
Lacrimal Gland Epithelial Cells Shape Immune Responses through the Modulation of Inflammasomes and Lipid Metabolism

 , ,
, ,  , ,
, ,  ,
,
Abstract
1. Introduction
2. Methods
2.1. Mice
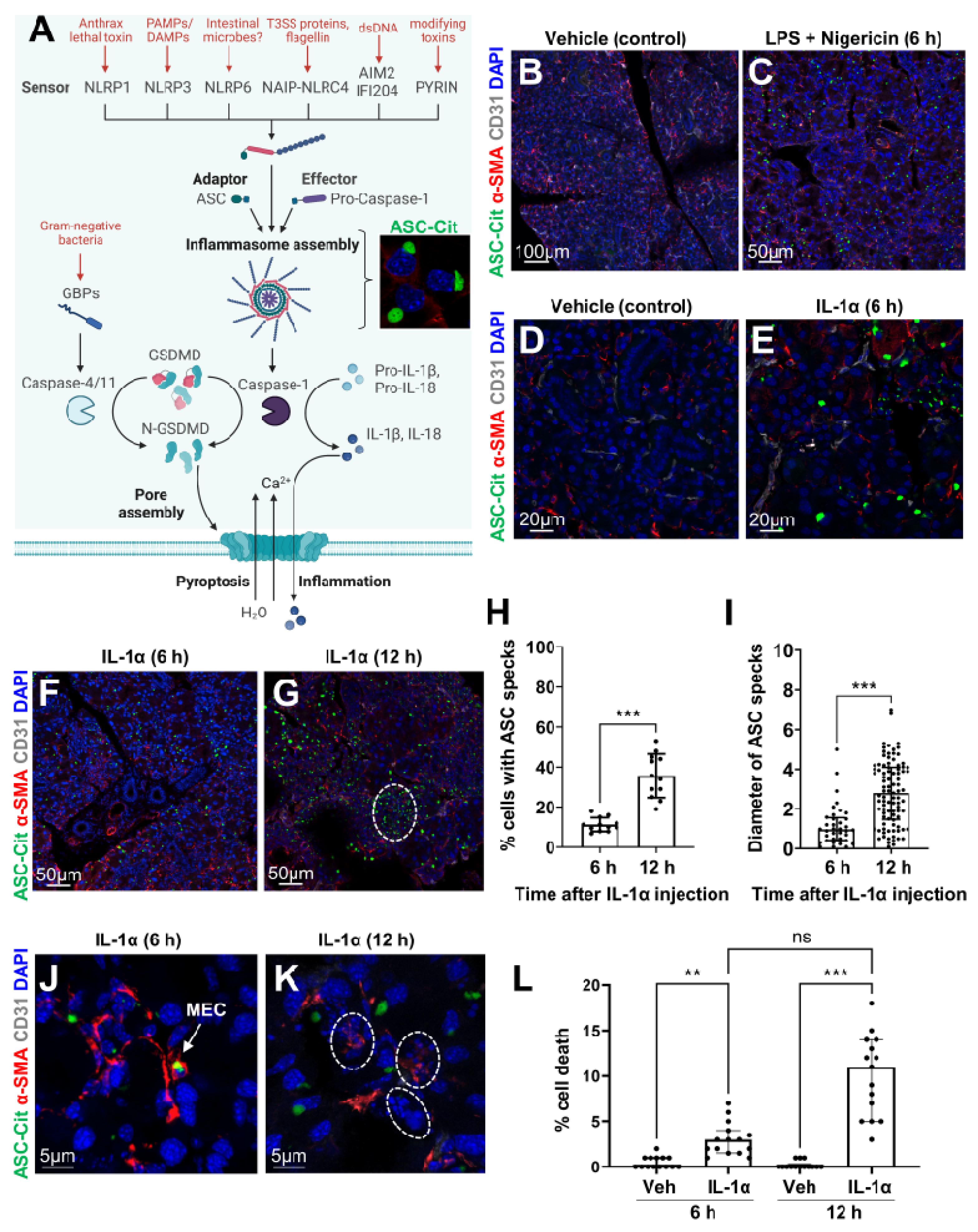
2.2. Induction of Inflammasome Formation with Lipopolysaccharide and Nigericin
2.3. LG Acute Injury
2.4. Frozen Section Preparation and Immunostaining
2.5. Western Blotting Analysis
2.6. qRT-PCR
2.7. RNA-Sequencing (RNA-seq) Data Analysis
2.8. Statistical Analysis
3. Results
3.1. Epithelial Cells Can Sense Microbial/Sterile Inflammatory Stimuli
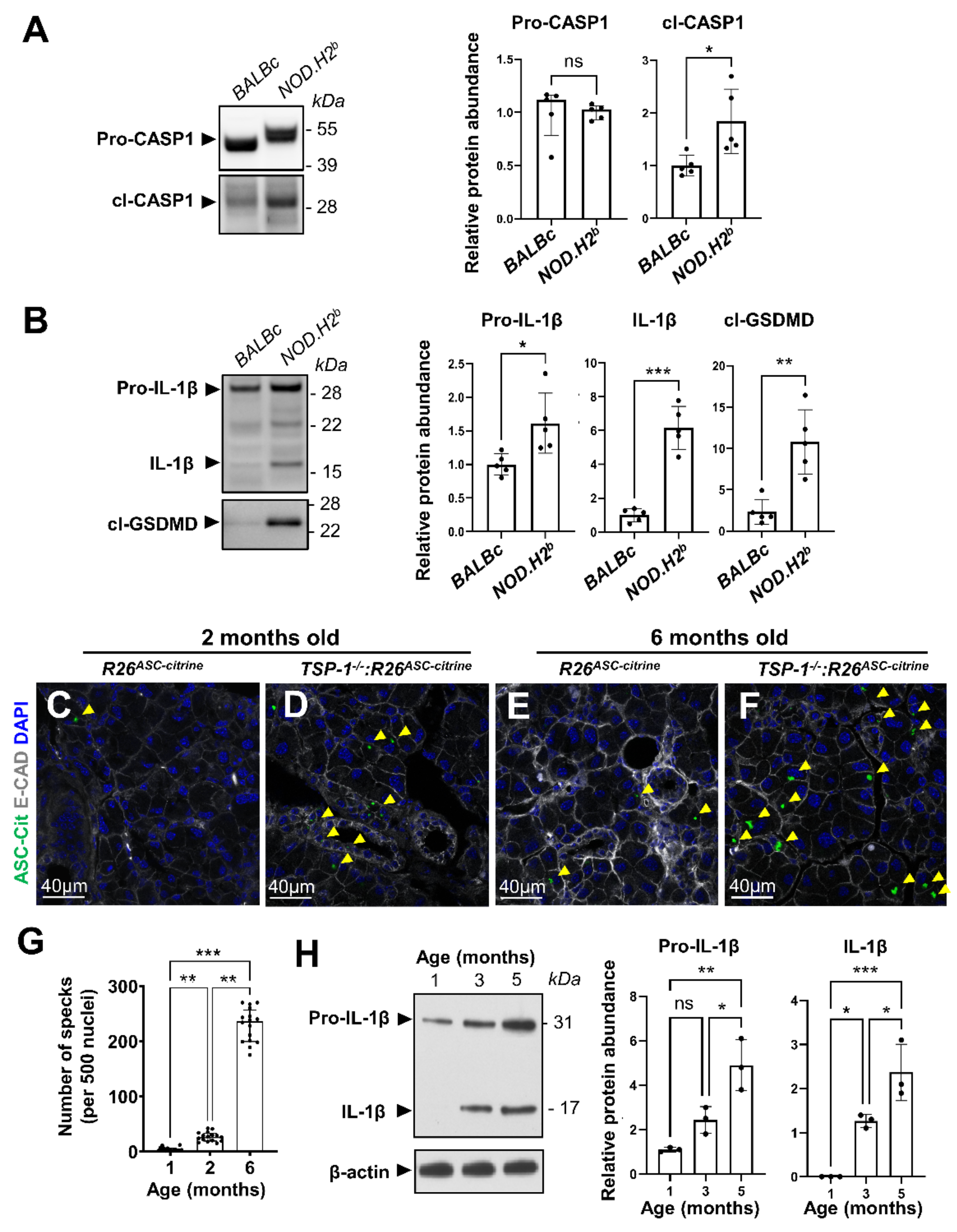
3.2. Inflammasome Activation during Chronic Inflammation
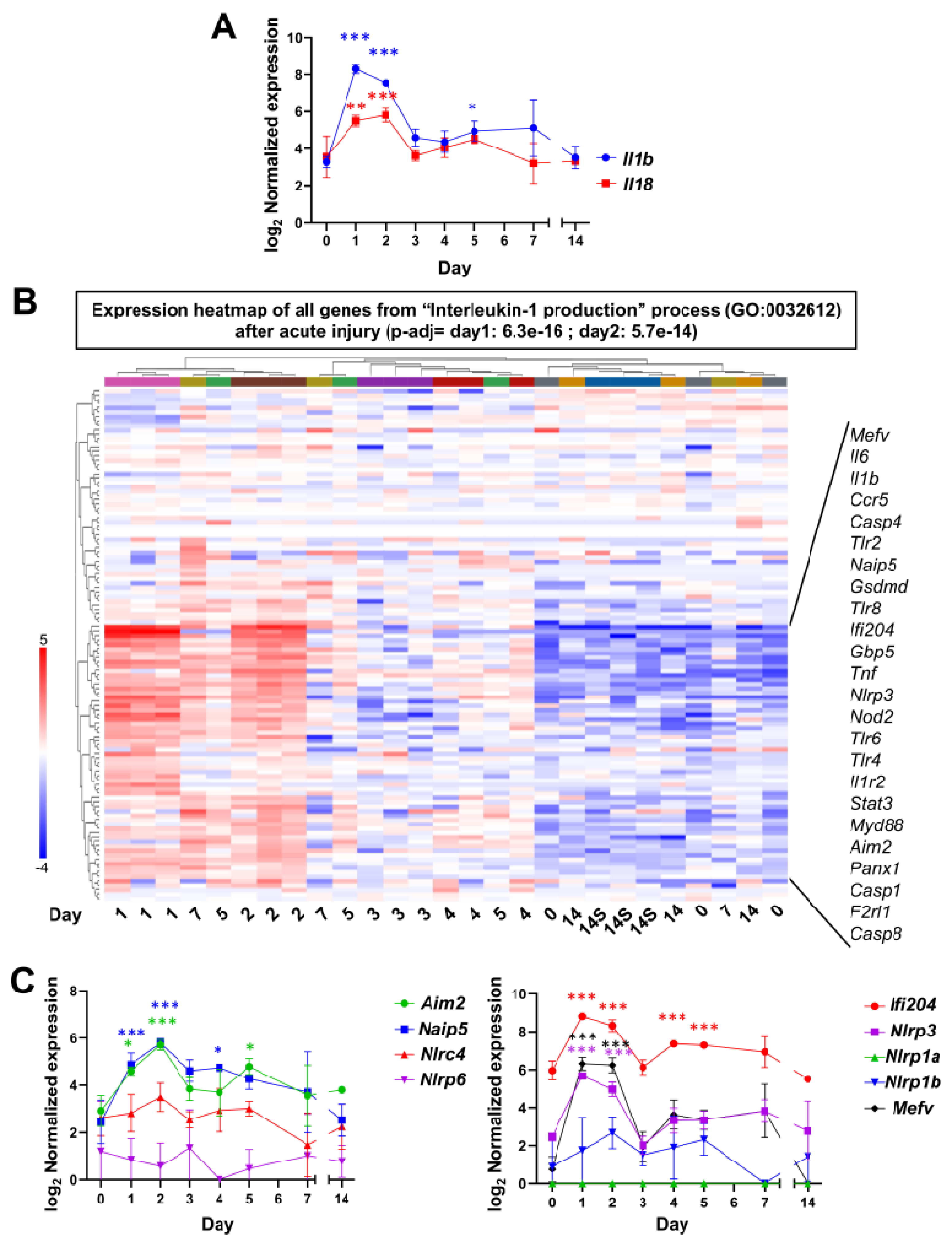
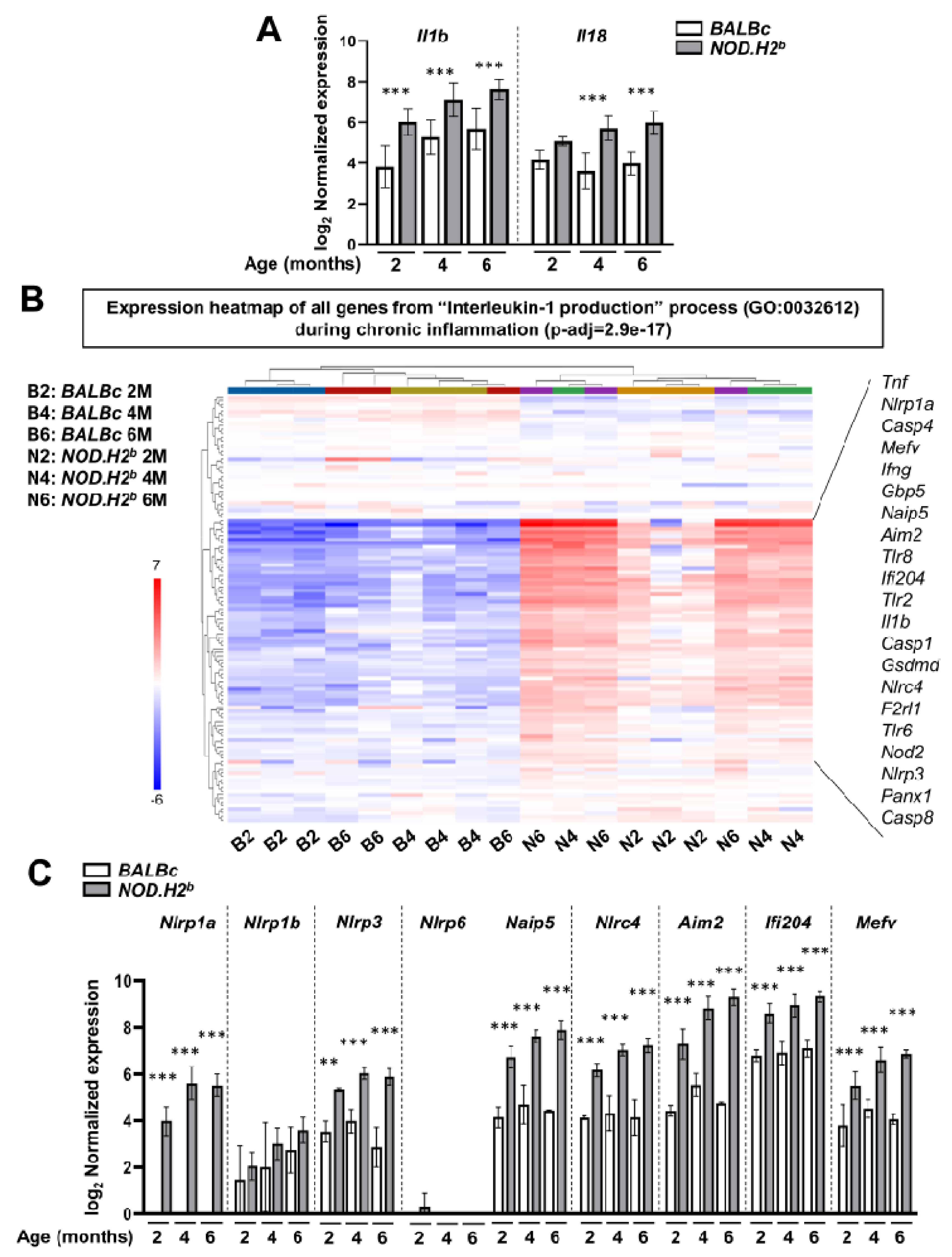
3.3. Acute Injury and Chronic Inflammation Upregulate Several Types of Inflammasomes
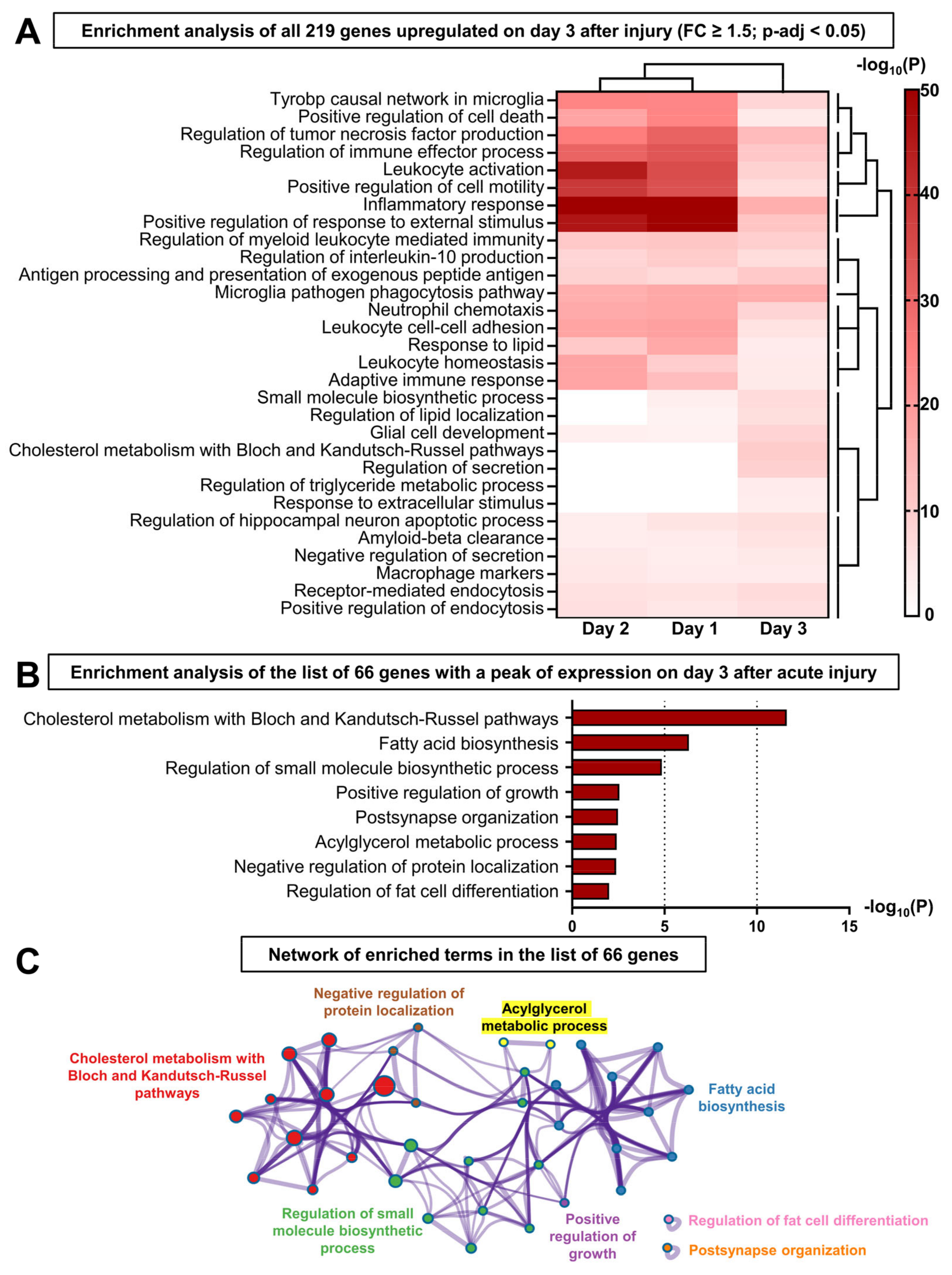
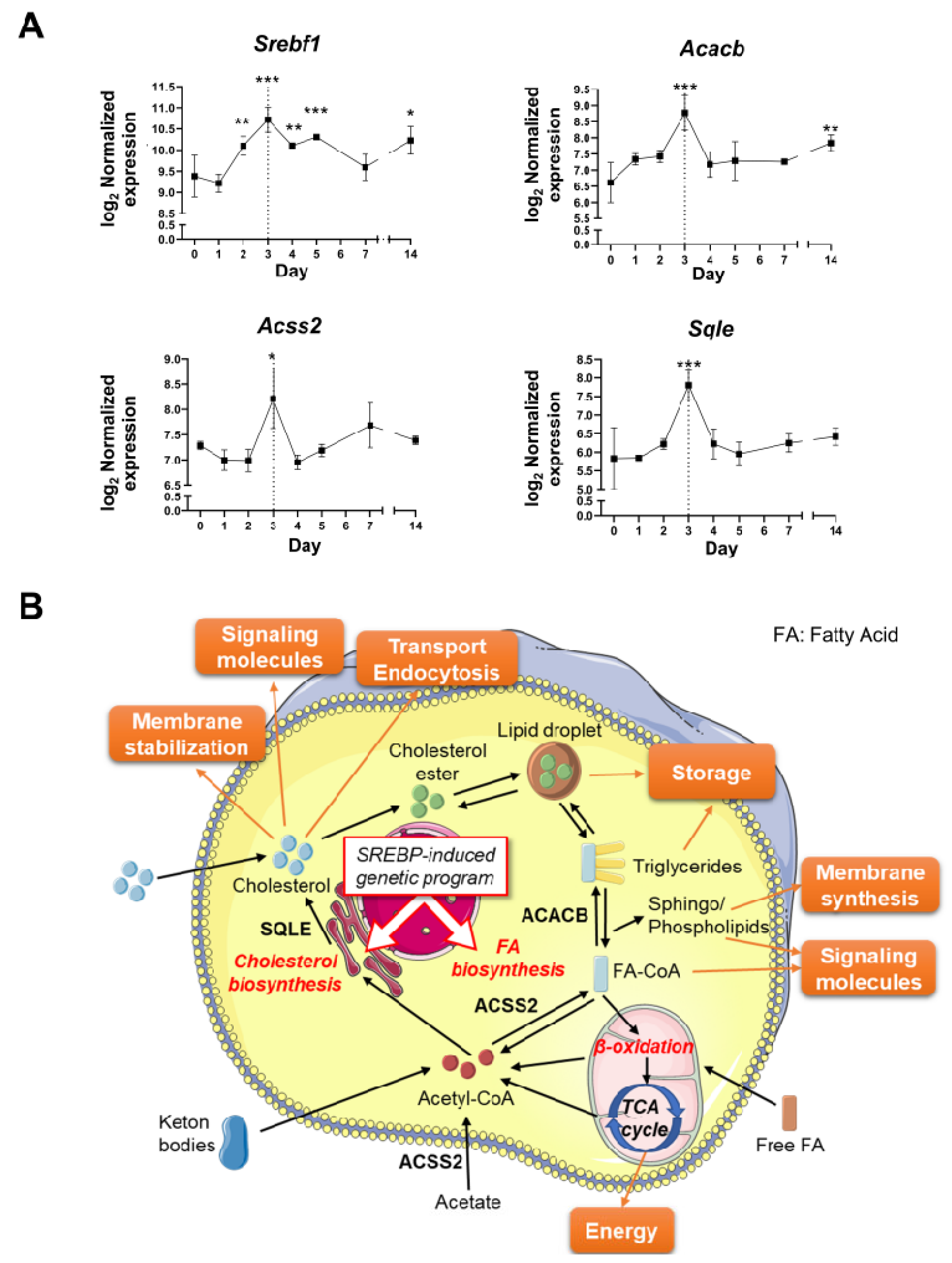
3.4. The Resolution of Inflammation and LG Regeneration following Acute Injury Is Concomitant with the Activation of Lipid Metabolism
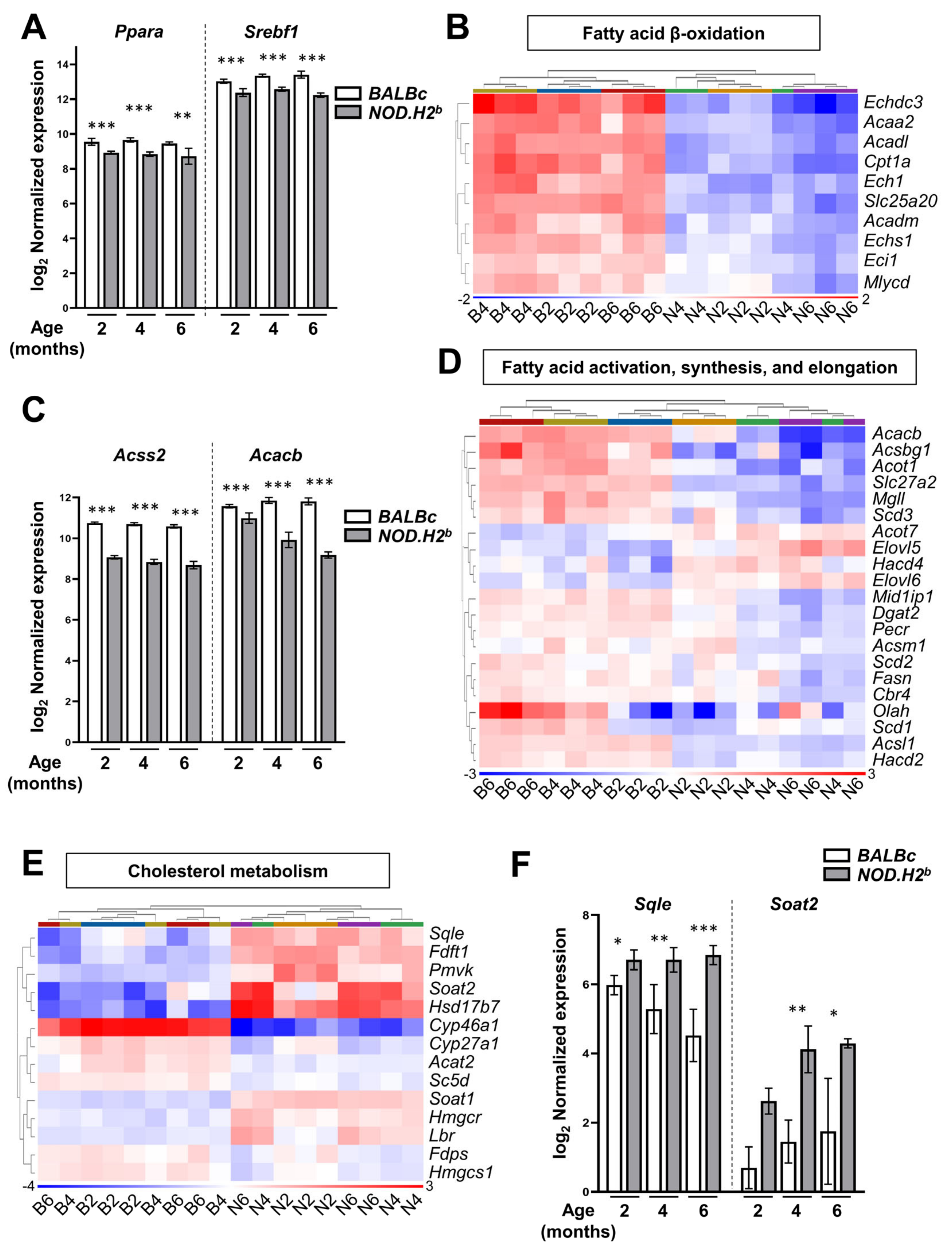
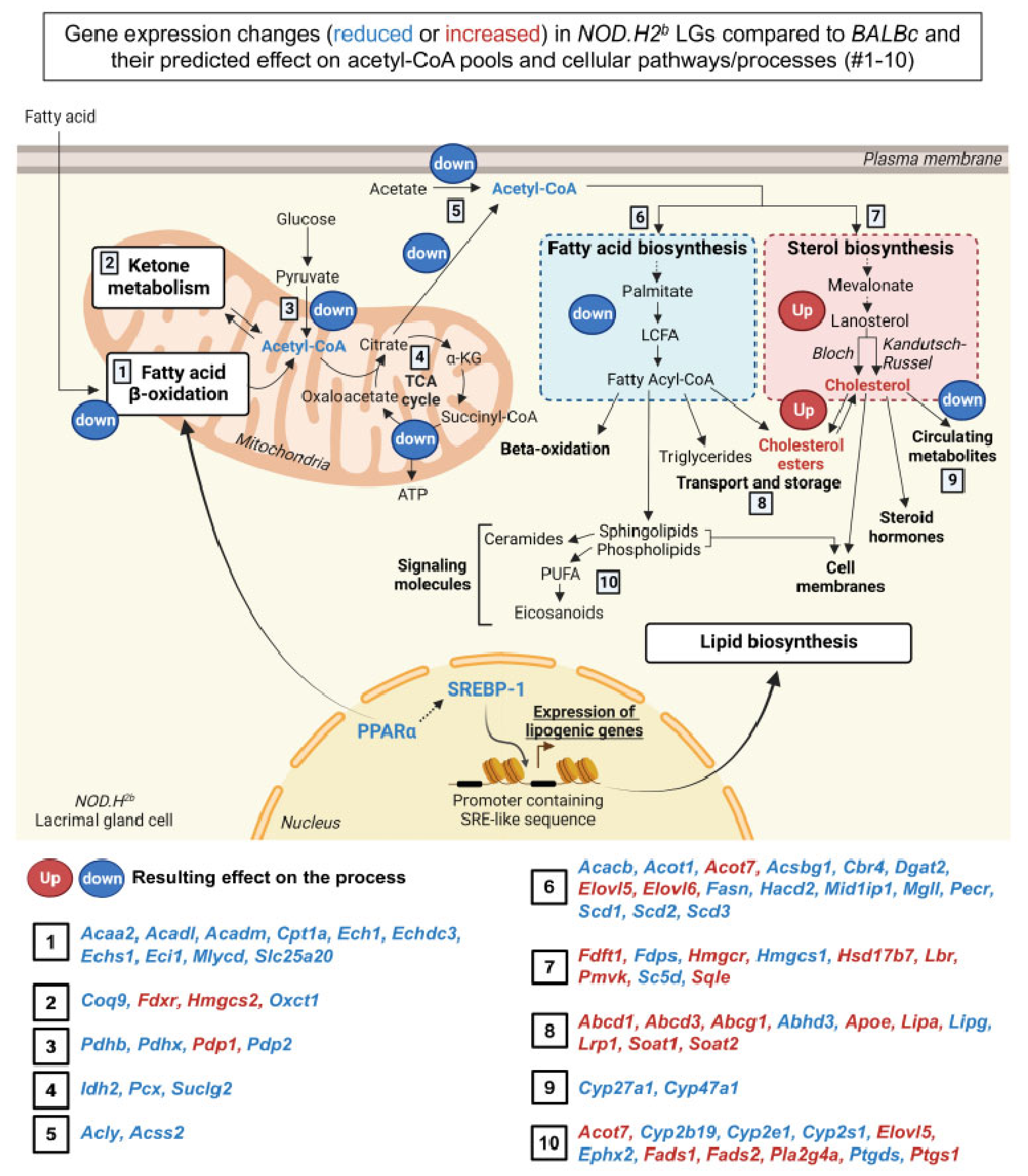
3.5. Lipid Metabolism Is Altered in Chronically Inflamed LG of NOD.H2b Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethics Approval and Consent to Participate
Abbreviations
| ACACB | Acetyl-CoA Carboxylase Beta |
| ACSS2 | Acyl-CoA Synthetase Short Chain Family Member 2 |
| ADDE | Aqueous deficiency dry eye |
| AIM2 | Absent in melanoma 2 |
| ALDH3B2 | Aldehyde Dehydrogenase 3 Family Member B2 |
| ANGT | Angiotensinogen |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| ATP | Adenosine triphosphate |
| CASP | Caspase |
| CoA | Coenzyme A |
| DAG | Diacylglycerol |
| DEG | Differentially expressed gene |
| DGKγ | Diacylglycerol Kinase Gamma |
| E-CAD | E-Cadherin |
| FA | Fatty acid |
| GBP | Guanylate-binding proteins |
| GSDMD | Gasdermin D |
| HMGCR | 3-Hydroxy-3-Methylglutaryl-CoA Reductase |
| IFI204 | Interferon-activable protein 204 |
| IFN | Interferon |
| IL | Interleukin |
| LG | Lacrimal gland |
| LPS | Lipopolysaccharide |
| M | Months old |
| MEC | Myoepithelial cell |
| MyD88 | Myeloid differentiation primary response 88 |
| NFκB | Nuclear factor kappa B |
| NLRP | nucleotide-binding oligomerization domain, leucine-rich repeat and Pyrin domain containing Proteins |
| PA | Phosphatidic acid |
| PANX1 | Pannexin-1 |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| pSS | Primary Sjögren’s syndrome |
| PUFA | Polyunsaturated fatty acid |
| PYHIN | Pyrin and HIN domain-containing protein |
| R26 | Rosa26 locus |
| RXRα | Retinoid X receptor alpha |
| SQLE | Squalene Epoxidase |
| SREBP1 | Sterol regulatory element-binding protein 1 |
| SS | Sjögren’s syndrome |
| SYK | Spleen Associated Tyrosine Kinase |
| TCA | Tricarboxylic acid |
| TLR | Toll-like receptor |
| TSP-1 | Thrombospondin-1 |
| VLCFA | Very long-chain fatty acid |
