
Unveil the Secret of the Bacteria and Phage Arms Race


Abstract
1. Introduction
2. Bacterial Anti-Phage Strategies
2.1. Blocking Phage Adsorption
2.2. Blocking Phage’s Injection
2.3. Interfering with Phage Replication
2.3.1. Restriction-Modification Systems
2.3.2. CRISPR-Cas Adaptive Immune Systems
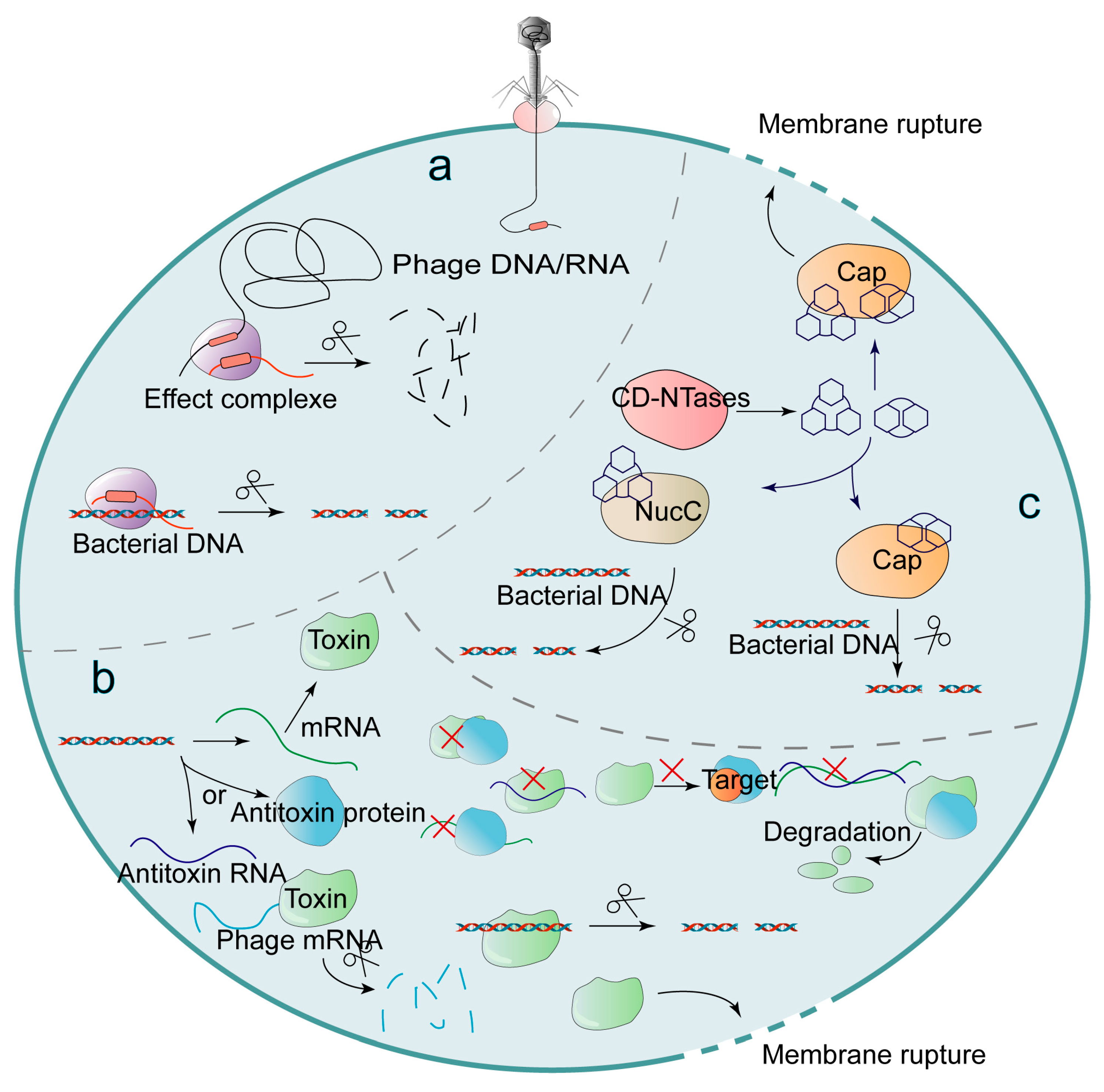
2.3.3. Abortive Infection
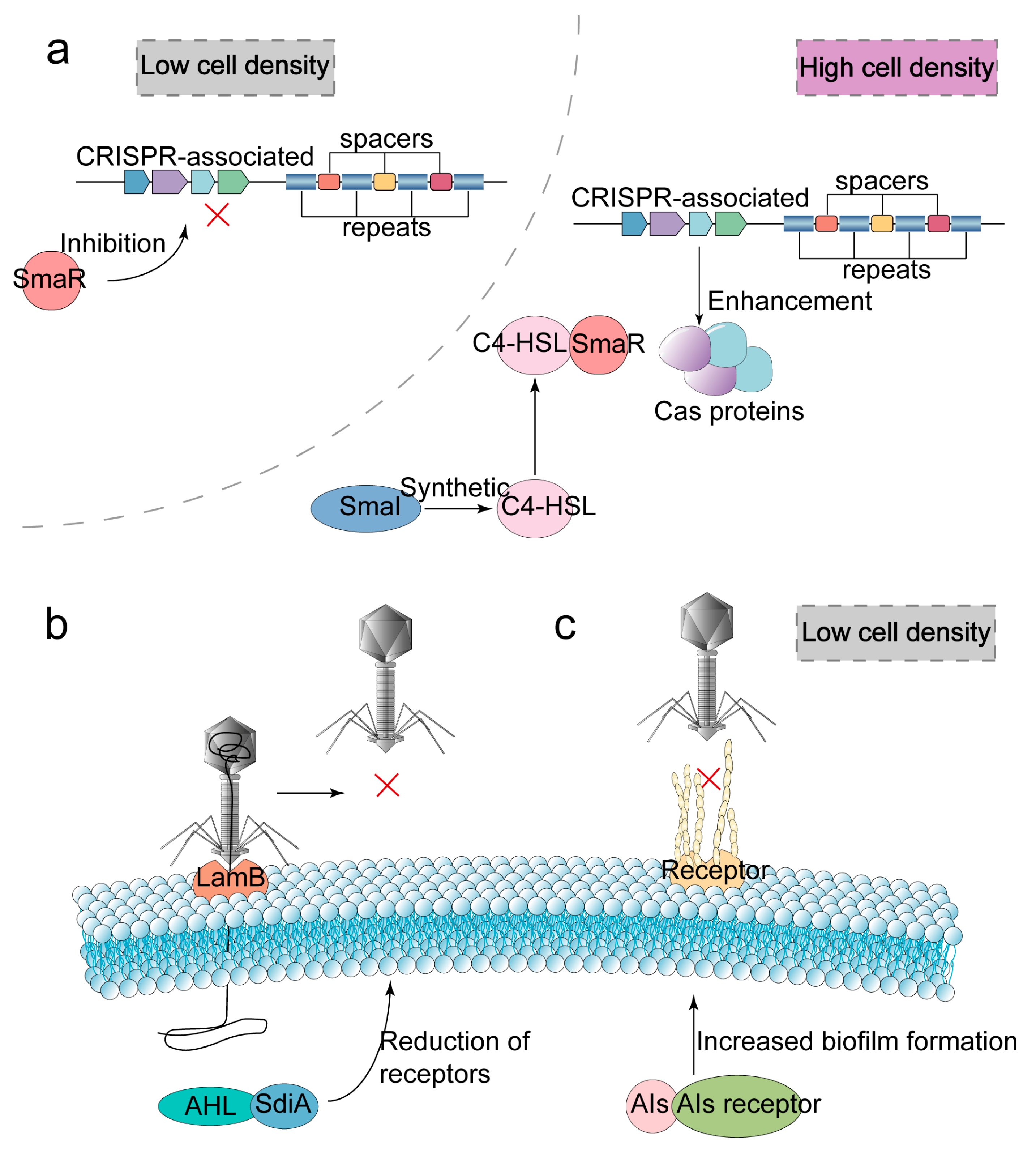
2.4. Quorum Sensing
3. Anti-Defense Strategies of Phages
3.1. Regaining the Ability to Identify and Adsorption Host
3.2. Anti-Defense Strategies for R-M Systems
3.3. Anti-Defense Strategies for CRISPR-Cas Systems
3.4. Anti-Defense Strategies for QS System
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feiner, R.; Argov, T.; Rabinovich, L.; Sigal, N.; Borovok, I.; Herskovits, A.A. A new perspective on lysogeny: Prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 2015, 13, 641–650. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.; Houchin, L.A.; Williamson, S.J.; Paul, J.H. Lysogeny in marine Synechococcus. Nature 2002, 415, 496. [Google Scholar] [CrossRef] [PubMed]
- Erez, Z.; Steinberger-Levy, I.; Shamir, M.; Doron, S.; Stokar-Avihail, A.; Peleg, Y.; Melamed, S.; Leavitt, A.; Savidor, A.; Albeck, S.; et al. Communication between viruses guides lysis-lysogeny decisions. Nature 2017, 541, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhang, H.; Gu, D.; Ma, Y.; Zhou, X. Identification of a novel bacterial receptor that binds tail tubular proteins and mediates phage infection of Vibrio parahaemolyticus. Emerg. Microbes Infect. 2020, 9, 855–867. [Google Scholar] [CrossRef]
- Lu, M.J.; Henning, U. Superinfection exclusion by T-even-type coliphages. Trends Microbiol. 1994, 2, 137–139. [Google Scholar] [CrossRef]
- Rao, D.N.; Dryden, D.T.; Bheemanaik, S. Type III restriction-modification enzymes: A historical perspective. Nucleic Acids Res. 2014, 42, 45–55. [Google Scholar] [CrossRef]
- Westra, E.R.; Swarts, D.C.; Staals, R.H.; Jore, M.M.; Brouns, S.J.; van der Oost, J. The CRISPRs, they are a-changin’: How prokaryotes generate adaptive immunity. Annu. Rev. Genet. 2012, 46, 311–339. [Google Scholar] [CrossRef]
- Lopatina, A.; Tal, N.; Sorek, R. Abortive Infection: Bacterial Suicide as an Antiviral Immune Strategy. Annu. Rev. Virol. 2020, 7, 371–384. [Google Scholar] [CrossRef]
- Bonsma-Fisher, M.; Soutière, D.; Goyal, S. How adaptive immunity constrains the composition and fate of large bacterial populations. Proc. Natl. Acad. Sci. USA 2018, 115, E7462–E7468. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Meyer, J.R.; Dobias, D.T.; Weitz, J.S.; Barrick, J.E.; Quick, R.T.; Lenski, R.E. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 2012, 335, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Flodman, K.; Corrêa, I.R., Jr.; Dai, N.; Weigele, P.; Xu, S.Y. In vitro Type II Restriction of Bacteriophage DNA With Modified Pyrimidines. Front. Microbiol. 2020, 11, 604618. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Cao, D.; Zhu, J.; Feng, H.; Luo, X.; Liu, S.; Yan, X.X.; Zhang, X.; Gao, P. Structural insights into assembly, operation and inhibition of a type I restriction-modification system. Nat. Microbiol. 2020, 5, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Jore, M.M.; Datsenko, K.A.; Semenova, A.; Westra, E.R.; Wanner, B.; van der Oost, J.; Brouns, S.J.; Severinov, K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. USA 2011, 108, 10098–10103. [Google Scholar] [CrossRef]
- Koonin, E.V.; Krupovic, M. Phages build anti-defence barriers. Nat. Microbiol. 2020, 5, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Xu, Y.; Zhu, T.; Li, N.; Qi, J.; Chai, Y.; Wu, M.; Zhang, X.; Shi, Y.; Wang, P.; et al. Alternate binding modes of anti-CRISPR viral suppressors AcrF1/2 to Csy surveillance complex revealed by cryo-EM structures. Cell Res. 2017, 27, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Botstein, K.; Lew, K.K.; Jarvik, V.; Swanson, C.A. Role of antirepressor in the bipartite control of repression and immunity by bacteriophage P22. J. Mol. Biol. 1975, 91, 439–462. [Google Scholar] [CrossRef]
- Silpe, J.E.; Bassler, B.L. Phage-Encoded LuxR-Type Receptors Responsive to Host-Produced Bacterial Quorum-Sensing Autoinducers. mBio 2019, 10, e00638-19. [Google Scholar] [CrossRef]
- Gómez, P.; Buckling, A. Bacteria-Phage Antagonistic Coevolution in Soil. Science 2011, 332, 106–109. [Google Scholar] [CrossRef]
- Attrill, E.L.; Claydon, R.; Łapińska, U.; Recker, M.; Meaden, S.; Brown, A.T.; Westra, E.R.; Harding, S.V.; Pagliara, S. Individual bacteria in structured environments rely on phenotypic resistance to phage. PLoS Biol. 2021, 19, e3001406. [Google Scholar] [CrossRef]
- Sumrall, E.T.; Shen, Y.; Keller, A.P.; Rismondo, J.; Pavlou, M.; Eugster, M.R.; Boulos, S.; Disson, O.; Thouvenot, P.; Kilcher, S.; et al. Phage resistance at the cost of virulence: Listeria monocytogenes serovar 4b requires galactosylated teichoic acids for InlB-mediated invasion. PLoS Pathog. 2019, 15, e1008032. [Google Scholar] [CrossRef]
