001). |
| Martinez-Lapiscina et al., 2016 [122] | 74 CIS, 664 RRMS, 83 SPMS and 58 PPMS | Patients with RNFL less than or equal to 87/88 μm had a double risk of disability worsening at any time from the first to the third year of follow-up (HR: 2.06, 95% CI = 1.36–3.11; p = 0.001). The risk increased almost 4 times from the 3rd to the 5th year of follow-up. |
| Rothman et al., 2019 [120] | 151 RRMS, 14 SPMS and 7 PPMS | There is an association between lower baseline total MV and a higher 10-year EDSS score, which was shown in the multivariable models (mean increase in EDSS of 0.75 per 1 mm3 loss in total MV (p = 0.02). Patients in the lowest tertile of baseline total MV had an average higher EDSS score at 10 years (mean difference = 0.86; 95% CI = 0.23–1.48) and had a 3.5 times increased chance of clinically significant EDSS worsening when compared with the patients in the highest tertile of baseline total MV (OR: 3.58; 95% CI = 1.30–9.82; p = 0.008). Using univariate models, it was possible to use RNFL to predict the 10-year EDSS progression. |
| Lambe et al., 2021 [121] | 92 RRMS and 25 PMS | The follow-up of the patients was an average of 10.4 years. Using multivariate models that excluded patients with prior ON, an average baseline GCIPL thickness <70 µm correlated with four times increased chance of significant EDSS worsening (p = 0.02) when compared with patients with an average GCIPL thickness ≥70 µm. |
4. Magnetic Resonance Imaging Biomarkers
4.1. Global and Regional Brain Atrophy
4.2. Chronic Active Lesions and Cortical Lesions
4.3. Spinal Cord Damage
4.4. Atrophied Lesion Volume
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levin, M.C.; Douglas, J.N.; Meyers, L.; Lee, S.; Shin, Y.; Gardner, L.A. Neurodegeneration in Multiple Sclerosis Involves Multiple Pathogenic Mechanisms. Degener. Neurol. Neuromuscul. Dis. 2014, 4, 49. [Google Scholar] [CrossRef]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J. Mitochondrial Dysfunction as a Cause of Axonal Degeneration in Multiple Sclerosis Patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J. The Molecular Basis of Neurodegeneration in Multiple Sclerosis. FEBS Lett. 2011, 585, 3715–3723. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.-A. Multiple Sclerosis: An Immune or Neurodegenerative Disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Dutta, R.; Trapp, B.D. Pathogenesis of Axonal and Neuronal Damage in Multiple Sclerosis. Neurology 2007, 68 (22 Suppl. 3), S22–S31. [Google Scholar] [CrossRef]
- Balasa, R.; Barcutean, L.; Mosora, O.; Manu, D. Reviewing the Significance of Blood–Brain Barrier Disruption in Multiple Sclerosis Pathology and Treatment. Int. J. Mol. Sci. 2021, 22, 8370. [Google Scholar] [CrossRef]
- Cree, B.A.; Arnold, D.L.; Chataway, J.; Chitnis, T.; Fox, R.J.; Ramajo, A.P.; Murphy, N.; Lassmann, H. Secondary Progressive Multiple Sclerosis: New Insights. Neurology 2021, 97, 378–388. [Google Scholar] [CrossRef]
- Sand, I.K.; Krieger, S.; Farrell, C.; Miller, A.E. Diagnostic Uncertainty during the Transition to Secondary Progressive Multiple Sclerosis. Mult. Scler. J. 2014, 20, 1654–1657. [Google Scholar] [CrossRef]
- Bogosian, A.; Morgan, M.; Moss-Morris, R. Multiple Challenges for People after Transitioning to Secondary Progressive Multiple Sclerosis: A Qualitative Study. BMJ Open 2019, 9, e026421. [Google Scholar] [CrossRef]
- Fisniku, L.; Brex, P.; Altmann, D.; Miszkiel, K.; Benton, C.; Lanyon, R.; Thompson, A.; Miller, D. Disability and T2 MRI Lesions: A 20-Year Follow-up of Patients with Relapse Onset of Multiple Sclerosis. Brain 2008, 131, 808–817. [Google Scholar] [CrossRef]
- Tremlett, H. Secondary Progressive Multiple Sclerosis. MS Focus 2009, 13, 13–14. [Google Scholar]
- Bălaşa, R.; Huţanu, A.; Bajko, Z.; Feier, C.; Pascu, I. Does the Serum IL-17 Titer Influence the Efficacy of Interferon-β Treatment in Multiple Sclerosis Patients? Revista Română Medicină Laborator Vol. 2011, 19, 381–389. [Google Scholar]
- Inojosa, H.; Proschmann, U.; Akgün, K.; Ziemssen, T. A focus on secondary progressive multiple sclerosis (SPMS): Challenges in diagnosis and definition. J. Neurol. 2021, 268, 1210–1221. [Google Scholar] [CrossRef]
- Davies, F.; Wood, F.; Brain, K.E.; Edwards, M.; Jones, R.; Wallbank, R.; Robertson, N.P.; Edwards, A. The transition to secondary progressive multiple sclerosis: An exploratory qualitative study of health professionals’ experiences. Int. J. MS Care 2016, 18, 257–264. [Google Scholar] [CrossRef]
- Sumowski, J.F.; Rocca, M.A.; Leavitt, V.M.; Riccitelli, G.; Comi, G.; DeLuca, J.; Filippi, M. Brain reserve and cognitive reserve in multiple sclerosis: What you’ve got and how you use it. Neurology 2013, 80, 2186–2193. [Google Scholar] [CrossRef]
- Dutta, R.; Trapp, B.D. Relapsing and progressive forms of multiple sclerosis: Insights from pathology. Curr. Opin. Neurol. 2014, 27, 271–278. [Google Scholar] [CrossRef]
- Davies, F.; Edwards, A.; Brain, K.E.; Edwards, M.; Jones, R.; Wallbank, R.; Robertson, N.P.; Wood, F. ’You are just left to get on with it’: Qualitative study of patient and carer experiences of the transition to secondary progressive multiple sclerosis. BMJ 2015, 5, e007674. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T. Siponimod versus Placebo in Secondary Progressive Multiple Sclerosis (EXPAND): A Double-Blind, Randomised, Phase 3 Study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Mey, G.M.; Mahajan, K.R.; DeSilva, T.M. Neurodegeneration in Multiple Sclerosis. WIREs Mech. Dis. 2022, 15, e1583. [Google Scholar] [CrossRef]
- Lorscheider, J.; Buzzard, K.; Jokubaitis, V.; Spelman, T.; Havrdova, E.; Horakova, D.; Trojano, M.; Izquierdo, G.; Girard, M.; Duquette, P.; et al. Defining secondary progressive multiple sclerosis. Brain 2016, 139 Pt 9, 2395–2405. [Google Scholar] [CrossRef]
- Sá, M.J.; Basílio, C.; Capela, C.; Cerqueira, J.J.; Mendes, I.; Morganho, A.; Correia de Sá, J.; Salgado, V.; Martins Silva, A.; Vale, J.; et al. Consensus for the Early Identification of Secondary Progressive Multiple Sclerosis in Portugal: A Delphi Panel. Acta Med. Port. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Oh, J.; Alikhani, K.; Bruno, T.; Devonshire, V.; Giacomini, P.S.; Giuliani, F.; Nakhaipour, H.R.; Schecter, R.; Larochelle, C. Diagnosis and management of secondary-progressive multiple sclerosis: Time for change. Neurodegener. Dis. Manag. 2019, 9, 301–317. [Google Scholar] [CrossRef]