| α-SMA | Alpha Smooth Muscle Actin |
References
- Stern, M.E.; Pflugfelder, S.C. Inflammation in dry eye. Ocul. Surf. 2004, 2, 124–130. [Google Scholar] [CrossRef]
- McMonnies, C.W. Aqueous deficiency is a contributor to evaporation-related dry eye disease. Eye Vis. 2020, 7, 6. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Garcia-Posadas, L.; Hodges, R.R.; Makarenkova, H.P.; Masli, S.; Dartt, D.A. Alteration in nerves and neurotransmitter stimulation of lacrimal gland secretion in the TSP-1-/- mouse model of aqueous deficiency dry eye. Mucosal Immunol. 2018, 11, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Boehm, N.; Funke, S.; Wiegand, M.; Wehrwein, N.; Pfeiffer, N.; Grus, F.H. Alterations in the tear proteome of dry eye patients—A matter of the clinical phenotype. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2385–2392. [Google Scholar] [CrossRef] [PubMed]
- Toth-Molnar, E.; Ding, C. New insight into lacrimal gland function: Role of the duct epithelium in tear secretion. Ocul. Surf. 2020, 18, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Voronov, D.; Gromova, A.; Liu, D.; Zoukhri, D.; Medvinsky, A.; Meech, R.; Makarenkova, H.P. Transcription factors Runx1 to 3 are expressed in the lacrimal gland epithelium and are involved in regulation of gland morphogenesis and regeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3115–3125. [Google Scholar] [CrossRef]
- Walcott, B. The Lacrimal Gland and Its Veil of Tears. News Physiol. Sci. 1998, 13, 97–103. [Google Scholar] [CrossRef]
- de Souza, R.G.; de Paiva, C.S.; Alves, M.R. Age-related Autoimmune Changes in Lacrimal Glands. Immune Netw. 2019, 19, e3. [Google Scholar] [CrossRef]
- Rocha, E.M.; Alves, M.; Rios, J.D.; Dartt, D.A. The aging lacrimal gland: Changes in structure and function. Ocul. Surf. 2008, 6, 162–174. [Google Scholar] [CrossRef]
- Lam, H.; Bleiden, L.; de Paiva, C.S.; Farley, W.; Stern, M.E.; Pflugfelder, S.C. Tear cytokine profiles in dysfunctional tear syndrome. Am. J. Ophthalmol. 2009, 147, 198–205.e1. [Google Scholar] [CrossRef]
- Zoukhri, D.; Macari, E.; Kublin, C.L. A single injection of interleukin-1 induces reversible aqueous-tear deficiency, lacrimal gland inflammation, and acinar and ductal cell proliferation. Exp. Eye Res. 2007, 84, 894–904. [Google Scholar] [CrossRef]
- Gromova, A.; Voronov, D.A.; Yoshida, M.; Thotakura, S.; Meech, R.; Dartt, D.A.; Makarenkova, H.P. Lacrimal Gland Repair Using Progenitor Cells. Stem Cells Transl. Med. 2017, 6, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Hawley, D.; Ding, J.; Thotakura, S.; Haskett, S.; Aluri, H.; Kublin, C.; Michel, A.; Clapisson, L.; Mingueneau, M.; Zoukhri, D. RNA-Seq and CyTOF immuno-profiling of regenerating lacrimal glands identifies a novel subset of cells expressing muscle-related proteins. PLoS ONE 2017, 12, e0179385. [Google Scholar] [CrossRef]