- Martínez, B.; Rodríguez, A.; Kulakauskas, S.; Chapot-Chartier, M.P. Cell wall homeostasis in lactic acid bacteria: Threats and defences. FEMS Microbiol. Rev. 2020, 44, 538–564. [Google Scholar] [CrossRef]
- Ongenae, V.; Briegel, A.; Claessen, D. Cell wall deficiency as an escape mechanism from phage infection. Open Biol. 2021, 11, 210199. [Google Scholar] [CrossRef] [PubMed]
- Olszak, T.; Danis-Wlodarczyk, K.; Arabski, M.; Gula, G.; Maciejewska, B.; Wasik, S.; Lood, C.; Higgins, G.; Harvey, B.J.; Lavigne, R.; et al. Pseudomonas aeruginosa PA5oct Jumbo Phage Impacts Planktonic and Biofilm Population and Reduces Its Host Virulence. Viruses 2019, 11, 1089. [Google Scholar] [CrossRef]
- Li, G.; Shen, M.; Yang, Y.; Le, S.; Li, M.; Wang, J.; Zhao, Y.; Tan, Y.; Hu, F.; Lu, S. Adaptation of Pseudomonas aeruginosa to Phage PaP1 Predation via O-Antigen Polymerase Mutation. Front. Microbiol. 2018, 9, 1170. [Google Scholar] [CrossRef] [PubMed]
- Gordillo Altamirano, F.; Forsyth, J.H.; Patwa, R.; Kostoulias, X.; Trim, M.; Subedi, D.; Archer, S.K.; Morris, F.C.; Oliveira, C.; Kielty, L.; et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 2021, 6, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Loh, B.; Gordillo Altamirano, F.; Yu, Y.; Hua, X.; Leptihn, S. Colistin-phage combinations decrease antibiotic resistance in Acinetobacter baumannii via changes in envelope architecture. Emerg. Microbes Infect. 2021, 10, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Looijesteijn, P.J.; Trapet, L.; de Vries, E.; Abee, T.; Hugenholtz, J. Physiological function of exopolysaccharides produced by Lactococcus lactis. Int. J. Food Microbiol. 2001, 64, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Angelin, J.; Kavitha, M. Exopolysaccharides from probiotic bacteria and their health potential. Int. J. Biol. Macromol. 2020, 162, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Riede, I.; Eschbach, M.L. Evidence that TraT interacts with OmpA of Escherichia coli. FEBS Lett. 1986, 205, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Nordström, K.; Forsgren, A. Effect of protein A on adsorption of bacteriophages to Staphylococcus aureus. J. Virol. 1974, 14, 198–202. [Google Scholar] [CrossRef]
- Toyofuku, M.; Nomura, N.; Eberl, L. Types and origins of bacterial membrane vesicles. Nat. Rev. Microbiol. 2019, 17, 13–24. [Google Scholar] [CrossRef]
- Augustyniak, D.; Olszak, T.; Drulis-Kawa, Z. Outer Membrane Vesicles (OMVs) of Pseudomonas aeruginosa Provide Passive Resistance but Not Sensitization to LPS-Specific Phages. Viruses 2022, 14, 121. [Google Scholar] [CrossRef]
- Singer, Z.S.; Ambrose, P.M.; Danino, T.; Rice, C.M. Quantitative measurements of early alphaviral replication dynamics in single cells reveals the basis for superinfection exclusion. Cell Syst. 2021, 12, 210–219.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Sun, R.; Guo, Q.; Zhang, S.; Meulia, T.; Halfmann, R.; Li, D.; Qu, F. A self-perpetuating repressive state of a viral replication protein blocks superinfection by the same virus. PLoS Pathog. 2017, 13, e1006253. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.Y.; Jang, H.J.; Bae, H.W.; Cho, Y.H. A phage protein that inhibits the bacterial ATPase required for type IV pilus assembly. Proc. Natl. Acad. Sci. USA 2014, 111, 11503–11508. [Google Scholar] [CrossRef]
- Shi, K.; Oakland, J.T.; Kurniawan, F.; Moeller, N.H.; Banerjee, S.; Aihara, H. Structural basis of superinfection exclusion by bacteriophage T4 Spackle. Commun. Biol. 2020, 3, 691. [Google Scholar] [CrossRef]
- Kirchberger, P.C.; Martinez, Z.A.; Luker, L.J.; Ochman, H. Defensive hypervariable regions confer superinfection exclusion in microviruses. Proc. Natl. Acad. Sci. USA 2021, 118, e2102786118. [Google Scholar] [CrossRef]
- Ranade, K.; Poteete, A.R. A switch in translation mediated by an antisense RNA. Genes Dev. 1993, 7, 1498–1507. [Google Scholar] [CrossRef]
- Kobayashi, I. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001, 29, 3742–3756. [Google Scholar] [CrossRef]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P. The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014, 42, 10618–10631. [Google Scholar] [CrossRef]
- Huang, X.; Wang, J.; Li, J.; Liu, Y.; Liu, X.; Li, Z.; Kurniyati, K.; Deng, Y.; Wang, G.; Ralph, J.D.; et al. Prevalence of phase variable epigenetic invertons among host-associated bacteria. Nucleic Acids Res. 2020, 48, 11468–11485. [Google Scholar] [CrossRef] [PubMed]
- Pleška, M.; Qian, L.; Okura, R.; Bergmiller, T.; Wakamoto, Y.; Kussell, E.; Guet, C.C. Bacterial Autoimmunity Due to a Restriction-Modification System. Curr. Biol. 2016, 26, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Morozova, N.; Sabantsev, A.; Bogdanova, E.; Fedorova, Y.; Maikova, A.; Vedyaykin, A.; Rodic, A.; Djordjevic, M.; Khodorkovskii, M.; Severinov, K. Temporal dynamics of methyltransferase and restriction endonuclease accumulation in individual cells after introducing a restriction-modification system. Nucleic Acids Res. 2016, 44, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Rezulak, M.; Borsuk, I.; Mruk, I. Natural C-independent expression of restriction endonuclease in a C protein-associated restriction-modification system. Nucleic Acids Res. 2016, 44, 2646–2660. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.; Werbowy, O.; Wons, E.; Dersch, S.; Hinrichs, R.; Graumann, P.L.; Mruk, I. Regulator-dependent temporal dynamics of a restriction-modification system’s gene expression upon entering new host cells: Single-cell and population studies. Nucleic Acids Res. 2021, 49, 3826–3840. [Google Scholar] [CrossRef] [PubMed]