- English, C.; Aloi, J.J. New FDA-approved disease-modifying therapies for multiple sclerosis. Clin. Ther. 2015, 37, 691–715. [Google Scholar] [CrossRef]
- Disanto, G.; Barro, C.; Benkert, P.; Naegelin, Y.; Schädelin, S.; Giardiello, A.; Zecca, C.; Blennow, K.; Zetterberg, H.; Leppert, D. Serum Neurofilament Light: A Biomarker of Neuronal Damage in Multiple Sclerosis. Ann. Neurol. 2017, 81, 857–870. [Google Scholar] [CrossRef]
- Barro, C.; Chitnis, T.; Weiner, H.L. Blood Neurofilament Light: A Critical Review of Its Application to Neurologic Disease. Ann. Clin. Transl. Neurol. 2020, 7, 2508–2523. [Google Scholar] [CrossRef]
- Rosengren, L.E.; Karlsson, J.; Karlsson, J.; Persson, L.I.; Wikkelsø, C. Patients with Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases Have Increased Levels of Neurofilament Protein in CSF. J. Neurochem. 1996, 67, 2013–2018. [Google Scholar] [CrossRef]
- Kuhle, J.; Plattner, K.; Bestwick, J.P.; Lindberg, R.L.; Ramagopalan, S.V.; Norgren, N.; Nissim, A.; Malaspina, A.; Leppert, D.; Giovannoni, G. A Comparative Study of CSF Neurofilament Light and Heavy Chain Protein in MS. Mult. Scler. J. 2013, 19, 1597–1603. [Google Scholar] [CrossRef]
- Lycke, J.; Karlsson, J.; Andersen, O.; Rosengren, L. Neurofilament Protein in Cerebrospinal Fluid: A Potential Marker of Activity in Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 1998, 64, 402–404. [Google Scholar] [CrossRef]
- Gunnarsson, M.; Malmeström, C.; Axelsson, M.; Sundström, P.; Dahle, C.; Vrethem, M.; Olsson, T.; Piehl, F.; Norgren, N.; Rosengren, L. Axonal Damage in Relapsing Multiple Sclerosis Is Markedly Reduced by Natalizumab. Ann. Neurol. 2011, 69, 83–89. [Google Scholar] [CrossRef]
- Kuhle, J.; Disanto, G.; Lorscheider, J.; Stites, T.; Chen, Y.; Dahlke, F.; Francis, G.; Shrinivasan, A.; Radue, E.-W.; Giovannoni, G. Fingolimod and CSF Neurofilament Light Chain Levels in Relapsing-Remitting Multiple Sclerosis. Neurology 2015, 84, 1639–1643. [Google Scholar] [CrossRef]
- Siller, N.; Kuhle, J.; Muthuraman, M.; Barro, C.; Uphaus, T.; Groppa, S.; Kappos, L.; Zipp, F.; Bittner, S. Serum Neurofilament Light Chain Is a Biomarker of Acute and Chronic Neuronal Damage in Early Multiple Sclerosis. Mult. Scler. J. 2019, 25, 678–686. [Google Scholar] [CrossRef]
- Guerrieri, S.; Comi, G.; Leocani, L. Optical Coherence Tomography and Visual Evoked Potentials as Prognostic and Monitoring Tools in Progressive Multiple Sclerosis. Front. Neurosci. 2021, 15, 806. [Google Scholar] [CrossRef]
- Kuhle, J.; Nourbakhsh, B.; Grant, D.; Morant, S.; Barro, C.; Yaldizli, Ö.; Pelletier, D.; Giovannoni, G.; Waubant, E.; Gnanapavan, S. Serum Neurofilament Is Associated with Progression of Brain Atrophy and Disability in Early MS. Neurology 2017, 88, 826–831. [Google Scholar] [CrossRef]
- Manouchehrinia, A.; Stridh, P.; Khademi, M.; Leppert, D.; Barro, C.; Michalak, Z.; Benkert, P.; Lycke, J.; Alfredsson, L.; Kappos, L. Plasma Neurofilament Light Levels Are Associated with Risk of Disability in Multiple Sclerosis. Neurology 2020, 94, e2457–e2467. [Google Scholar] [CrossRef]
- Kuhle, J.; Kropshofer, H.; Haering, D.A.; Kundu, U.; Meinert, R.; Barro, C.; Dahlke, F.; Tomic, D.; Leppert, D.; Kappos, L. Blood Neurofilament Light Chain as a Biomarker of MS Disease Activity and Treatment Response. Neurology 2019, 92, e1007–e1015. [Google Scholar] [CrossRef]
- Barro, C.; Benkert, P.; Disanto, G.; Tsagkas, C.; Amann, M.; Naegelin, Y.; Leppert, D.; Gobbi, C.; Granziera, C.; Yaldizli, Ö. Serum Neurofilament as a Predictor of Disease Worsening and Brain and Spinal Cord Atrophy in Multiple Sclerosis. Brain 2018, 141, 2382–2391. [Google Scholar] [CrossRef]
- Thebault, S.; Abdoli, M.; Fereshtehnejad, S.-M.; Tessier, D.; Tabard-Cossa, V.; Freedman, M.S. Serum Neurofilament Light Chain Predicts Long Term Clinical Outcomes in Multiple Sclerosis. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Chitnis, T.; Gonzalez, C.; Healy, B.C.; Saxena, S.; Rosso, M.; Barro, C.; Michalak, Z.; Paul, A.; Kivisakk, P.; Diaz-Cruz, C. Neurofilament Light Chain Serum Levels Correlate with 10-year MRI Outcomes in Multiple Sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 1478–1491. [Google Scholar] [CrossRef]
- Leppert, D.; Kropshofer, H.; Häring, D.A.; Dahlke, F.; Patil, A.; Meinert, R.; Tomic, D.; Kappos, L.; Kuhle, J. Blood Neurofilament Light in Progressive Multiple Sclerosis: Post Hoc Analysis of 2 Randomized Controlled Trials. Neurology 2022, 98, e2120–e2131. [Google Scholar] [CrossRef]
- Uzunköprü, C.; Yüceyar, N.; Yilmaz, S.G.; Afrashi, F.; Ekmekçi, Ö.; Taşkiran, D. Retinal Nerve Fiber Layer Thickness Correlates with Serum and Cerebrospinal Fluid Neurofilament Levels and is Associated with Current Disability in Multiple Sclerosis. Noro Psikiyatr. Ars 2021, 58, 34–40. [Google Scholar] [CrossRef]
- Williams, T.; Zetterberg, H.; Chataway, J. Neurofilaments in progressive multiple sclerosis: A systematic review. J. Neurol. 2021, 268, 3212–3222. [Google Scholar] [CrossRef]
- Blum, A.; Lindberg, R.; Leppert, P.; Kuster, P.; Muller-Lenke, N.; Regeniter, A.; Radu, E.W.; Kappos, L.; Kuhle, J. The relation of quantitative MRI measures, CSF markers and disability in multiple sclerosis. Mult. Scler. 2010, 16, S190. [Google Scholar]
- Uzunköprü, C.; Yuceyar, N.; Taskiran, D.; Yilmaz, S.G.; Afrashi, F.; Ekmekci, O. Correlation of neurofilaments and nitrotyrosine with retinal nerve fiber layer thickness and disability in different phases of multiple sclerosis. Mult. Scler. 2014, 20, 184. [Google Scholar]
- Kaymakamzade, B.; Kurne, A.T.; Tumani, H.; Lehmensiek, V.; Sayat, G.; Karabudak, R. Evaluation of neurofilament heavy chain levels in progressive multiple sclerosis patients: Preliminary results. J. Neurol. 2014, 261, S453. [Google Scholar]
- Gresle, M.M.; Liu, Y.; Dagley, L.F.; Haartsen, J.; Pearson, F.; Purcell, A.W.; Laverick, L.; Petzold, A.; Lucas, R.M.; Van der Walt, A.; et al. Serum phosphorylated neurofilament-heavy chain levels in multiple sclerosis patients. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1209–1213. [Google Scholar] [CrossRef]
- Petzold, A.; Eikelenboom, M.J.; Keir, G.; Grant, D.; Lazeron, R.; Polman, C.; Uitdehaag, B.; Thompson, E.; Giovannoni, G. Axonal damage accumulates in the progressive phase of multiple sclerosis: Three year follow up study. J. Neurol. Neurosurg. Psychiatry 2005, 76, 206–211. [Google Scholar] [CrossRef]