- Basova, L.V.; Tang, X.; Umasume, T.; Gromova, A.; Zyrianova, T.; Shmushkovich, T.; Wolfson, A.; Hawley, D.; Zoukhri, D.; Shestopalov, V.I.; et al. Manipulation of Panx1 Activity Increases the Engraftment of Transplanted Lacrimal Gland Epithelial Progenitor Cells. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5654–5665. [Google Scholar] [CrossRef]
- Makarenkova, H.P.; Shah, S.B.; Shestopalov, V.I. The two faces of pannexins: New roles in inflammation and repair. J. Inflamm. Res. 2018, 11, 273–288. [Google Scholar] [CrossRef]
- Evavold, C.L.; Kagan, J.C. Inflammasomes: Threat-Assessment Organelles of the Innate Immune System. Immunity 2019, 51, 609–624. [Google Scholar] [CrossRef]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e6. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Takahashi, M. NLRP3 in myocardial ischaemia-reperfusion injury: Inflammasome-dependent or -independent role in different cell types. Cardiovasc. Res. 2013, 99, 4–5. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- van Hout, G.P.J.; Bosch, L. The Inflammasomes in Cardiovascular Disease. Exp. Suppl. 2018, 108, 9–40. [Google Scholar] [CrossRef] [PubMed]
- Barabino, S.; Chen, Y.; Chauhan, S.; Dana, R. Ocular surface immunity: Homeostatic mechanisms and their disruption in dry eye disease. Prog. Retin. Eye Res. 2012, 31, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell Longev. 2020, 2020, 4063562. [Google Scholar] [CrossRef]
- Lin, L.R.; Xiao, Y.; Liu, W.; Chen, Y.Y.; Zhu, X.Z.; Gao, Z.X.; Gao, K.; Tong, M.L.; Zhang, H.L.; Li, S.L.; et al. Development of tissue inflammation accompanied by NLRP3 inflammasome activation in rabbits infected with Treponema pallidum strain Nichols. BMC Infect. Dis. 2018, 18, 101. [Google Scholar] [CrossRef]
- Niu, L.; Zhang, S.; Wu, J.; Chen, L.; Wang, Y. Upregulation of NLRP3 Inflammasome in the Tears and Ocular Surface of Dry Eye Patients. PLoS ONE 2015, 10, e0126277. [Google Scholar] [CrossRef]
- Vakrakou, A.G.; Svolaki, I.P.; Evangelou, K.; Gorgoulis, V.G.; Manoussakis, M.N. Cell-autonomous epithelial activation of AIM2 (absent in melanoma-2) inflammasome by cytoplasmic DNA accumulations in primary Sjogren’s syndrome. J. Autoimmun. 2020, 108, 102381. [Google Scholar] [CrossRef]
- Oh, J.Y.; Ko, J.H.; Ryu, J.S.; Lee, H.J.; Kim, M.K.; Wee, W.R. Transcription Profiling of NOD-like Receptors in the Human Cornea with Disease. Ocul. Immunol. Inflamm. 2017, 25, 364–369. [Google Scholar] [CrossRef]
- Baldini, C.; Rossi, C.; Ferro, F.; Santini, E.; Seccia, V.; Donati, V.; Solini, A. The P2X7 receptor-inflammasome complex has a role in modulating the inflammatory response in primary Sjogren’s syndrome. J. Intern. Med. 2013, 274, 480–489. [Google Scholar] [CrossRef]
- Hong, S.M.; Lee, J.; Jang, S.G.; Lee, J.; Cho, M.L.; Kwok, S.K.; Park, S.H. Type I Interferon Increases Inflammasomes Associated Pyroptosis in the Salivary Glands of Patients with Primary Sjogren’s Syndrome. Immune Netw. 2020, 20, e39. [Google Scholar] [CrossRef]