- Mruk, I.; Blumenthal, R.M. Real-time kinetics of restriction-modification gene expression after entry into a new host cell. Nucleic Acids Res. 2008, 36, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chesne, M.T.; Terns, R.M.; Terns, M.P. Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res. 2015, 43, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, M.È.; Villion, M.; Magadán, A.H.; Moineau, S. CRISPR-Cas and restriction-modification systems are compatible and increase phage resistance. Nat. Commun. 2013, 4, 2087. [Google Scholar] [CrossRef]
- Dong, Y.; Ma, K.; Cao, Q.; Huang, H.; Nie, M.; Liu, G.; Jiang, M.; Lu, C.; Liu, Y. CRISPR-dependent endogenous gene regulation is required for virulence in piscine Streptococcus agalactiae. Emerg. Microbes Infect. 2021, 10, 2113–2124. [Google Scholar] [CrossRef]
- Soto-Perez, P.; Bisanz, J.E.; Berry, J.D.; Lam, K.N.; Bondy-Denomy, J.; Turnbaugh, P.J. CRISPR-Cas System of a Prevalent Human Gut Bacterium Reveals Hyper-targeting against Phages in a Human Virome Catalog. Cell Host Microbe 2019, 26, 325–335.e5. [Google Scholar] [CrossRef] [PubMed]
- Faure, G.; Shmakov, S.A.; Yan, W.X.; Cheng, D.R.; Scott, D.A.; Peters, J.E.; Makarova, K.S.; Koonin, E.V. CRISPR-Cas in mobile genetic elements: Counter-defence and beyond. Nat. Rev. Microbiol. 2019, 17, 513–525. [Google Scholar] [CrossRef]
- Modell, J.W.; Jiang, W.; Marraffini, L.A. CRISPR-Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature 2017, 544, 101–104. [Google Scholar] [CrossRef]
- van Houte, S.; Ekroth, A.K.; Broniewski, J.M.; Chabas, H.; Ashby, B.; Bondy-Denomy, J.; Gandon, S.; Boots, M.; Paterson, S.; Buckling, A.; et al. The diversity-generating benefits of a prokaryotic adaptive immune system. Nature 2016, 532, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Sontheimer, E.J. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 2010, 463, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, F.L.; Walinga, H.; Dutilh, B.E.; Brouns, S. Prophages are associated with extensive CRISPR-Cas auto-immunity. Nucleic Acids Res. 2020, 48, 12074–12084. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Leon, L.M.; Mendoza, S.D.; Bondy-Denomy, J. How bacteria control the CRISPR-Cas arsenal. Curr. Opin. Microbiol. 2018, 42, 87–95. [Google Scholar] [CrossRef]
- Zhang, N.; Jing, X.; Liu, Y.; Chen, M.; Zhu, X.; Jiang, J.; Wang, H.; Li, X.; Hao, P. Interfering with retrotransposition by two types of CRISPR effectors: Cas12a and Cas13a. Cell Discov. 2020, 6, 30. [Google Scholar] [CrossRef]
- Stella, S.; Alcón, P.; Montoya, G. Class 2 CRISPR-Cas RNA-guided endonucleases: Swiss Army knives of genome editing. Nat. Struct. Mol. Biol. 2017, 24, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Meaden, S.; Biswas, A.; Arkhipova, K.; Morales, S.E.; Dutilh, B.E.; Westra, E.R.; Fineran, P.C. High viral abundance and low diversity are associated with increased CRISPR-Cas prevalence across microbial ecosystems. Curr. Biol. 2022, 32, 220–227.e5. [Google Scholar] [CrossRef] [PubMed]
- Alseth, E.O.; Pursey, E.; Luján, A.M.; McLeod, I.; Rollie, C.; Westra, E.R. Bacterial biodiversity drives the evolution of CRISPR-based phage resistance. Nature 2019, 574, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Heler, R.; Wright, A.V.; Vucelja, M.; Doudna, J.A.; Marraffini, L.A. Spacer Acquisition Rates Determine the Immunological Diversity of the Type II CRISPR-Cas Immune Response. Cell Host Microbe 2019, 25, 242–249.e3. [Google Scholar] [CrossRef]
- Dimitriu, T.; Kurilovich, E.; Łapińska, U.; Severinov, K.; Pagliara, S.; Szczelkun, M.D.; Westra, E.R. Bacteriostatic antibiotics promote CRISPR-Cas adaptive immunity by enabling increased spacer acquisition. Cell Host Microbe 2022, 30, 31–40.e5. [Google Scholar] [CrossRef]
- Strotskaya, A.; Savitskaya, E.; Metlitskaya, A.; Morozova, N.; Datsenko, K.A.; Semenova, E.; Severinov, K. The action of Escherichia coli CRISPR-Cas system on lytic bacteriophages with different lifestyles and development strategies. Nucleic Acids Res. 2017, 45, 1946–1957. [Google Scholar] [CrossRef]
- Watson, B.; Vercoe, R.B.; Salmond, G.; Westra, E.R.; Staals, R.; Fineran, P.C. Type I-F CRISPR-Cas resistance against virulent phages results in abortive infection and provides population-level immunity. Nat. Commun. 2019, 10, 5526. [Google Scholar] [CrossRef]
- Varble, A.; Marraffini, L.A. Three New Cs for CRISPR: Collateral, Communicate, Cooperate. Trends Genet. 2019, 35, 446–456. [Google Scholar] [CrossRef]