- Kuhle, J.; Leppert, D.; Petzold, A.; Regeniter, A.; Schindler, C.; Mehling, M.; Anthony, D.C.; Kappos, L.; Lindberg, R.L.P. Neurofilament heavy chain in CSF correlates with relapses and disability in multiple sclerosis. Neurology 2011, 76, 1206–1213. [Google Scholar] [CrossRef]
- Vorobyeva, A.; Fominykh, V.; Zakharova, M.; Gulyaeva, N. Biochemical markers of neurodegeneration in multiple sclerosis. Mult. Scler. 2013, 19, 369. [Google Scholar]
- Altintas, A.; Akkas, S.Y.; Lehmensiek, V.; Tumani, H. CSF proteomic profile in primary progressive multiple sclerosis. J. Neurol. 2014, 261, S443. [Google Scholar]
- Rommer, P.S.; Kamin, F.; Petzold, A.; Tumani, H.; Abu-Mugheisib, M.; Koehler, W.; Hoffmann, F.; Winkelmann, A.; Benecke, R.; Zettl, U.K. Effects of repeated intrathecal triamcinolone-acetonide application on cerebrospinal fluid biomarkers of axonal damage and glial activity in multiple sclerosis patients. Mol. Diagn. Ther. 2014, 18, 631–637. [Google Scholar] [CrossRef]
- Herrera, M.I.; Kölliker-Frers, R.A.; Otero-Losada, M.; Perez Lloret, S.; Filippo, M.; Tau, J.; Capani, F.; Villa, A.M. A Pilot Cross-Sectional Study to Investigate the Biomarker Potential of Phosphorylated Neurofilament-H and Immune Mediators of Disability in Patients With 5 Year Relapsing-Remitting Multiple Sclerosis. Front. Neurol. 2019, 10, 1046. [Google Scholar] [CrossRef]
- Gao, Y.; Han, D.; Feng, J. MicroRNA in Multiple Sclerosis. Clin. Chim. Acta 2021, 516, 92–99. [Google Scholar] [CrossRef]
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. [Google Scholar] [CrossRef]
- Pietrasik, S.; Dziedzic, A.; Miller, E.; Starosta, M.; Saluk-Bijak, J. Circulating MiRNAs as Potential Biomarkers Distinguishing Relapsing–Remitting from Secondary Progressive Multiple Sclerosis. A Review. Int. J. Mol. Sci. 2021, 22, 11887. [Google Scholar] [CrossRef]
- Ferrante, M.; Conti, G.O. Environment and Neurodegenerative Diseases: An Update on MiRNA Role. Microrna 2017, 6, 157–165. [Google Scholar] [CrossRef]
- Martinez, B.; Peplow, P. MicroRNAs in Blood and Cerebrospinal Fluid as Diagnostic Biomarkers of Multiple Sclerosis and to Monitor Disease Progression. Neural Regen. Res. 2020, 15, 606–619. [Google Scholar]
- Qiu, L.; Tan, E.K.; Zeng, L. MicroRNAs and Neurodegenerative Diseases. microRNA Med. Evid. 2015, 888, 85–105. [Google Scholar]
- Haghikia, A.; Haghikia, A.; Hellwig, K.; Baraniskin, A.; Holzmann, A.; Décard, B.F.; Thum, T.; Gold, R. Regulated MicroRNAs in the CSF of Patients with Multiple Sclerosis: A Case-Control Study. Neurology 2012, 79, 2166–2170. [Google Scholar] [CrossRef]
- Kramer, S.; Haghikia, A.; Bang, C.; Scherf, K.; Pfanne, A.; Duscha, A.; Kaisler, J.; Gisevius, B.; Gold, R.; Thum, T. Elevated Levels of MiR-181c and MiR-633 in the CSF of Patients with MS: A Validation Study. Neurol.-Neuroimmunol. Neuroinflammation 2019, 6, e623. [Google Scholar] [CrossRef]
- Gandhi, R.; Healy, B.; Gholipour, T.; Egorova, S.; Musallam, A.; Hussain, M.S.; Nejad, P.; Patel, B.; Hei, H.; Khoury, S. Circulating MicroRNAs as Biomarkers for Disease Staging in Multiple Sclerosis. Ann. Neurol. 2013, 73, 729–740. [Google Scholar] [CrossRef]
- Tsuchida, A.; Ohno, S.; Wu, W.; Borjigin, N.; Fujita, K.; Aoki, T.; Ueda, S.; Takanashi, M.; Kuroda, M. MiR-92 Is a Key Oncogenic Component of the MiR-17–92 Cluster in Colon Cancer. Cancer Sci. 2011, 102, 2264–2271. [Google Scholar] [CrossRef]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative Disease and Autoimmunity in Mice with Increased MiR-17-92 Expression in Lymphocytes. Nat. Immunol. 2008, 9, 405–414. [Google Scholar] [CrossRef]
- Tripathi, A.; Pandit, I.; Perles, A.; Zhou, Y.; Cheng, F.; Dutta, R. Identifying MiRNAs in Multiple Sclerosis Gray Matter Lesions That Correlate with Atrophy Measures. Ann. Clin. Transl. Neurol. 2021, 8, 1279–1291. [Google Scholar] [CrossRef]
- Fritsche, L.; Teuber-Hanselmann, S.; Soub, D.; Harnisch, K.; Mairinger, F.; Junker, A. MicroRNA profiles of MS gray matter lesions identify modulators of the synaptic protein synaptotagmin-7. Brain Pathol. 2020, 30, 524–540. [Google Scholar] [CrossRef]
- Regev, K.; Healy, B.C.; Khalid, F.; Paul, A.; Chu, R.; Tauhid, S.; Tummala, S.; Diaz-Cruz, C.; Raheja, R.; Mazzola, M.A. Association between Serum MicroRNAs and Magnetic Resonance Imaging Measures of Multiple Sclerosis Severity. JAMA Neurol. 2017, 74, 275–285. [Google Scholar] [CrossRef]
- Keller, A.; Leidinger, P.; Steinmeyer, F.; Stähler, C.; Franke, A.; Hemmrich-Stanisak, G.; Kappel, A.; Wright, I.; Dörr, J.; Paul, F. Comprehensive Analysis of MicroRNA Profiles in Multiple Sclerosis Including Next-Generation Sequencing. Mult. Scler. J. 2014, 20, 295–303. [Google Scholar] [CrossRef]
- Regev, K.; Paul, A.; Healy, B.; von Glenn, F.; Diaz-Cruz, C.; Gholipour, T.; Mazzola, M.A.; Raheja, R.; Nejad, P.; Glanz, B.I. Comprehensive Evaluation of Serum MicroRNAs as Biomarkers in Multiple Sclerosis. Neurol.-Neuroimmunol. Neuroinflammation 2016, 3, e267. [Google Scholar] [CrossRef]
- Min, S.; Li, L.; Zhang, M.; Zhang, Y.; Liang, X.; Xie, Y.; He, Q.; Li, Y.; Sun, J.; Liu, Q. TGF-β-Associated MiR-27a Inhibits Dendritic Cell-Mediated Differentiation of Th1 and Th17 Cells by TAB3, P38 MAPK, MAP2K4 and MAP2K7. Genes Immun. 2012, 13, 621–631. [Google Scholar] [CrossRef]
- Regev, K.; Healy, B.C.; Paul, A.; Diaz-Cruz, C.; Mazzola, M.A.; Raheja, R.; Glanz, B.I.; Kivisäkk, P.; Chitnis, T.; Jagodic, M. Identification of MS-Specific Serum MiRNAs in an International Multicenter Study. Neurol.-Neuroimmunol. Neuroinflammation 2018, 5, e491. [Google Scholar] [CrossRef]
- Sharaf-Eldin, W.E.; Kishk, N.A.; Gad, Y.Z.; Hassan, H.; Ali, M.A.; Zaki, M.S.; Mohamed, M.R.; Essawi, M.L. Extracellular MiR-145, MiR-223 and MiR-326 Expression Signature Allow for Differential Diagnosis of Immune-Mediated Neuroinflammatory Diseases. J. Neurol. Sci. 2017, 383, 188–198. [Google Scholar] [CrossRef]
- Ebrahimkhani, S.; Vafaee, F.; Young, P.E.; Hur, S.S.J.; Hawke, S.; Devenney, E.; Beadnall, H.; Barnett, M.H.; Suter, C.M.; Buckland, M.E. Exosomal microRNA signatures in multiple sclerosis reflect disease status. Sci. Rep. 2017, 7, 14293. [Google Scholar] [CrossRef]
- Khedr, A.M.; Shaker, O.G.; Hassan, A.; Hussein, M.; Kamal, Y.S.; Azouz, T.A. MicroRNA-22 Level in Patients with Multiple Sclerosis and Its Relationship with Vitamin D and Vitamin D Receptor Levels. Neuroimmunomodulation 2022, 29, 128–134. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; El-Mehdawy, K.M.; Seleem, M.; El-Sawalhi, M.M.; Shaheen, A.A. Serum ROCK2, MiR-300 and MiR-450b-5p Levels in Two Different Clinical Phenotypes of Multiple Sclerosis: Relation to Patient Disability and Disease Progression. J. Neuroimmunol. 2020, 347, 577356. [Google Scholar] [CrossRef]