- Kopitar-Jerala, N. The Role of Interferons in Inflammation and Inflammasome Activation. Front. Immunol. 2017, 8, 873. [Google Scholar] [CrossRef]
- Haque, M.; Siegel, R.J.; Fox, D.A.; Ahmed, S. Interferon-stimulated GTPases in autoimmune and inflammatory diseases: Promising role for the guanylate-binding protein (GBP) family. Rheumatology 2021, 60, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Vestal, D.J. The guanylate-binding proteins (GBPs): Proinflammatory cytokine-induced members of the dynamin superfamily with unique GTPase activity. J. Interferon Cytokine Res. 2005, 25, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.C.; Casciola-Rosen, L.; Berger, A.E.; Kapsogeorgou, E.K.; Cheadle, C.; Tzioufas, A.G.; Baer, A.N.; Rosen, A. Precise probes of type II interferon activity define the origin of interferon signatures in target tissues in rheumatic diseases. Proc. Natl. Acad. Sci. USA 2012, 109, 17609–17614. [Google Scholar] [CrossRef]
- Hu, S.; Gao, K.; Pollard, R.; Arellano-Garcia, M.; Zhou, H.; Zhang, L.; Elashoff, D.; Kallenberg, C.G.; Vissink, A.; Wong, D.T. Preclinical validation of salivary biomarkers for primary Sjogren’s syndrome. Arthritis Care Res. 2010, 62, 1633–1638. [Google Scholar] [CrossRef]
- Hjelmervik, T.O.; Petersen, K.; Jonassen, I.; Jonsson, R.; Bolstad, A.I. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjogren’s syndrome patients from healthy control subjects. Arthritis Rheum. 2005, 52, 1534–1544. [Google Scholar] [CrossRef]
- Tzeng, T.C.; Schattgen, S.; Monks, B.; Wang, D.; Cerny, A.; Latz, E.; Fitzgerald, K.; Golenbock, D.T. A Fluorescent Reporter Mouse for Inflammasome Assembly Demonstrates an Important Role for Cell-Bound and Free ASC Specks during In Vivo Infection. Cell Rep. 2016, 16, 571–582. [Google Scholar] [CrossRef]
- Mauduit, O.; Delcroix, V.; Umazume, T.; de Paiva, C.S.; Dartt, D.A.; Makarenkova, H.P. Spatial transcriptomics of the lacrimal gland features macrophage activity and epithelium metabolism as key alterations during chronic inflammation. Front. Immunol. 2022, 13, 1011125. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Cullen, S.P.; Kearney, C.J.; Clancy, D.M.; Martin, S.J. Diverse Activators of the NLRP3 Inflammasome Promote IL-1beta Secretion by Triggering Necrosis. Cell Rep. 2015, 11, 1535–1548. [Google Scholar] [CrossRef]
- Robinson, C.P.; Yamachika, S.; Bounous, D.I.; Brayer, J.; Jonsson, R.; Holmdahl, R.; Peck, A.B.; Humphreys-Beher, M.G. A novel NOD-derived murine model of primary Sjogren’s syndrome. Arthritis Rheum. 1998, 41, 150–156. [Google Scholar] [CrossRef]