- Wen, Y.; Behiels, E.; Devreese, B. Toxin-Antitoxin systems: Their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 2014, 70, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, P.; Sandhya, S.; Nadig, K.; Paul, S.; Srinivasan, N.; Rothweiler, U.; Singh, M. Identification, functional characterization, assembly and structure of ToxIN type III toxin-antitoxin complex from E. coli. Nucleic Acids Res. 2022, 50, 1687–1700. [Google Scholar] [CrossRef]
- Guegler, C.K.; Laub, M.T. Shutoff of host transcription triggers a toxin-antitoxin system to cleave phage RNA and abort infection. Mol. Cell 2021, 81, 2361–2373.e9. [Google Scholar] [CrossRef] [PubMed]
- Brantl, S.; Jahn, N. sRNAs in bacterial type I and type III toxin-antitoxin systems. FEMS Microbiol. Rev. 2015, 39, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Quiroga, C.; Chen, Q.; McAnulty, M.J.; Benedik, M.J.; Wood, T.K.; Wang, X. RalR (a DNase) and RalA (a small RNA) form a type I toxin-antitoxin system in Escherichia coli. Nucleic Acids Res. 2014, 42, 6448–6462. [Google Scholar] [CrossRef]
- Song, S.; Wood, T.K. A Primary Physiological Role of Toxin/Antitoxin Systems Is Phage Inhibition. Front. Microbiol. 2020, 11, 1895. [Google Scholar] [CrossRef]
- Jurėnas, D.; Fraikin, N.; Goormaghtigh, F.; De Bruyn, P.; Vandervelde, A.; Zedek, S.; Jové, T.; Charlier, D.; Loris, R.; Van Melderen, L. Bistable Expression of a Toxin-Antitoxin System Located in a Cryptic Prophage of Escherichia coli O157:H7. mBio 2021, 12, e0294721. [Google Scholar] [CrossRef]
- Culviner, P.H.; Nocedal, I.; Fortune, S.M.; Laub, M.T. Global Analysis of the Specificities and Targets of Endoribonucleases from Escherichia coli Toxin-Antitoxin Systems. mBio 2021, 12, e0201221. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, G.; Charlier, D.; Wilmaerts, D.; Michiels, J.; Loris, R. Alternative dimerization is required for activity and inhibition of the HEPN ribonuclease RnlA. Nucleic Acids Res 2021, 49, 7164–7178. [Google Scholar] [CrossRef]
- Rao, F.; Short, F.L.; Voss, J.E.; Blower, T.R.; Orme, A.L.; Whittaker, T.E.; Luisi, B.F.; Salmond, G.P. Co-evolution of quaternary organization and novel RNA tertiary interactions revealed in the crystal structure of a bacterial protein-RNA toxin-antitoxin system. Nucleic Acids Res. 2015, 43, 9529–9540. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Short, F.L.; Rao, F.; Mizuguchi, K.; Pei, X.Y.; Fineran, P.C.; Luisi, B.F.; Salmond, G.P. Identification and classification of bacterial Type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 2012, 40, 6158–6173. [Google Scholar] [CrossRef] [PubMed]
- Dy, R.L.; Przybilski, R.; Semeijn, K.; Salmond, G.P.; Fineran, P.C. A widespread bacteriophage abortive infection system functions through a Type IV toxin-antitoxin mechanism. Nucleic Acids Res. 2014, 42, 4590–4605. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lord, D.M.; Cheng, H.Y.; Osbourne, D.O.; Hong, S.H.; Sanchez-Torres, V.; Quiroga, C.; Zheng, K.; Herrmann, T.; Peti, W.; et al. A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 2012, 8, 855–861. [Google Scholar] [CrossRef]
- Aakre, C.D.; Phung, T.N.; Huang, D.; Laub, M.T. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the β sliding clamp. Mol. Cell 2013, 52, 617–628. [Google Scholar] [CrossRef]
- Maikova, A.; Peltier, J.; Boudry, P.; Hajnsdorf, E.; Kint, N.; Monot, M.; Poquet, I.; Martin-Verstraete, I.; Dupuy, B.; Soutourina, O. Discovery of new type I toxin-antitoxin systems adjacent to CRISPR arrays in Clostridium difficile. Nucleic Acids Res. 2018, 46, 4733–4751. [Google Scholar] [CrossRef]
- Govande, A.A.; Duncan-Lowey, B.; Eaglesham, J.B.; Whiteley, A.T.; Kranzusch, P.J. Molecular basis of CD-NTase nucleotide selection in CBASS anti-phage defense. Cell Rep. 2021, 35, 109206. [Google Scholar] [CrossRef]
- Fatma, S.; Chakravarti, A.; Zeng, X.; Huang, R.H. Molecular mechanisms of the CdnG-Cap5 antiphage defense system employing 3’,2’-cGAMP as the second messenger. Nat. Commun. 2021, 12, 6381. [Google Scholar] [CrossRef]
- Duncan-Lowey, B.; McNamara-Bordewick, N.K.; Tal, N.; Sorek, R.; Kranzusch, P.J. Effector-mediated membrane disruption controls cell death in CBASS antiphage defense. Mol. Cell 2021, 81, 5039–5051.e5. [Google Scholar] [CrossRef]
- Lau, R.K.; Ye, Q.; Birkholz, E.A.; Berg, K.R.; Patel, L.; Mathews, I.T.; Watrous, J.D.; Ego, K.; Whiteley, A.T.; Lowey, B.; et al. Structure and Mechanism of a Cyclic Trinucleotide-Activated Bacterial Endonuclease Mediating Bacteriophage Immunity. Mol. Cell 2020, 77, 723–733.e6. [Google Scholar] [CrossRef]
- Lowey, B.; Whiteley, A.T.; Keszei, A.; Morehouse, B.R.; Mathews, I.T.; Antine, S.P.; Cabrera, V.J.; Kashin, D.; Niemann, P.; Jain, M.; et al. CBASS Immunity Uses CARF-Related Effectors to Sense 3’-5’- and 2’-5’-Linked Cyclic Oligonucleotide Signals and Protect Bacteria from Phage Infection. Cell 2020, 182, 38–49.e17. [Google Scholar] [CrossRef]
- Millman, A.; Melamed, S.; Amitai, G.; Sorek, R. Diversity and classification of cyclic-oligonucleotide-based anti-phage signalling systems. Nat. Microbiol. 2020, 5, 1608–1615. [Google Scholar] [CrossRef]