- Magner, W.J.; Weinstock-Guttman, B.; Rho, M.; Hojnacki, D.; Ghazi, R.; Ramanathan, M.; Tomasi, T.B. Dicer and MicroRNA Expression in Multiple Sclerosis and Response to Interferon Therapy. J. Neuroimmunol. 2016, 292, 68–78. [Google Scholar] [CrossRef]
- Abdelhak, A.; Huss, A.; Kassubek, J.; Tumani, H.; Otto, M. Serum GFAP as a Biomarker for Disease Severity in Multiple Sclerosis. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Sun, M.; Liu, N.; Xie, Q.; Li, X.; Sun, J.; Wang, H.; Wang, M. A Candidate Biomarker of Glial Fibrillary Acidic Protein in CSF and Blood in Differentiating Multiple Sclerosis and Its Subtypes: A Systematic Review and Meta-Analysis. Mult. Scler. Relat. Disord. 2021, 51, 102870. [Google Scholar] [CrossRef]
- Huss, A.; Otto, M.; Senel, M.; Ludolph, A.C.; Abdelhak, A.; Tumani, H. A Score Based on NfL and Glial Markers May Differentiate between Relapsing–Remitting and Progressive MS Course. Front. Neurol. 2020, 11, 608. [Google Scholar] [CrossRef]
- Madeddu, R.; Farace, C.; Tolu, P.; Solinas, G.; Asara, Y.; Sotgiu, M.A.; Delogu, L.G.; Prados, J.C.; Sotgiu, S.; Montella, A. Cytoskeletal Proteins in the Cerebrospinal Fluid as Biomarker of Multiple Sclerosis. Neurol. Sci. 2013, 34, 181–186. [Google Scholar] [CrossRef]
- Mañé-Martínez, M.A.; Olsson, B.; Bau, L.; Matas, E.; Cobo-Calvo, Á.; Andreasson, U.; Blennow, K.; Romero-Pinel, L.; Martínez-Yélamos, S.; Zetterberg, H. Glial and Neuronal Markers in Cerebrospinal Fluid in Different Types of Multiple Sclerosis. J. Neuroimmunol. 2016, 299, 112–117. [Google Scholar] [CrossRef]
- Petzold, A.; Eikelenboom, M.; Gveric, D.; Keir, G.; Chapman, M.; Lazeron, R.; Cuzner, M.; Polman, C.; Uitdehaag, B.; Thompson, E. Markers for Different Glial Cell Responses in Multiple Sclerosis: Clinical and Pathological Correlations. Brain 2002, 125, 1462–1473. [Google Scholar] [CrossRef]
- Ayrignac, X.; Le Bars, E.; Duflos, C.; Hirtz, C.; Maleska Maceski, A.; Carra-Dallière, C.; Charif, M.; Pinna, F.; Prin, P.; Menjot de Champfleur, N. Serum GFAP in Multiple Sclerosis: Correlation with Disease Type and MRI Markers of Disease Severity. Sci. Rep. 2020, 10, 1–5. [Google Scholar] [CrossRef]
- Lim, C.K.; Bilgin, A.; Lovejoy, D.B.; Tan, V.; Bustamante, S.; Taylor, B.V.; Bessede, A.; Brew, B.J.; Guillemin, G.J. Kynurenine Pathway Metabolomics Predicts and Provides Mechanistic Insight into Multiple Sclerosis Progression. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Biernacki, T.; Sandi, D.; Bencsik, K.; Vécsei, L. Kynurenines in the Pathogenesis of Multiple Sclerosis: Therapeutic Perspectives. Cells 2020, 9, 1564. [Google Scholar] [CrossRef]
- Sandi, D.; Fricska-Nagy, Z.; Bencsik, K.; Vécsei, L. Neurodegeneration in Multiple Sclerosis: Symptoms of Silent Progression, Biomarkers and Neuroprotective Therapy—Kynurenines Are Important Players. Molecules 2021, 26, 3423. [Google Scholar] [CrossRef]
- Aeinehband, S.; Brenner, P.; Ståhl, S.; Bhat, M.; Fidock, M.D.; Khademi, M.; Olsson, T.; Engberg, G.; Jokinen, J.; Erhardt, S. Cerebrospinal Fluid Kynurenines in Multiple Sclerosis; Relation to Disease Course and Neurocognitive Symptoms. Brain Behav. Immun. 2016, 51, 47–55. [Google Scholar] [CrossRef]
- Hinsinger, G.; Galéotti, N.; Nabholz, N.; Urbach, S.; Rigau, V.; Demattei, C.; Lehman, S.; Camu, W.; Labauge, P.; Castelnovo, G.; et al. Chitinase 3-like proteins as diagnostic and prognostic biomarkers of multiple sclerosis. Mult. Scler. 2015, 21, 1251–1261. [Google Scholar] [CrossRef]
- Sellebjerg, F.; Börnsen, L.; Ammitzbøll, C.; Nielsen, J.E.; Vinther-Jensen, T.; Hjermind, L.E.; von Essen, M.; Ratzer, R.L.; Sørensen, P.S.; Christensen, J.R. Defining active progressive multiple sclerosis. Mult. Scler. 2017, 23, 1727–1735. [Google Scholar] [CrossRef]
- Martínez, M.A.M.; Olsson, B.; Bau, L.; Matas, E.; Cobo Calvo, A.; Andreasson, U.; Blennow, K.; Romero-Pine, L.; Martínez-Yélamo, S.; Zetterberg, H. Glial and neuronal markers in cerebrospinal fluid predict progression in multiple sclerosis. Mult. Scler. 2015, 21, 550–561. [Google Scholar] [CrossRef]
- Cantó, E.; Reverter, F.; Morcillo-Suárez, C.; Matesanz, F.; Fernandez, O.; Izquierdo, G.; Vandenbroek, K.; Rodríguez-Antigüedad, A.; Urcelay, E.; Arroyo, R.; et al. Chitinase 3-like 1 plasma levels are increased in patients with progressive forms of multiple sclerosis. Mult. Scler. 2012, 18, 983–990. [Google Scholar] [CrossRef]
- Quintana, E.; Coll, C.; Salavedra-Pont, J.; Muñoz-San Martín, M.; Robles-Cedeño, R.; Tomàs-Roig, J.; Buxó, M.; Matute-Blanch, C.; Villar, L.M.; Montalban, X.; et al. Cognitive impairment in early stages of multiple sclerosis is associated with high cerebrospinal fluid levels of chitinase 3-like 1 and neurofilament light chain. Eur. J. Neurol. 2018, 25, 1189–1191. [Google Scholar] [CrossRef]
- Floro, S.; Carandini, T.; Pietroboni, A.M.; De Riz, M.A.; Scarpini, E.; Galimberti, D. Role of Chitinase 3-like 1 as a Biomarker in Multiple Sclerosis: A Systematic Review and Meta-analysis. Neurol.-Neuroimmunol. Neuroinflammation 2022, 9, e1164. [Google Scholar] [CrossRef]
- Lamancová, P.; Urban, P.; Mašlanková, J.; Rabajdová, M.; Mareková, M. Correlation of selected serum protein levels with the degree of disability and NEDA-3 status in multiple sclerosis phenotypes. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 3933–3941. [Google Scholar]
- Gil-Perotin, S.; Castillo-Villalba, J.; Cubas-Nuñez, L.; Gasque, R.; Hervas, D.; Gomez-Mateu, J.; Alcala, C.; Perez-Miralles, F.; Gascon, F.; Dominguez, J.; et al. Combined Cerebrospinal Fluid Neurofilament Light Chain Protein and Chitinase-3 Like-1 Levels in Defining Disease Course and Prognosis in Multiple Sclerosis. Front. Neurol. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef]
- Cubas-Núñez, L.; Gil-Perotín, S.; Castillo-Villalba, J.; López, V.; Solís Tarazona, L.; Gasqué-Rubio, R.; Carratalá-Boscá, S.; Alcalá-Vicente, C.; Pérez-Miralles, F.; Lassmann, H.; et al. Potential Role of CHI3L1+ Astrocytes in Progression in MS. Neurol.-Neuroimmunol. Neuroinflammation 2021, 8, e972. [Google Scholar] [CrossRef]
- LoPresti, P. Serum-Based Biomarkers in Neurodegeneration and Multiple Sclerosis. Biomedicines 2022, 10, 1077. [Google Scholar] [CrossRef]
- Anderson, J.M.; Patani, R.; Reynolds, R.; Nicholas, R.; Compston, A.; Spillantini, M.G.; Chandran, S. Evidence for Abnormal Tau Phosphorylation in Early Aggressive Multiple Sclerosis. Acta Neuropathol. 2009, 117, 583–589. [Google Scholar] [CrossRef]