- Szyszko, E.A.; Aqrawi, L.A.; Jonsson, R.; Brokstad, K.A.; Skarstein, K. Non-proliferating plasma cells detected in the salivary glands and bone marrow of autoimmune NOD.B10.H2b mice, a model for primary Sjogren’s syndrome. Autoimmunity 2016, 49, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Damianou, A.; Di Daniel, E.; Kessler, B.M. Inflammasome activation controlled by the interplay between post-translational modifications: Emerging drug target opportunities. Cell Commun. Signal. 2021, 19, 23. [Google Scholar] [CrossRef] [PubMed]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Shatos, M.A.; Hodges, R.R.; Morinaga, M.; McNay, D.E.; Islam, R.; Bhattacharya, S.; Li, D.; Turpie, B.; Makarenkova, H.P.; Masli, S.; et al. Alteration in cellular turnover and progenitor cell population in lacrimal glands from thrombospondin 1-/- mice, a model of dry eye. Exp. Eye Res. 2016, 153, 27–41. [Google Scholar] [CrossRef]
- Turpie, B.; Yoshimura, T.; Gulati, A.; Rios, J.D.; Dartt, D.A.; Masli, S. Sjogren’s syndrome-like ocular surface disease in thrombospondin-1 deficient mice. Am. J. Pathol. 2009, 175, 1136–1147. [Google Scholar] [CrossRef]
- Thinwa, J.; Segovia, J.A.; Bose, S.; Dube, P.H. Integrin-mediated first signal for inflammasome activation in intestinal epithelial cells. J. Immunol. 2014, 193, 1373–1382. [Google Scholar] [CrossRef]
- Jarry, A.; Malard, F.; Bou-Hanna, C.; Meurette, G.; Mohty, M.; Mosnier, J.F.; Laboisse, C.L.; Bossard, C. Interferon-Alpha Promotes Th1 Response and Epithelial Apoptosis via Inflammasome Activation in Human Intestinal Mucosa. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 72–81. [Google Scholar] [CrossRef]
- Stutz, A.; Horvath, G.L.; Monks, B.G.; Latz, E. ASC speck formation as a readout for inflammasome activation. Methods Mol. Biol. 2013, 1040, 91–101. [Google Scholar] [CrossRef]
- Su, Q.; Li, L.; Sun, Y.; Yang, H.; Ye, Z.; Zhao, J. Effects of the TLR4/Myd88/NF-kappaB Signaling Pathway on NLRP3 Inflammasome in Coronary Microembolization-Induced Myocardial Injury. Cell Physiol. Biochem. 2018, 47, 1497–1508. [Google Scholar] [CrossRef]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; Tsai, S.Y.; Chang, T.H.; Berton, M.T.; Morris, I.R.; Allen, I.C.; Ting, J.P.; Bose, S. TLR2/MyD88/NF-kappaB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS ONE 2012, 7, e29695. [Google Scholar] [CrossRef]
- Rallabhandi, P.; Nhu, Q.M.; Toshchakov, V.Y.; Piao, W.; Medvedev, A.E.; Hollenberg, M.D.; Fasano, A.; Vogel, S.N. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: A novel paradigm for receptor cooperativity. J. Biol. Chem. 2008, 283, 24314–24325. [Google Scholar] [CrossRef]
- Nhu, Q.M.; Shirey, K.; Teijaro, J.R.; Farber, D.L.; Netzel-Arnett, S.; Antalis, T.M.; Fasano, A.; Vogel, S.N. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol. 2010, 3, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Tenthorey, J.L.; Haloupek, N.; Lopez-Blanco, J.R.; Grob, P.; Adamson, E.; Hartenian, E.; Lind, N.A.; Bourgeois, N.M.; Chacon, P.; Nogales, E.; et al. The structural basis of flagellin detection by NAIP5: A strategy to limit pathogen immune evasion. Science 2017, 358, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Sastalla, I.; Crown, D.; Masters, S.L.; McKenzie, A.; Leppla, S.H.; Moayeri, M. Transcriptional analysis of the three Nlrp1 paralogs in mice. BMC Genom. 2013, 14, 188. [Google Scholar] [CrossRef] [PubMed]