- Ye, Q.; Lau, R.K.; Mathews, I.T.; Birkholz, E.A.; Watrous, J.D.; Azimi, C.S.; Pogliano, J.; Jain, M.; Corbett, K.D. HORMA Domain Proteins and a Trip13-like ATPase Regulate Bacterial cGAS-like Enzymes to Mediate Bacteriophage Immunity. Mol. Cell 2020, 77, 709–722.e7. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.; How, K.Y.; Tee, K.K.; Chan, K.G. Characterization and Transcriptome Studies of Autoinducer Synthase Gene from Multidrug Resistant Acinetobacter baumannii Strain 863. Genes 2019, 10, 282. [Google Scholar] [CrossRef] [PubMed]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Mumford, R.; Friman, V.P. Bacterial competition and quorum-sensing signalling shape the eco-evolutionary outcomes of model in vitro phage therapy. Evol. Appl. 2017, 10, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ni, Z.; Tang, J.; Ding, Y.; Wang, X.; Li, F. The abaI/abaR Quorum Sensing System Effects on Pathogenicity in Acinetobacter baumannii. Front. Microbiol. 2021, 12, 679241. [Google Scholar] [CrossRef]
- Høyland-Kroghsbo, N.M.; Paczkowski, J.; Mukherjee, S.; Broniewski, J.; Westra, E.; Bondy-Denomy, J.; Bassler, B.L. Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proc. Natl. Acad. Sci. USA 2017, 114, 131–135. [Google Scholar] [CrossRef]
- Semenova, E.; Severinov, K. Come Together: CRISPR-Cas Immunity Senses the Quorum. Mol. Cell 2016, 64, 1013–1015. [Google Scholar] [CrossRef]
- Patterson, A.G.; Jackson, S.A.; Taylor, C.; Evans, G.B.; Salmond, G.; Przybilski, R.; Staals, R.; Fineran, P.C. Quorum Sensing Controls Adaptive Immunity through the Regulation of Multiple CRISPR-Cas Systems. Mol. Cell 2016, 64, 1102–1108. [Google Scholar] [CrossRef]
- Høyland-Kroghsbo, N.M.; Maerkedahl, R.B.; Svenningsen, S.L. A quorum-sensing-induced bacteriophage defense mechanism. mBio 2013, 4, e00362. [Google Scholar] [CrossRef]
- Saipriya, K.; Swathi, C.H.; Ratnakar, K.S.; Sritharan, V. Quorum-sensing system in Acinetobacter baumannii: A potential target for new drug development. J. Appl. Microbiol. 2020, 128, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Hansen, M.F.; de Carvalho, L.N.; Røder, H.L.; Burmølle, M.; Middelboe, M.; Svenningsen, S.L. High cell densities favor lysogeny: Induction of an H20 prophage is repressed by quorum sensing and enhances biofilm formation in Vibrio anguillarum. ISME J. 2020, 14, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Andrews, B.; Fields, S. Balance between promiscuity and specificity in phage λ host range. ISME J. 2021, 15, 2195–2205. [Google Scholar] [CrossRef]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and Function of Phage Encoded Depolymerases. Front. Microbiol. 2019, 10, 2949. [Google Scholar] [CrossRef]
- Liu, Y.; Mi, Z.; Mi, L.; Huang, Y.; Li, P.; Liu, H.; Yuan, X.; Niu, W.; Jiang, N.; Bai, C.; et al. Identification and characterization of capsule depolymerase Dpo48 from Acinetobacter baumannii phage IME200. PeerJ 2019, 7, e61732. [Google Scholar] [CrossRef]
- Liu, Y.; Leung, S.; Huang, Y.; Guo, Y.; Jiang, N.; Li, P.; Chen, J.; Wang, R.; Bai, C.; Mi, Z.; et al. Identification of Two Depolymerases From Phage IME205 and Their Antivirulent Functions on K47 Capsule of Klebsiella pneumoniae. Front. Microbiol. 2020, 11, 218. [Google Scholar] [CrossRef]
- Tkhilaishvili, T.; Lombardi, L.; Klatt, A.B.; Trampuz, A.; Di Luca, M. Bacteriophage Sb-1 enhances antibiotic activity against biofilm, degrades exopolysaccharide matrix and targets persisters of Staphylococcus aureus. Int. J. Antimicrob. Agents 2018, 52, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Mao, J.; Xie, J. Bacteriophage polysaccharide depolymerases and biomedical applications. BioDrugs 2014, 28, 265–274. [Google Scholar] [CrossRef]
- Karambelkar, S.; Udupa, S.; Gowthami, V.N.; Ramachandra, S.G.; Swapna, G.; Nagaraja, V. Emergence of a novel immune-evasion strategy from an ancestral protein fold in bacteriophage Mu. Nucleic Acids Res. 2020, 48, 5294–5305. [Google Scholar] [CrossRef] [PubMed]
- Kot, W.; Olsen, N.S.; Nielsen, T.K.; Hutinet, G.; de Crécy-Lagard, V.; Cui, L.; Dedon, P.C.; Carstens, A.B.; Moineau, S.; Swairjo, M.A.; et al. Detection of preQ0 deazaguanine modifications in bacteriophage CAjan DNA using Nanopore sequencing reveals same hypermodification at two distinct DNA motifs. Nucleic Acids Res. 2020, 48, 10383–10396. [Google Scholar] [CrossRef]
- Hutinet, G.; Kot, W.; Cui, L.; Hillebrand, R.; Balamkundu, S.; Gnanakalai, S.; Neelakandan, R.; Carstens, A.B.; Fa Lui, C.; Tremblay, D.; et al. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 2019, 10, 5442. [Google Scholar] [CrossRef]
- Atanasiu, C.; Su, T.J.; Sturrock, S.S.; Dryden, D.T. Interaction of the ocr gene 0.3 protein of bacteriophage T7 with EcoKI restriction/modification enzyme. Nucleic Acids Res. 2002, 30, 3936–3944. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.A.; Stephanou, A.S.; Kanwar, N.; Dawson, A.; Cooper, L.P.; Chen, K.; Nutley, M.; Cooper, A.; Blakely, G.W.; Dryden, D.T. Exploring the DNA mimicry of the Ocr protein of phage T7. Nucleic Acids Res. 2012, 40, 8129–8143. [Google Scholar] [CrossRef]