- Anderson, J.M.; Patani, R.; Reynolds, R.; Nicholas, R.; Compston, A.; Spillantini, M.G.; Chandran, S. Abnormal Tau Phosphorylation in Primary Progressive Multiple Sclerosis. Acta Neuropathol. 2010, 119, 591–600. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Shobeiri, P.; Saghazadeh, A.; Teunissen, C.E.; Burman, J.; Szalardy, L.; Klivenyi, P.; Bartos, A.; Fernandes, A.; Rezaei, N. Neuronal and Glial CSF Biomarkers in Multiple Sclerosis: A Systematic Review and Meta-Analysis. Rev. Neurosci. 2021, 32, 573–595. [Google Scholar] [CrossRef]
- Van Lierop, Z.Y.; Wieske, L.; Koel-Simmelink, M.J.; Chatterjee, M.; Dekker, I.; Leurs, C.E.; Willemse, E.A.; Moraal, B.; Barkhof, F.; Eftimov, F. Serum Contactin-1 as a Biomarker of Long-Term Disease Progression in Natalizumab-Treated Multiple Sclerosis. Mult. Scler. J. 2022, 28, 102–110. [Google Scholar] [CrossRef]
- Chatterjee, M.; Koel-Simmelink, M.J.; Verberk, I.M.; Killestein, J.; Vrenken, H.; Enzinger, C.; Ropele, S.; Fazekas, F.; Khalil, M.; Teunissen, C.E. Contactin-1 and Contactin-2 in Cerebrospinal Fluid as Potential Biomarkers for Axonal Domain Dysfunction in Multiple Sclerosis. Mult. Scler. J.–Exp. Transl. Clin. 2018, 4, 2055217318819535. [Google Scholar] [CrossRef]
- Berman, S.; Backner, Y.; Krupnik, R.; Paul, F.; Petrou, P.; Karussis, D.; Levin, N.; Mezer, A.A. Conduction Delays in the Visual Pathways of Progressive Multiple Sclerosis Patients Covary with Brain Structure. NeuroImage 2020, 221, 117204. [Google Scholar] [CrossRef]
- Costello, F. Vision Disturbances in Multiple Sclerosis; Thieme Medical Publishers: New York, NY, USA, 2016; Volume 36, pp. 185–195. [Google Scholar]
- Siger, M.; Dzięgielewski, K.; Jasek, L.; Bieniek, M.; Nicpan, A.; Nawrocki, J.; Selmaj, K. Optical coherence tomography in multiple sclerosis: Thickness of the retinal nerve fiber layer as a potential measure of axonal loss and brain atrophy. J. Neurol. 2008, 255, 1555–1560. [Google Scholar] [CrossRef]
- Albrecht, P.; Frohlich, R.; Hartung, H.P.; Kieseier, B.C.; Methner, A. Optical coherence tomography measures axonal loss in multiple sclerosis independently of optic neuritis. J. Neurol. 2007, 254, 1595–1596. [Google Scholar] [CrossRef]
- Alonso, R.; Gonzalez-Moron, D.; Garcea, O. Optical Coherence Tomography as a Biomarker of Neurodegeneration in Multiple Sclerosis: A Review. Mult. Scler. Relat. Disord. 2018, 22, 77–82. [Google Scholar] [CrossRef]
- De Stefano, N.; Narayanan, S.; Francis, G.S.; Arnaoutelis, R.; Tartaglia, M.C.; Antel, J.P.; Matthews, P.M.; Arnold, D.L. Evidence of Axonal Damage in the Early Stages of Multiple Sclerosis and Its Relevance to Disability. Arch. Neurol. 2001, 58, 65–70. [Google Scholar] [CrossRef]
- Blumenthal, E.; Parikh, R.; Pe’er, J.; Naik, M.; Kaliner, E.; Cohen, M.; Prabakaran, S.; Kogan, M.; Thomas, R. Retinal Nerve Fibre Layer Imaging Compared with Histological Measurements in a Human Eye. Eye 2009, 23, 171–175. [Google Scholar] [CrossRef]
- Behbehani, R.; Abu Al-Hassan, A.; Al-Salahat, A.; Sriraman, D.; Oakley, J.; Alroughani, R. Optical Coherence Tomography Segmentation Analysis in Relapsing Remitting versus Progressive Multiple Sclerosis. PLoS ONE 2017, 12, e0172120. [Google Scholar] [CrossRef]
- Bjartmar, C.; Wujek, J.; Trapp, B. Axonal Loss in the Pathology of MS: Consequences for Understanding the Progressive Phase of the Disease. J. Neurol. Sci. 2003, 206, 165–171. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Goodin, D.S.; Boscardin, W.J.; Nolan, R.; Cuneo, A.; Green, A.J. Retinal Axonal Loss Begins Early in the Course of Multiple Sclerosis and Is Similar between Progressive Phenotypes. PLoS ONE 2012, 7, e36847. [Google Scholar] [CrossRef]
- Pulicken, M.; Gordon-Lipkin, E.; Balcer, L.; Frohman, E.; Cutter, G.; Calabresi, P. Optical Coherence Tomography and Disease Subtype in Multiple Sclerosis. Neurology 2007, 69, 2085–2092. [Google Scholar] [CrossRef]
- Oberwahrenbrock, T.; Schippling, S.; Ringelstein, M.; Kaufhold, F.; Zimmermann, H.; Keser, N.; Young, K.L.; Harmel, J.; Hartung, H.-P.; Martin, R. Retinal Damage in Multiple Sclerosis Disease Subtypes Measured by High-Resolution Optical Coherence Tomography. Mult. Scler. Int. 2012, 2012, 530305. [Google Scholar] [CrossRef]
- Skirková, M.; Mikula, P.; Maretta, M.; Fedičová, M.; Vitková, M.; Frigová, L.; Szilasi, J.; Moravská, M.; Horňák, M.; Szilasiová, J. Associations of Optical Coherence Tomography with Disability and Brain MRI Volumetry in Patients with Multiple Sclerosis. Neurol. Neurochir. Pol. 2022, 56, 326–332. [Google Scholar] [CrossRef]
- Glasner, P.; Sabisz, A.; Chylińska, M.; Komendziński, J.; Wyszomirski, A.; Karaszewski, B. Retinal Nerve Fiber and Ganglion Cell Complex Layer Thicknesses Mirror Brain Atrophy in Patients with Relapsing-Remitting Multiple Sclerosis. Restor. Neurol. Neurosci. 2022, 40, 35–42. [Google Scholar] [CrossRef]
- Mizell, R.; Chen, H.; Lambe, J.; Saidha, S.; Harrison, D.M. Association of Retinal Atrophy with Cortical Lesions and Leptomeningeal Enhancement in Multiple Sclerosis on 7T MRI. Mult. Scler. J. 2022, 28, 393–405. [Google Scholar] [CrossRef]
- Bsteh, G.; Berek, K.; Hegen, H.; Teuchner, B.; Buchmann, A.; Voortman, M.M.; Auer, M.; Wurth, S.; Zinganell, A.; Di Pauli, F. Serum Neurofilament Levels Correlate with Retinal Nerve Fiber Layer Thinning in Multiple Sclerosis. Mult. Scler. J. 2020, 26, 1682–1690. [Google Scholar] [CrossRef]
- Rothman, A.; Murphy, O.C.; Fitzgerald, K.C.; Button, J.; Gordon-Lipkin, E.; Ratchford, J.N.; Newsome, S.D.; Mowry, E.M.; Sotirchos, E.S.; Syc-Mazurek, S.B. Retinal Measurements Predict 10-year Disability in Multiple Sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 222–232. [Google Scholar] [CrossRef]
- Lambe, J.; Fitzgerald, K.C.; Murphy, O.C.; Filippatou, A.G.; Sotirchos, E.S.; Kalaitzidis, G.; Vasileiou, E.; Pellegrini, N.; Ogbuokiri, E.; Toliver, B. Association of Spectral-Domain OCT with Long-Term Disability Worsening in Multiple Sclerosis. Neurology 2021, 96, e2058–e2069. [Google Scholar] [CrossRef]
- Martinez-Lapiscina, E.H.; Arnow, S.; Wilson, J.A.; Saidha, S.; Preiningerova, J.L.; Oberwahrenbrock, T.; Brandt, A.U.; Pablo, L.E.; Guerrieri, S.; Gonzalez, I. Retinal Thickness Measured with Optical Coherence Tomography and Risk of Disability Worsening in Multiple Sclerosis: A Cohort Study. Lancet Neurol. 2016, 15, 574–584. [Google Scholar] [CrossRef]