- Cornut, M.; Bourdonnay, E.; Henry, T. Transcriptional Regulation of Inflammasomes. Int. J. Mol. Sci. 2020, 21, 8087. [Google Scholar] [CrossRef]
- Tretina, K.; Park, E.S.; Maminska, A.; MacMicking, J.D. Interferon-induced guanylate-binding proteins: Guardians of host defense in health and disease. J. Exp. Med. 2019, 216, 482–500. [Google Scholar] [CrossRef]
- Qiao, Y.; Tian, X.; Men, L.; Li, S.; Chen, Y.; Xue, M.; Hu, Y.; Zhou, P.; Long, G.; Shi, Y.; et al. Spleen tyrosine kinase promotes NLR family pyrin domain containing 3 inflammasomemediated IL1beta secretion via cJun Nterminal kinase activation and cell apoptosis during diabetic nephropathy. Mol. Med. Rep. 2018, 18, 1995–2008. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Knight, B.L.; Hebbachi, A.; Hauton, D.; Brown, A.M.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. A role for PPARalpha in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 2005, 389, 413–421. [Google Scholar] [CrossRef]
- van der Meer, D.L.; Degenhardt, T.; Vaisanen, S.; de Groot, P.J.; Heinaniemi, M.; de Vries, S.C.; Muller, M.; Carlberg, C.; Kersten, S. Profiling of promoter occupancy by PPARalpha in human hepatoma cells via ChIP-chip analysis. Nucleic Acids Res. 2010, 38, 2839–2850. [Google Scholar] [CrossRef]
- Wu, K.; Joffre, C.; Li, X.; MacVeigh-Aloni, M.; Hom, M.; Hwang, J.; Ding, C.; Gregoire, S.; Bretillon, L.; Zhong, J.F.; et al. Altered expression of genes functioning in lipid homeostasis is associated with lipid deposition in NOD mouse lacrimal gland. Exp. Eye Res. 2009, 89, 319–332. [Google Scholar] [CrossRef]
- Griswold, A.R.; Huang, H.C.; Bachovchin, D.A. The NLRP1 Inflammasome Induces Pyroptosis in Human Corneal Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2022, 63, 2. [Google Scholar] [CrossRef]
- Hamada, A.; Torre, C.; Drancourt, M.; Ghigo, E. Trained Immunity Carried by Non-immune Cells. Front. Microbiol. 2018, 9, 3225. [Google Scholar] [CrossRef]
- Vijmasi, T.; Chen, F.Y.; Chen, Y.T.; Gallup, M.; McNamara, N. Topical administration of interleukin-1 receptor antagonist as a therapy for aqueous-deficient dry eye in autoimmune disease. Mol. Vis. 2013, 19, 1957–1965. [Google Scholar]
- de Torre-Minguela, C.; Mesa Del Castillo, P.; Pelegrin, P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 43. [Google Scholar] [CrossRef]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef]
- Kiripolsky, J.; McCabe, L.G.; Gaile, D.P.; Kramer, J.M. Myd88 is required for disease development in a primary Sjogren’s syndrome mouse model. J. Leukoc. Biol. 2017, 102, 1411–1420. [Google Scholar] [CrossRef]
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327. [Google Scholar] [CrossRef]