- Isaev, A.; Drobiazko, A.; Sierro, N.; Gordeeva, J.; Yosef, I.; Qimron, U.; Ivanov, N.V.; Severinov, K. Phage T7 DNA mimic protein Ocr is a potent inhibitor of BREX defence. Nucleic Acids Res. 2020, 48, 5397–5406. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Kotta-Loizou, I.; Jovanovic, M.; Liu, X.; Dryden, D.T.; Buck, M.; Zhang, X. Structural basis of transcription inhibition by the DNA mimic protein Ocr of bacteriophage T7. Elife 2020, 9, e52125. [Google Scholar] [CrossRef]
- Malone, L.M.; Warring, S.L.; Jackson, S.A.; Warnecke, C.; Gardner, P.P.; Gumy, L.F.; Fineran, P.C. A jumbo phage that forms a nucleus-like structure evades CRISPR-Cas DNA targeting but is vulnerable to type III RNA-based immunity. Nat. Microbiol. 2020, 5, 48–55. [Google Scholar] [CrossRef]
- Mendoza, S.D.; Nieweglowska, E.S.; Govindarajan, S.; Leon, L.M.; Berry, J.D.; Tiwari, A.; Chaikeeratisak, V.; Pogliano, J.; Agard, D.A.; Bondy-Denomy, J. A bacteriophage nucleus-like compartment shields DNA from CRISPR nucleases. Nature 2020, 577, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Vlot, M.; Houkes, J.; Lochs, S.; Swarts, D.C.; Zheng, P.; Kunne, T.; Mohanraju, P.; Anders, C.; Jinek, M.; van der Oost, J.; et al. Bacteriophage DNA glucosylation impairs target DNA binding by type I and II but not by type V CRISPR-Cas effector complexes. Nucleic Acids Res. 2018, 46, 873–885. [Google Scholar] [CrossRef]
- Jackson, S.A.; Birkholz, N.; Malone, L.M.; Fineran, P.C. Imprecise Spacer Acquisition Generates CRISPR-Cas Immune Diversity through Primed Adaptation. Cell Host Microbe 2019, 25, 250–260.e4. [Google Scholar] [CrossRef]
- Gabel, C.; Li, Z.; Zhang, H.; Chang, L. Structural basis for inhibition of the type I-F CRISPR-Cas surveillance complex by AcrIF4, AcrIF7 and AcrIF14. Nucleic Acids Res. 2021, 49, 584–594. [Google Scholar] [CrossRef]
- Kim, I.; Koo, J.; An, S.Y.; Hong, S.; Ka, D.; Kim, E.H.; Bae, E.; Suh, J.Y. Structural and mechanistic insights into the CRISPR inhibition of AcrIF7. Nucleic Acids Res. 2020, 48, 9959–9968. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.T.; Trost, C.N.; Müller-Esparza, H.; Randau, L.; Davidson, A.R. Anti-CRISPR AcrIF9 functions by inducing the CRISPR-Cas complex to bind DNA non-specifically. Nucleic Acids Res. 2021, 49, 3381–3393. [Google Scholar] [CrossRef]
- Niu, Y.; Yang, L.; Gao, T.; Dong, C.; Zhang, B.; Yin, P.; Hopp, A.K.; Li, D.; Gan, R.; Wang, H.; et al. A Type I-F Anti-CRISPR Protein Inhibits the CRISPR-Cas Surveillance Complex by ADP-Ribosylation. Mol. Cell 2020, 80, 512–524.e5. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Garcia, B.; Strum, S.; Du, M.; Rollins, M.F.; Hidalgo-Reyes, Y.; Wiedenheft, B.; Maxwell, K.L.; Davidson, A.R. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 2015, 526, 136–139. [Google Scholar] [CrossRef]
- Osuna, B.A.; Karambelkar, S.; Mahendra, C.; Christie, K.A.; Garcia, B.; Davidson, A.R.; Kleinstiver, B.P.; Kilcher, S.; Bondy-Denomy, J. Listeria Phages Induce Cas9 Degradation to Protect Lysogenic Genomes. Cell Host Microbe 2020, 28, 31–40.e9. [Google Scholar] [CrossRef]
- Dong, D.; Guo, M.; Wang, S.; Zhu, Y.; Wang, S.; Xiong, Z.; Yang, J.; Xu, Z.; Huang, Z. Structural basis of CRISPR-SpyCas9 inhibition by an anti-CRISPR protein. Nature 2017, 546, 436–439. [Google Scholar] [CrossRef]
- Liu, L.; Yin, M.; Wang, M.; Wang, Y. Phage AcrIIA2 DNA Mimicry: Structural Basis of the CRISPR and Anti-CRISPR Arms Race. Mol. Cell 2019, 73, 611–620.e3. [Google Scholar] [CrossRef]
- Yang, H.; Patel, D.J. Inhibition Mechanism of an Anti-CRISPR Suppressor AcrIIA4 Targeting SpyCas9. Mol. Cell 2017, 67, 117–127.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, Y.; Lu, Z.; Huang, Z. Structural basis of Staphylococcus aureus Cas9 inhibition by AcrIIA14. Nucleic Acids Res. 2021, 49, 6587–6595. [Google Scholar] [CrossRef] [PubMed]
- Pawluk, A.; Amrani, N.; Zhang, Y.; Garcia, B.; Hidalgo-Reyes, Y.; Lee, J.; Edraki, A.; Shah, M.; Sontheimer, E.J.; Maxwell, K.L.; et al. Naturally Occurring Off-Switches for CRISPR-Cas9. Cell 2016, 167, 1829–1838.e9. [Google Scholar] [CrossRef] [PubMed]
- Thavalingam, A.; Cheng, Z.; Garcia, B.; Huang, X.; Shah, M.; Sun, W.; Wang, M.; Harrington, L.; Hwang, S.; Hidalgo-Reyes, Y.; et al. Inhibition of CRISPR-Cas9 ribonucleoprotein complex assembly by anti-CRISPR AcrIIC2. Nat. Commun. 2019, 10, 2806. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.B.; Doxzen, K.W.; Ma, E.; Liu, J.J.; Knott, G.J.; Edraki, A.; Garcia, B.; Amrani, N.; Chen, J.S.; Cofsky, J.C.; et al. A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell 2017, 170, 1224–1233.e15. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yang, J.; Cheng, Z.; Amrani, N.; Liu, C.; Wang, K.; Ibraheim, R.; Edraki, A.; Huang, X.; Wang, M.; et al. Structures of Neisseria meningitidis Cas9 Complexes in Catalytically Poised and Anti-CRISPR-Inhibited States. Mol. Cell 2019, 76, 938–952.e5. [Google Scholar] [CrossRef]