- Yurtogullari, S.; Erbahceci, I. Inner and Outer Retina Findings Determined by Optical Coherence Tomography in Different Subtypes of Multiple Sclerosis. Niger. J. Clin. Pract. 2022, 25, 1069. [Google Scholar] [CrossRef]
- Pisa, M.; Ratti, F.; Vabanesi, M.; Radaelli, M.; Guerrieri, S.; Moiola, L.; Martinelli, V.; Comi, G.; Leocani, L. Subclinical Neurodegeneration in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorder Revealed by Optical Coherence Tomography. Mult. Scler. J. 2020, 26, 1197–1206. [Google Scholar] [CrossRef]
- Balk, L.; Tewarie, P.; Killestein, J.; Polman, C.; Uitdehaag, B.; Petzold, A. Disease Course Heterogeneity and OCT in Multiple Sclerosis. Mult. Scler. J. 2014, 20, 1198–1206. [Google Scholar] [CrossRef]
- Jankowska-Lech, I.; Wasyluk, J.; Palasik, W.; Terelak-Borys, B.; Grabska-Liberek, I. Peripapillary Retinal Nerve Fiber Layer Thickness Measured by Optical Coherence Tomography in Different Clinical Subtypes of Multiple Sclerosis. Mult. Scler. Relat. Disord. 2019, 27, 260–268. [Google Scholar] [CrossRef]
- Estiasari, R.; Diwyacitta, A.; Sidik, M.; Rida Ariarini, N.N.; Sitorus, F.; Marwadhani, S.S.; Maharani, K.; Imran, D.; Arpandy, R.A.; Pangeran, D. Evaluation of Retinal Structure and Optic Nerve Function Changes in Multiple Sclerosis: Longitudinal Study with 1-Year Follow-Up. Neurol. Res. Int. 2021, 2021, 5573839. [Google Scholar] [CrossRef]
- Jakimovski, D.; Zivadinov, R.; Vaughn, C.B.; Ozel, O.; Weinstock-Guttman, B. Clinical Effects Associated with Five-Year Retinal Nerve Fiber Layer Thinning in Multiple Sclerosis. J. Neurol. Sci. 2021, 427, 117552. [Google Scholar] [CrossRef]
- Cellerino, M.; Priano, L.; Bruschi, N.; Boffa, G.; Petracca, M.; Novi, G.; Lapucci, C.; Sbragia, E.; Uccelli, A.; Inglese, M. Relationship Between Retinal Layer Thickness and Disability Worsening in Relapsing-Remitting and Progressive Multiple Sclerosis. J. Neuro-Ophthalmol. 2021, 41, 329–334. [Google Scholar] [CrossRef]
- Sotirchos, E.S.; Gonzalez Caldito, N.; Filippatou, A.; Fitzgerald, K.C.; Murphy, O.C.; Lambe, J.; Nguyen, J.; Button, J.; Ogbuokiri, E.; Crainiceanu, C.M. Progressive Multiple Sclerosis Is Associated with Faster and Specific Retinal Layer Atrophy. Ann. Neurol. 2020, 87, 885–896. [Google Scholar] [CrossRef]
- Eslami, F.; Ghiasian, M.; Khanlarzade, E.; Moradi, E. Retinal Nerve Fiber Layer Thickness and Total Macular Volume in Multiple Sclerosis Subtypes and Their Relationship with Severity of Disease, a Cross-Sectional Study. Eye Brain 2020, 12, 15. [Google Scholar] [CrossRef]
- Berek, K.; Hegen, H.; Hocher, J.; Auer, M.; Di Pauli, F.; Krajnc, N.; Angermann, R.; Barket, R.; Zinganell, A.; Riedl, K. Retinal Layer Thinning as a Biomarker of Long-Term Disability Progression in Multiple Sclerosis. Mult. Scler. J. 2022, 28, 1871–1880. [Google Scholar] [CrossRef]
- Balıkçı, A.; Parmak Yener, N.; Seferoğlu, M. Optical Coherence Tomography and Optical Coherence Tomography Angiography Findings in Multiple Sclerosis Patients. Neuro-Ophthalmol. 2022, 46, 19–33. [Google Scholar] [CrossRef]
- Piedrabuena, R.; Bittar, M. Optical Coherence Tomography and Visual Evoked Potential and Its Relationship with Neurological Disability in Patients with Relapsing-Remitting Multiple Sclerosis. Mult. Scler. Relat. Disord. 2022, 57, 103420. [Google Scholar] [CrossRef]
- Barreiro-González, A.; Sanz, M.T.; Carratalà-Boscà, S.; Pérez-Miralles, F.; Alcalá, C.; Carreres-Polo, J.; España-Gregori, E.; Casanova, B. Design and Validation of an Expanded Disability Status Scale Model in Multiple Sclerosis. Eur. Neurol. 2022, 85, 112–121. [Google Scholar] [CrossRef]
- Bsteh, G.; Hegen, H.; Teuchner, B.; Amprosi, M.; Berek, K.; Ladstätter, F.; Wurth, S.; Auer, M.; Di Pauli, F.; Deisenhammer, F.; et al. Peripapillary retinal nerve fibre layer as measured by optical coherence tomography is a prognostic biomarker not only for physical but also for cognitive disability progression in multiple sclerosis. Mult. Scler. 2019, 25, 196–203. [Google Scholar] [CrossRef]
- Schurz, N.; Sariaslani, L.; Altmann, P.; Leutmezer, F.; Mitsch, C.; Pemp, B.; Rommer, P.; Zrzavy, T.; Berger, T.; Bsteh, G. Evaluation of Retinal Layer Thickness Parameters as Biomarkers in a Real-World Multiple Sclerosis Cohort. Eye Brain 2021, 13, 59. [Google Scholar] [CrossRef]
- Cilingir, V.; Batur, M. First Measured Retinal Nerve Fiber Layer Thickness in RRMS Can Be Used as a Biomarker for the Course of the Disease: Threshold Value Discussions. J. Neurol. 2021, 268, 2858–2865. [Google Scholar] [CrossRef]
- Bitirgen, G.; Akpinar, Z.; Uca, A.U.; Ozkagnici, A.; Petropoulos, I.N.; Malik, R.A. Progressive Loss of Corneal and Retinal Nerve Fibers in Patients with Multiple Sclerosis: A 2-Year Follow-up Study. Transl. Vis. Sci. Technol. 2020, 9, 37. [Google Scholar] [CrossRef]
- Cilingir, V.; Batur, M. Axonal Degeneration Independent of Inflammatory Activity: Is It More Intense in the Early Stages of Relapsing-Remitting Multiple Sclerosis Disease? Eur. Neurol. 2020, 83, 508–516. [Google Scholar] [CrossRef]
- Bsteh, G.; Berek, K.; Hegen, H.; Altmann, P.; Wurth, S.; Auer, M.; Zinganell, A.; Di Pauli, F.; Rommer, P.; Leutmezer, F. Macular Ganglion Cell–Inner Plexiform Layer Thinning as a Biomarker of Disability Progression in Relapsing Multiple Sclerosis. Mult. Scler. J. 2021, 27, 684–694. [Google Scholar] [CrossRef]
- Vidal-Jordana, A.; Pareto, D.; Cabello, S.; Alberich, M.; Rio, J.; Tintore, M.; Auger, C.; Montalban, X.; Rovira, A.; Sastre-Garriga, J. Optical Coherence Tomography Measures Correlate with Brain and Spinal Cord Atrophy and Multiple Sclerosis Disease-related Disability. Eur. J. Neurol. 2020, 27, 2225–2232. [Google Scholar] [CrossRef]
- Koraysha, N.A.; Kishk, N.; Hassan, A.; El Gendy, N.M.S.; Shehata, H.S.; Al-Azayem, S.A.; Kamal, Y.S. Evaluating Optic Nerve Diameter as a Possible Biomarker for Disability in Patients with Multiple Sclerosis. Neuropsychiatr. Dis. Treat. 2019, 15, 2571. [Google Scholar] [CrossRef]
- Cordano, C.; Nourbakhsh, B.; Devereux, M.; Damotte, V.; Bennett, D.; Hauser, S.L.; Cree, B.A.; Gelfand, J.M.; Green, A.J. PRNFL as a Marker of Disability Worsening in the Medium/Long Term in Patients with MS. Neurol.-Neuroimmunol. Neuroinflammation 2019, 6, e533. [Google Scholar] [CrossRef]
- Garcia-Martin, E.; Ara, J.R.; Martin, J.; Almarcegui, C.; Dolz, I.; Vilades, E.; Gil-Arribas, L.; Fernandez, F.J.; Polo, V.; Larrosa, J.M. Retinal and Optic Nerve Degeneration in Patients with Multiple Sclerosis Followed up for 5 Years. Ophthalmology 2017, 124, 688–696. [Google Scholar] [CrossRef]