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.; Mitsuyama, M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Huang, D.-Y.; Wang, J.-S.; Lin, Y.-L.; Hsieh, S.-L.; Huang, K.-C.; Lin, W.-W. Syk is involved in NLRP3 inflammasome-mediated caspase-1 activation through adaptor ASC phosphorylation and enhanced oligomerization. J. Leukoc. Biol. 2015, 97, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Del Papa, N.; Minniti, A.; Lorini, M.; Carbonelli, V.; Maglione, W.; Pignataro, F.; Montano, N.; Caporali, R.; Vitali, C. The Role of Interferons in the Pathogenesis of Sjogren’s Syndrome and Future Therapeutic Perspectives. Biomolecules 2021, 11, 251. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef]
- Navia-Pelaez, J.M.; Choi, S.H.; Dos Santos Aggum Capettini, L.; Xia, Y.; Gonen, A.; Agatisa-Boyle, C.; Delay, L.; Goncalves Dos Santos, G.; Catroli, G.F.; Kim, J.; et al. Normalization of cholesterol metabolism in spinal microglia alleviates neuropathic pain. J. Exp. Med. 2021, 218, e20202059. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Spann, N.J.; Link, V.M.; Muse, E.D.; Strid, T.; Edillor, C.; Kolar, M.J.; Matsuzaka, T.; Hayakawa, S.; Tao, J.; et al. SREBP1 Contributes to Resolution of Pro-inflammatory TLR4 Signaling by Reprogramming Fatty Acid Metabolism. Cell Metab. 2017, 25, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; van der Goot, F.G. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 2006, 126, 1135–1145. [Google Scholar] [CrossRef]
- Ding, C.; MacVeigh, M.; Pidgeon, M.; da Costa, S.R.; Wu, K.; Hamm-Alvarez, S.F.; Schechter, J.E. Unique ultrastructure of exorbital lacrimal glands in male NOD and BALB/c mice. Curr. Eye Res. 2006, 31, 13–22. [Google Scholar] [CrossRef]
- Meyers, A.K.; Zhu, X. The NLRP3 Inflammasome: Metabolic Regulation and Contribution to Inflammaging. Cells 2020, 9, 1808. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Reboldi, A.; Dang, E.V.; McDonald, J.G.; Liang, G.; Russell, D.W.; Cyster, J.G. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 2014, 345, 679–684. [Google Scholar] [CrossRef]
- Izumi, M.; Eguchi, K.; Uetani, M.; Nakamura, H.; Takagi, Y.; Hayashi, K.; Nakamura, T. MR features of the lacrimal gland in Sjogren’s syndrome. AJR Am. J. Roentgenol. 1998, 170, 1661–1666. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Crovella, S.; Celsi, F. Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link. Int. J. Mol. Sci. 2015, 16, 16067–16084. [Google Scholar] [CrossRef]
- He, X.; Zhao, Z.; Wang, S.; Kang, J.; Zhang, M.; Bu, J.; Cai, X.; Jia, C.; Li, Y.; Li, K.; et al. High-Fat Diet-Induced Functional and Pathologic Changes in Lacrimal Gland. Am. J. Pathol. 2020, 190, 2387–2402. [Google Scholar] [CrossRef]
- Wang, S.; He, X.; Li, Q.; Zhang, Y.; Hu, J.; Zong, R.; Zhuang, J.; Quantock, A.J.; Gao, Y.; Li, W.; et al. Obstructive Sleep Apnea Affects Lacrimal Gland Function. Investig. Ophthalmol. Vis. Sci. 2022, 63, 3. [Google Scholar] [CrossRef]