- Athukoralage, J.S.; McMahon, S.A.; Zhang, C.; Grüschow, S.; Graham, S.; Krupovic, M.; Whitaker, R.J.; Gloster, T.M.; White, M.F. An anti-CRISPR viral ring nuclease subverts type III CRISPR immunity. Nature 2020, 577, 572–575. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Z.; Daczkowski, C.M.; Gabel, C.; Mesecar, A.D.; Chang, L. Structural Basis for the Inhibition of CRISPR-Cas12a by Anti-CRISPR Proteins. Cell Host Microbe 2019, 25, 815–826.e4. [Google Scholar] [CrossRef]
- Meeske, A.J.; Jia, N.; Cassel, A.K.; Kozlova, A.; Liao, J.; Wiedmann, M.; Patel, D.J.; Marraffini, L.A. A phage-encoded anti-CRISPR enables complete evasion of type VI-A CRISPR-Cas immunity. Science 2020, 369, 54–59. [Google Scholar] [CrossRef]
- Lin, P.; Qin, S.; Pu, Q.; Wang, Z.; Wu, Q.; Gao, P.; Schettler, J.; Guo, K.; Li, R.; Li, G.; et al. CRISPR-Cas13 Inhibitors Block RNA Editing in Bacteria and Mammalian Cells. Mol. Cell 2020, 78, 850–861.e5. [Google Scholar] [CrossRef]
- Chevallereau, A.; Meaden, S.; Fradet, O.; Landsberger, M.; Maestri, A.; Biswas, A.; Gandon, S.; van Houte, S.; Westra, E.R. Exploitation of the Cooperative Behaviors of Anti-CRISPR Phages. Cell Host Microbe 2020, 27, 189–198.e6. [Google Scholar] [CrossRef]
- Birkholz, N.; Fagerlund, R.D.; Smith, L.M.; Jackson, S.A.; Fineran, P.C. The autoregulator Aca2 mediates anti-CRISPR repression. Nucleic Acids Res. 2019, 47, 9658–9665. [Google Scholar] [CrossRef]
- Stanley, S.Y.; Borges, A.L.; Chen, K.H.; Swaney, D.L.; Krogan, N.J.; Bondy-Denomy, J.; Davidson, A.R. Anti-CRISPR-Associated Proteins Are Crucial Repressors of Anti-CRISPR Transcription. Cell 2019, 178, 1452–1464.e13. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Rodríguez, A.; García, P. Phage or foe: An insight into the impact of viral predation on microbial communities. ISME J. 2018, 12, 1171–1179. [Google Scholar] [CrossRef]
- Silpe, J.E.; Bassler, B.L. A Host-Produced Quorum-Sensing Autoinducer Controls a Phage Lysis-Lysogeny Decision. Cell 2019, 176, 268–280.e13. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.L. Phages Tune in to Host Cell Quorum Sensing. Cell 2019, 176, 7–8. [Google Scholar] [CrossRef]
- Harms, A.; Diard, M. Crowd Controlled-Host Quorum Sensing Drives Phage Decision. Cell Host Microbe 2019, 25, 179–181. [Google Scholar] [CrossRef]
- Lemos Rocha, L.F.; Blokesch, M. A Vibriophage Takes Antirepression to the Next Level. Cell Host Microbe 2020, 27, 493–495. [Google Scholar] [CrossRef] [PubMed]
- van Kessel, J.C.; Mukherjee, S. Another battle won in the phage-host arms race: Pseudomonas phage blocks quorum sensing regulator LasR. Mol. Cell 2021, 81, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Taylor, V.L.; Bona, D.; Tsao, Y.; Stanley, S.Y.; Pimentel-Elardo, S.M.; McCallum, M.; Bondy-Denomy, J.; Howell, P.L.; Nodwell, J.R.; et al. A phage-encoded anti-activator inhibits quorum sensing in Pseudomonas aeruginosa. Mol. Cell 2021, 81, 571–583.e6. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, H.; Zimmermann-Kogadeeva, M.; Zimmermann, M.; Sauer, U.; De Smet, J.; Muchez, L.; Lissens, M.; Staes, I.; Voet, M.; Wagemans, J.; et al. Metabolic reprogramming of Pseudomonas aeruginosa by phage-based quorum sensing modulation. Cell Rep. 2022, 38, 110372. [Google Scholar] [CrossRef]
- Ning, B.; Yu, T.; Zhang, S.; Huang, Z.; Tian, D.; Lin, Z.; Niu, A.; Golden, N.; Hensley, K.; Threeton, B.; et al. A smartphone-read ultrasensitive and quantitative saliva test for COVID-19. Sci. Adv. 2021, 7, eabe3703. [Google Scholar] [CrossRef]
- Roberts, A.; Barrangou, R. Applications of CRISPR-Cas systems in lactic acid bacteria. FEMS Microbiol. Rev. 2020, 44, 523–537. [Google Scholar] [CrossRef]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of biofilm formation by T7 bacteriophages producing quorum-quenching enzymes. Appl. Environ. Microbiol. 2014, 80, 5340–5348. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.A.; Magill, D.; Millen, A.; Horvath, P.; Fremaux, C. Dairy lactococcal and streptococcal phage-host interactions: An industrial perspective in an evolving phage landscape. FEMS Microbiol. Rev. 2020, 44, 909–932. [Google Scholar] [CrossRef] [PubMed]
- Egido, J.E.; Costa, A.R.; Aparicio-Maldonado, C.; Haas, P.J.; Brouns, S. Mechanisms and clinical importance of bacteriophage resistance. FEMS Microbiol. Rev. 2021, 46, fuab048. [Google Scholar] [CrossRef]
- Arya, S.; Todman, H.; Baker, M.; Hooton, S.; Millard, A.; Kreft, J.U.; Hobman, J.L.; Stekel, D.J. A generalised model for generalised transduction: The importance of co-evolution and stochasticity in phage mediated antimicrobial resistance transfer. FEMS Microbiol. Ecol. 2020, 96, fiaa100. [Google Scholar] [CrossRef] [PubMed]
- Cano, E.J.; Caflisch, K.M.; Bollyky, P.L.; Van Belleghem, J.D.; Patel, R.; Fackler, J.; Brownstein, M.J.; Horne, B.; Biswas, B.; Henry, M.; et al. Phage Therapy for Limb-threatening Prosthetic Knee Klebsiella pneumoniae Infection: Case Report and In Vitro Characterization of Anti-biofilm Activity. Clin. Infect. Dis. 2021, 73, e144–e151. [Google Scholar] [CrossRef] [PubMed]