- Albrecht, P.; Ringelstein, M.; Müller, A.; Keser, N.; Dietlein, T.; Lappas, A.; Foerster, A.; Hartung, H.; Aktas, O.; Methner, A. Degeneration of Retinal Layers in Multiple Sclerosis Subtypes Quantified by Optical Coherence Tomography. Mult. Scler. J. 2012, 18, 1422–1429. [Google Scholar] [CrossRef]
- Bsteh, G.; Hegen, H.; Altmann, P.; Auer, M.; Berek, K.; Pauli, F.D.; Wurth, S.; Zinganell, A.; Rommer, P.; Deisenhammer, F. Retinal Layer Thinning Is Reflecting Disability Progression Independent of Relapse Activity in Multiple Sclerosis. Mult. Scler. J.–Exp. Transl. Clin. 2020, 6, 2055217320966344. [Google Scholar] [CrossRef]
- Bsteh, G.; Hegen, H.; Teuchner, B.; Berek, K.; Wurth, S.; Auer, M.; Di Pauli, F.; Deisenhammer, F.; Berger, T. Peripapillary Retinal Nerve Fibre Layer Thinning Rate as a Biomarker Discriminating Stable and Progressing Relapsing–Remitting Multiple Sclerosis. Eur. J. Neurol. 2019, 26, 865–871. [Google Scholar] [CrossRef]
- Tavazzi, E.; Zivadinov, R.; Dwyer, M.G.; Jakimovski, D.; Singhal, T.; Weinstock-Guttman, B.; Bergsland, N. MRI Biomarkers of Disease Progression and Conversion to Secondary-Progressive Multiple Sclerosis. Expert Rev. Neurother. 2020, 20, 821–834. [Google Scholar] [CrossRef]
- Chard, D.; Trip, S.A. Resolving the Clinico-Radiological Paradox in Multiple Sclerosis. F1000Research 2017, 6, 1828. [Google Scholar] [CrossRef]
- Dekker, I.; Eijlers, A.; Popescu, V.; Balk, L.; Vrenken, H.; Wattjes, M.; Uitdehaag, B.; Killestein, J.; Geurts, J.; Barkhof, F. Predicting Clinical Progression in Multiple Sclerosis after 6 and 12 Years. Eur. J. Neurol. 2019, 26, 893–902. [Google Scholar] [CrossRef]
- Ontaneda, D.; Tallantyre, E.; Kalincik, T.; Planchon, S.M.; Evangelou, N. Early Highly Effective versus Escalation Treatment Approaches in Relapsing Multiple Sclerosis. Lancet Neurol. 2019, 18, 973–980. [Google Scholar] [CrossRef]
- Zivadinov, R.; Horakova, D.; Bergsland, N.; Hagemeier, J.; Ramasamy, D.; Uher, T.; Vaneckova, M.; Havrdova, E.; Dwyer, M. A Serial 10-Year Follow-up Study of Atrophied Brain Lesion Volume and Disability Progression in Patients with Relapsing-Remitting MS. Am. J. Neuroradiol. 2019, 40, 446–452. [Google Scholar] [CrossRef]
- Bermel, R.A.; Bakshi, R. The Measurement and Clinical Relevance of Brain Atrophy in Multiple Sclerosis. Lancet Neurol. 2006, 5, 158–170. [Google Scholar] [CrossRef]
- Zivadinov, R.; Jakimovski, D.; Gandhi, S.; Ahmed, R.; Dwyer, M.G.; Horakova, D.; Weinstock-Guttman, B.; Benedict, R.R.; Vaneckova, M.; Barnett, M. Clinical Relevance of Brain Atrophy Assessment in Multiple Sclerosis. Implications for Its Use in a Clinical Routine. Expert Rev. Neurother. 2016, 16, 777–793. [Google Scholar] [CrossRef]
- Treabǎ, C.A.; Bǎlaşa, R.; Podeanu, D.M.; Simu, I.P.; Buruian, M.M. Cerebral Lesions of Multiple Sclerosis: Is Gadolinium Always Irreplaceable in Assessing Lesion Activity? Diagn. Interv. Radiol. 2014, 20, 178. [Google Scholar] [CrossRef]
- Ghione, E.; Bergsland, N.; Dwyer, M.G.; Hagemeier, J.; Jakimovski, D.; Paunkoski, I.; Ramasamy, D.P.; Silva, D.; Carl, E.; Hojnacki, D. Brain Atrophy Is Associated with Disability Progression in Patients with MS Followed in a Clinical Routine. Am. J. Neuroradiol. 2018, 39, 2237–2242. [Google Scholar] [CrossRef]
- University of California, San Francisco MS-EPIC Team; Cree, B.A.; Hollenbach, J.A.; Bove, R.; Kirkish, G.; Sacco, S.; Caverzasi, E.; Bischof, A.; Gundel, T.; Zhu, A.H. Silent Progression in Disease Activity–Free Relapsing Multiple Sclerosis. Ann. Neurol. 2019, 85, 653–666. [Google Scholar]
- Guevara, C.; Garrido, C.; Martinez, M.; Farias, G.A.; Orellana, P.; Soruco, W.; Alarcón, P.; Diaz, V.; Silva, C.; Kempton, M.J. Prospective Assessment of No Evidence of Disease Activity-4 Status in Early Disease Stages of Multiple Sclerosis in Routine Clinical Practice. Front. Neurol. 2019, 10, 788. [Google Scholar] [CrossRef]
- Smeets, D.; Ribbens, A.; Sima, D.M.; Cambron, M.; Horakova, D.; Jain, S.; Maertens, A.; Van Vlierberghe, E.; Terzopoulos, V.; Van Binst, A. Reliable Measurements of Brain Atrophy in Individual Patients with Multiple Sclerosis. Brain Behav. 2016, 6, e00518. [Google Scholar] [CrossRef]
- Eshaghi, A.; Prados, F.; Brownlee, W.J.; Altmann, D.R.; Tur, C.; Cardoso, M.J.; De Angelis, F.; van de Pavert, S.H.; Cawley, N.; De Stefano, N. Deep Gray Matter Volume Loss Drives Disability Worsening in Multiple Sclerosis. Ann. Neurol. 2018, 83, 210–222. [Google Scholar] [CrossRef]
- Filippi, M.; Rocca, M. MR Imaging of Gray Matter Involvement in Multiple Sclerosis: Implications for Understanding Disease Pathophysiology and Monitoring Treatment Efficacy. Am. J. Neuroradiol. 2010, 31, 1171–1177. [Google Scholar] [CrossRef]
- Hänninen, K.; Viitala, M.; Paavilainen, T.; Karhu, J.O.; Rinne, J.; Koikkalainen, J.; Lötjönen, J.; Soilu-Hänninen, M. Thalamic Atrophy without Whole Brain Atrophy Is Associated with Absence of 2-Year NEDA in Multiple Sclerosis. Front. Neurol. 2019, 10, 459. [Google Scholar] [CrossRef]
- De Stefano, N.; Stromillo, M.L.; Giorgio, A.; Bartolozzi, M.L.; Battaglini, M.; Baldini, M.; Portaccio, E.; Amato, M.P.; Sormani, M.P. Establishing Pathological Cut-Offs of Brain Atrophy Rates in Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 93–99. [Google Scholar] [CrossRef]
- Preziosa, P.; Pagani, E.; Meani, A.; Moiola, L.; Rodegher, M.; Filippi, M.; Rocca, M.A. Slowly Expanding Lesions Predict 9-Year Multiple Sclerosis Disease Progression. Neurol.-Neuroimmunol. Neuroinflammation 2022, 9, e1139. [Google Scholar] [CrossRef]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W. Clinical and Pathological Insights into the Dynamic Nature of the White Matter Multiple Sclerosis Plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef]
- Luchetti, S.; Fransen, N.L.; van Eden, C.G.; Ramaglia, V.; Mason, M.; Huitinga, I. Progressive Multiple Sclerosis Patients Show Substantial Lesion Activity That Correlates with Clinical Disease Severity and Sex: A Retrospective Autopsy Cohort Analysis. Acta Neuropathol. 2018, 135, 511–528. [Google Scholar] [CrossRef]
- Kolb, H.; Al-Louzi, O.; Beck, E.S.; Sati, P.; Absinta, M.; Reich, D.S. From pathology to MRI and back: Clinically relevant biomarkers of multiple sclerosis lesions. Neuroimage Clin. 2022, 36, 103194. [Google Scholar] [CrossRef]