- Guo, X.; Dang, W.; Li, N.; Wang, Y.; Sun, D.; Nian, H.; Wei, R. PPAR-alpha Agonist Fenofibrate Ameliorates Sjogren Syndrome-Like Dacryoadenitis by Modulating Th1/Th17 and Treg Cell Responses in NOD Mice. Investig. Ophthalmol. Vis. Sci. 2022, 63, 12. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Pringle, S.; Bootsma, H.; Kroese, F.G.M. Epithelial-immune cell interplay in primary Sjogren syndrome salivary gland pathogenesis. Nat. Rev. Rheumatol. 2021, 17, 333–348. [Google Scholar] [CrossRef]
- Pflugfelder, S.C.; de Paiva, C.S.; Li, D.Q.; Stern, M.E. Epithelial-immune cell interaction in dry eye. Cornea 2008, 27 (Suppl. 1), S9–S11. [Google Scholar] [CrossRef]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer Sequence |
|---|---|
| Nlrp3 | Forward: 5′-AGAAGAGACCACGGCAGAAG-3′ |
| Reverse: 5′-CCTTGGACCAGGTTCAGTGT-3′ | |
| Nlrc4 | Forward: 5′-CTTGGCCAGGAGAGCCTTG-3′ |
| Reverse: 5′-GGGCTCGTCTGTTGTTCCTT-3′ | |
| Aim2 | Forward: 5′-GATTCAAAGTGCAGGTGCGG-3′ |
| Reverse: 5′-TCTGAGGCTTAGCTTGAGGAC-3′ | |
| Casp1 | Forward: 5′-GCGAAGCATACTTTCAGTTTC-3′ |
| Reverse: 5′-TCTCCTTCAGGACCTTGTCG-3′ | |
| Casp4(11) | Forward: 5′-AGGAGCCCACTCCTACAGAG-3′ |
| Reverse: 5′-AAGGTTGCCCGATCAATGGT-3′ | |
| Actb | Forward: 5′-AGAGGGAAATCGTGCGTGAC-3′ |
| Reverse: 5′-CAATAGTGATGACCTGGCCGT-3′ | |
| Gapdh | Forward: 5′-CGTCCCGTAGACAAAATG GT-3′ |
| Reverse: 5′-TTGATGGCAACAATCTCC AC-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delcroix, V.; Mauduit, O.; Yang, M.; Srivastava, A.; Umazume, T.; de Paiva, C.S.; Shestopalov, V.I.; Dartt, D.A.; Makarenkova, H.P. Lacrimal Gland Epithelial Cells Shape Immune Responses through the Modulation of Inflammasomes and Lipid Metabolism. Int. J. Mol. Sci. 2023, 24, 4309. https://doi.org/10.3390/ijms24054309
Delcroix V, Mauduit O, Yang M, Srivastava A, Umazume T, de Paiva CS, Shestopalov VI, Dartt DA, Makarenkova HP. Lacrimal Gland Epithelial Cells Shape Immune Responses through the Modulation of Inflammasomes and Lipid Metabolism. International Journal of Molecular Sciences. 2023; 24(5):4309. https://doi.org/10.3390/ijms24054309
Chicago/Turabian StyleDelcroix, Vanessa, Olivier Mauduit, Menglu Yang, Amrita Srivastava, Takeshi Umazume, Cintia S. de Paiva, Valery I. Shestopalov, Darlene A. Dartt, and Helen P. Makarenkova. 2023. "Lacrimal Gland Epithelial Cells Shape Immune Responses through the Modulation of Inflammasomes and Lipid Metabolism" International Journal of Molecular Sciences 24, no. 5: 4309. https://doi.org/10.3390/ijms24054309
APA StyleDelcroix, V., Mauduit, O., Yang, M., Srivastava, A., Umazume, T., de Paiva, C. S., Shestopalov, V. I., Dartt, D. A., & Makarenkova, H. P. (2023). Lacrimal Gland Epithelial Cells Shape Immune Responses through the Modulation of Inflammasomes and Lipid Metabolism. International Journal of Molecular Sciences, 24(5), 4309. https://doi.org/10.3390/ijms24054309