- Torres-Barceló, C. Phage Therapy Faces Evolutionary Challenges. Viruses 2018, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Vaitekenas, A.; Tai, A.S.; Ramsay, J.P.; Stick, S.M.; Kicic, A. Pseudomonas aeruginosa Resistance to Bacteriophages and Its Prevention by Strategic Therapeutic Cocktail Formulation. Antibiotics 2021, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Schilcher, K.; Horswill, A.R. Staphylococcal Biofilm Development: Structure, Regulation, and Treatment Strategies. Microbiol. Mol. Biol. Rev. 2020, 84, e00026-19. [Google Scholar] [CrossRef] [PubMed]
- Hesse, S.; Rajaure, M.; Wall, E.; Johnson, J.; Bliskovsky, V.; Gottesman, S.; Adhya, S. Phage Resistance in Multidrug-Resistant Klebsiella pneumoniae ST258 Evolves via Diverse Mutations That Culminate in Impaired Adsorption. mBio 2020, 11, e02530-19. [Google Scholar] [CrossRef]
- Davies, E.V.; James, C.E.; Williams, D.; O’Brien, S.; Fothergill, J.L.; Haldenby, S.; Paterson, S.; Winstanley, C.; Brockhurst, M.A. Temperate phages both mediate and drive adaptive evolution in pathogen biofilms. Proc. Natl. Acad. Sci. USA 2016, 113, 8266–8271. [Google Scholar] [CrossRef]
- Irwin, N.; Pittis, A.A.; Richards, T.A.; Keeling, P.J. Systematic evaluation of horizontal gene transfer between eukaryotes and viruses. Nat. Microbiol. 2022, 7, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Maciá, M.D.; Oliver, A.; Schachar, I.; Buckling, A. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 2007, 450, 1079–1081. [Google Scholar] [CrossRef]
- Paterson, S.; Vogwill, T.; Buckling, A.; Benmayor, R.; Spiers, A.J.; Thomson, N.R.; Quail, M.; Smith, F.; Walker, D.; Libberton, B.; et al. Antagonistic coevolution accelerates molecular evolution. Nature 2010, 464, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenberghe, J.; Santamaría, R.I.; Bustos, P.; Juárez, S.; Ducci, M.A.; Figueroa Fleming, T.; Etcheverry, A.V.; González, V. Spatial patterns in phage-Rhizobium coevolutionary interactions across regions of common bean domestication. ISME J. 2021, 15, 2092–2106. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Anti-Phage Strategies | Description of the Strategies |
| Blocking phage adsorption | Loss or structural change of receptors. |
| Mask phage receptors by physical barriers such as outer membrane vesicles (OMVs), masking proteins or extracellular polymeric substances (EPS). | |
| Blocking phage injection | Avoid bacterial re-infestation by another identical or highly similar phage after infection by a lysogenic phage through superinfection exclusion (Sie). |
| Interfering with phage replication | Cleavage of phosphodiester bonds of unmethylated phage DNA by the restricting modification (R-M) systems. |
| Recognition and cleavage of phage DNA/RNA by the effector complex consisting of Cas protein and guide RNA (gRNA) of CRISPR-Cas system. | |
| Bacteria inhibit their metabolism through abortive infection (Abi) leading to their own growth arrest or death, thus avoiding the maturation and release of phages. | |
| Quorum sensing (QS) | Synergistic CRISPR-Cas systems stimulate the expression of cas genes and promote recognition and cleavage of target genes under conditions of high population density. |
| Reduce the expression of phage receptors or increase biofilm production to mask phage receptors under conditions of low cell density. | |
| Anti-defense strategies of phages | Description of the strategies |
| Regaining the ability to identify and adsorb host | Mutate the receptor binding proteins (RBPs). |
| Degrade EPS by depolymerase. | |
| Anti-defense strategies for R-M systems | Modify the genes of phages. Evolve to overcome classical restriction (Ocr) protein and the restriction of DNA A (ArdA) protein that can inhibit the R-M complex. |
| Anti-defense strategies for CRISPR-Cas systems | Construct nucleus-like compartments to shield nuclease. |
| Mutate the target sequence to block the recognition of effector complexes. | |
| Through anti-CRISPR (Acr) protein to inhibit recognition or cleaving of foreign nucleic acids by effector complexes. | |
| Anti-defense strategies for QS system | Expression of anti-repressor that can bind cI repressor, thereby allowing the phage to enter the lysis cycle. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Fan, H.; Tong, Y. Unveil the Secret of the Bacteria and Phage Arms Race. Int. J. Mol. Sci. 2023, 24, 4363. https://doi.org/10.3390/ijms24054363
Wang Y, Fan H, Tong Y. Unveil the Secret of the Bacteria and Phage Arms Race. International Journal of Molecular Sciences. 2023; 24(5):4363. https://doi.org/10.3390/ijms24054363
Chicago/Turabian StyleWang, Yuer, Huahao Fan, and Yigang Tong. 2023. "Unveil the Secret of the Bacteria and Phage Arms Race" International Journal of Molecular Sciences 24, no. 5: 4363. https://doi.org/10.3390/ijms24054363
APA StyleWang, Y., Fan, H., & Tong, Y. (2023). Unveil the Secret of the Bacteria and Phage Arms Race. International Journal of Molecular Sciences, 24(5), 4363. https://doi.org/10.3390/ijms24054363