- Absinta, M.; Sati, P.; Masuzzo, F.; Nair, G.; Sethi, V.; Kolb, H.; Ohayon, J.; Wu, T.; Cortese, I.C.; Reich, D.S. Association of Chronic Active Multiple Sclerosis Lesions with Disability in Vivo. JAMA Neurol. 2019, 76, 1474–1483. [Google Scholar] [CrossRef]
- Treaba, C.A.; Conti, A.; Klawiter, E.C.; Barletta, V.T.; Herranz, E.; Mehndiratta, A.; Russo, A.W.; Sloane, J.A.; Kinkel, R.P.; Toschi, N. Cortical and Phase Rim Lesions on 7 T MRI as Markers of Multiple Sclerosis Disease Progression. Brain Commun. 2021, 3, fcab134. [Google Scholar] [CrossRef]
- Absinta, M.; Sati, P.; Schindler, M.; Leibovitch, E.C.; Ohayon, J.; Wu, T.; Meani, A.; Filippi, M.; Jacobson, S.; Cortese, I.C. Persistent 7-Tesla Phase Rim Predicts Poor Outcome in New Multiple Sclerosis Patient Lesions. J. Clin. Investig. 2016, 126, 2597–2609. [Google Scholar] [CrossRef]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Höftberger, R.; Berger, T.; Auff, E.; Leutmezer, F.; Trattnig, S.; Lassmann, H. Slow Expansion of Multiple Sclerosis Iron Rim Lesions: Pathology and 7 T Magnetic Resonance Imaging. Acta Neuropathol. 2017, 133, 25–42. [Google Scholar] [CrossRef]
- Treaba, C.A.; Herranz, E.; Barletta, V.T.; Mehndiratta, A.; Ouellette, R.; Sloane, J.A.; Klawiter, E.C.; Kinkel, R.P.; Mainero, C. The Relevance of Multiple Sclerosis Cortical Lesions on Cortical Thinning and Their Clinical Impact as Assessed by 7.0-T MRI. J. Neurol. 2021, 268, 2473–2481. [Google Scholar] [CrossRef]
- Curti, E.; Graziuso, S.; Tsantes, E.; Crisi, G.; Granella, F. Correlation between Cortical Lesions and Cognitive Impairment in Multiple Sclerosis. Brain Behav. 2018, 8, e00955. [Google Scholar] [CrossRef]
- Calabrese, M.; Poretto, V.; Favaretto, A.; Alessio, S.; Bernardi, V.; Romualdi, C.; Rinaldi, F.; Perini, P.; Gallo, P. Cortical Lesion Load Associates with Progression of Disability in Multiple Sclerosis. Brain 2012, 135, 2952–2961. [Google Scholar] [CrossRef]
- Blindenbacher, N.; Brunner, E.; Asseyer, S.; Scheel, M.; Siebert, N.; Rasche, L.; Bellmann-Strobl, J.; Brandt, A.; Ruprecht, K.; Meier, D. Evaluation of the ‘Ring Sign’and the ‘Core Sign’as a Magnetic Resonance Imaging Marker of Disease Activity and Progression in Clinically Isolated Syndrome and Early Multiple Sclerosis. Mult. Scler. J.–Exp. Transl. Clin. 2020, 6, 2055217320915480. [Google Scholar]
- Hemond, C.C.; Reich, D.S.; Dundamadappa, S.K. Paramagnetic Rim Lesions in Multiple Sclerosis: Comparison of Visualization at 1.5-T and 3-T MRI. AJR Am. J. Roentgenol. 2022, 219, 120–131. [Google Scholar] [CrossRef]
- Casserly, C.; Seyman, E.E.; Alcaide-Leon, P.; Guenette, M.; Lyons, C.; Sankar, S.; Svendrovski, A.; Baral, S.; Oh, J. Spinal Cord Atrophy in Multiple Sclerosis: A Systematic Review and Meta-analysis. J. Neuroimaging 2018, 28, 556–586. [Google Scholar] [CrossRef]
- De Stefano, N.; Giorgio, A.; Battaglini, M.; Rovaris, M.; Sormani, M.; Barkhof, F.; Korteweg, T.; Enzinger, C.; Fazekas, F.; Calabrese, M. Assessing Brain Atrophy Rates in a Large Population of Untreated Multiple Sclerosis Subtypes. Neurology 2010, 74, 1868–1876. [Google Scholar] [CrossRef]
- Daams, M.; Weiler, F.; Steenwijk, M.D.; Hahn, H.K.; Geurts, J.J.; Vrenken, H.; Van Schijndel, R.A.; Balk, L.J.; Tewarie, P.K.; Tillema, J.-M. Mean Upper Cervical Cord Area (MUCCA) Measurement in Long-Standing Multiple Sclerosis: Relation to Brain Findings and Clinical Disability. Mult. Scler. J. 2014, 20, 1860–1865. [Google Scholar] [CrossRef]
- Zurawski, J.; Glanz, B.I.; Healy, B.C.; Tauhid, S.; Khalid, F.; Chitnis, T.; Weiner, H.L.; Bakshi, R. The Impact of Cervical Spinal Cord Atrophy on Quality of Life in Multiple Sclerosis. J. Neurol. Sci. 2019, 403, 38–43. [Google Scholar] [CrossRef]
- Yiannakas, M.C.; Mustafa, A.M.; De Leener, B.; Kearney, H.; Tur, C.; Altmann, D.R.; De Angelis, F.; Plantone, D.; Ciccarelli, O.; Miller, D.H. Fully Automated Segmentation of the Cervical Cord from T1-Weighted MRI Using PropSeg: Application to Multiple Sclerosis. NeuroImage Clin. 2016, 10, 71–77. [Google Scholar] [CrossRef]
- Rocca, M.A.; Valsasina, P.; Meani, A.; Gobbi, C.; Zecca, C.; Barkhof, F.; Schoonheim, M.M.; Strijbis, E.M.; Vrenken, H.; Gallo, A. Spinal Cord Lesions and Brain Grey Matter Atrophy Independently Predict Clinical Worsening in Definite Multiple Sclerosis: A 5-Year, Multicentre Study. J. Neurol. Neurosurg. Psychiatry 2022, 94, 10–18. [Google Scholar] [CrossRef]
- Bischof, A.; Papinutto, N.; Keshavan, A.; Rajesh, A.; Kirkish, G.; Zhang, X.; Mallott, J.M.; Asteggiano, C.; Sacco, S.; Gundel, T.J. Spinal Cord Atrophy Predicts Progressive Disease in Relapsing Multiple Sclerosis. Ann. Neurol. 2022, 91, 268–281. [Google Scholar] [CrossRef]
- Dwyer, M.G.; Bergsland, N.; Ramasamy, D.P.; Jakimovski, D.; Weinstock-Guttman, B.; Zivadinov, R. Atrophied Brain Lesion Volume: A New Imaging Biomarker in Multiple Sclerosis. J. Neuroimaging 2018, 28, 490–495. [Google Scholar] [CrossRef]
- Genovese, A.V.; Hagemeier, J.; Bergsland, N.; Jakimovski, D.; Dwyer, M.G.; Ramasamy, D.P.; Lizarraga, A.A.; Hojnacki, D.; Kolb, C.; Weinstock-Guttman, B. Atrophied Brain T2 Lesion Volume at MRI Is Associated with Disability Progression and Conversion to Secondary Progressive Multiple Sclerosis. Radiology 2019, 293, 424. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maier, S.; Barcutean, L.; Andone, S.; Manu, D.; Sarmasan, E.; Bajko, Z.; Balasa, R. Recent Progress in the Identification of Early Transition Biomarkers from Relapsing-Remitting to Progressive Multiple Sclerosis. Int. J. Mol. Sci. 2023, 24, 4375. https://doi.org/10.3390/ijms24054375
Maier S, Barcutean L, Andone S, Manu D, Sarmasan E, Bajko Z, Balasa R. Recent Progress in the Identification of Early Transition Biomarkers from Relapsing-Remitting to Progressive Multiple Sclerosis. International Journal of Molecular Sciences. 2023; 24(5):4375. https://doi.org/10.3390/ijms24054375
Chicago/Turabian StyleMaier, Smaranda, Laura Barcutean, Sebastian Andone, Doina Manu, Emanuela Sarmasan, Zoltan Bajko, and Rodica Balasa. 2023. "Recent Progress in the Identification of Early Transition Biomarkers from Relapsing-Remitting to Progressive Multiple Sclerosis" International Journal of Molecular Sciences 24, no. 5: 4375. https://doi.org/10.3390/ijms24054375
APA StyleMaier, S., Barcutean, L., Andone, S., Manu, D., Sarmasan, E., Bajko, Z., & Balasa, R. (2023). Recent Progress in the Identification of Early Transition Biomarkers from Relapsing-Remitting to Progressive Multiple Sclerosis. International Journal of Molecular Sciences, 24(5), 4375. https://doi.org/10.3390/ijms24054375