
Aberrantly Expressed MicroRNAs in Cancer-Associated Fibroblasts and Their Target Oncogenic Signatures in Hepatocellular Carcinoma

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
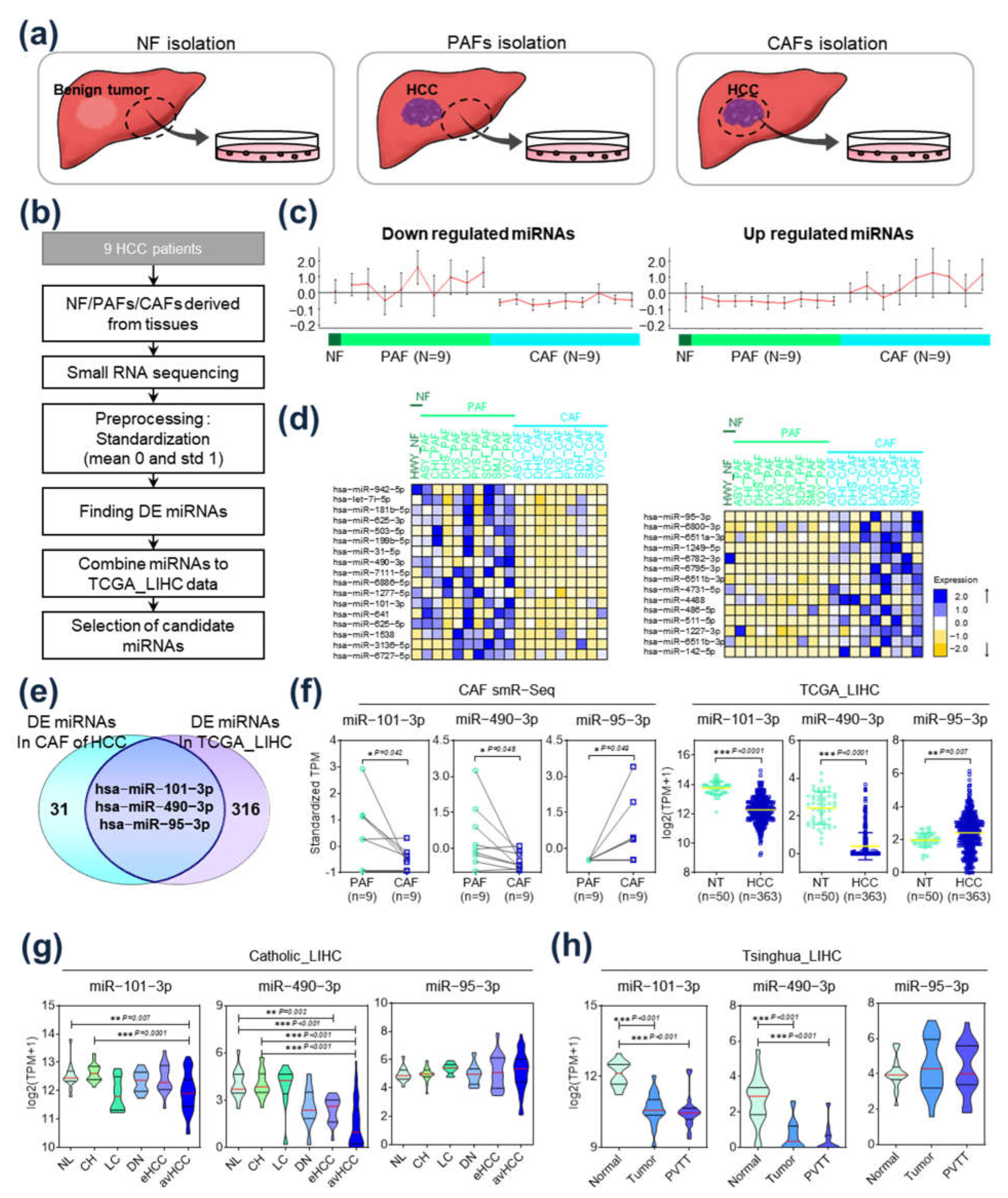
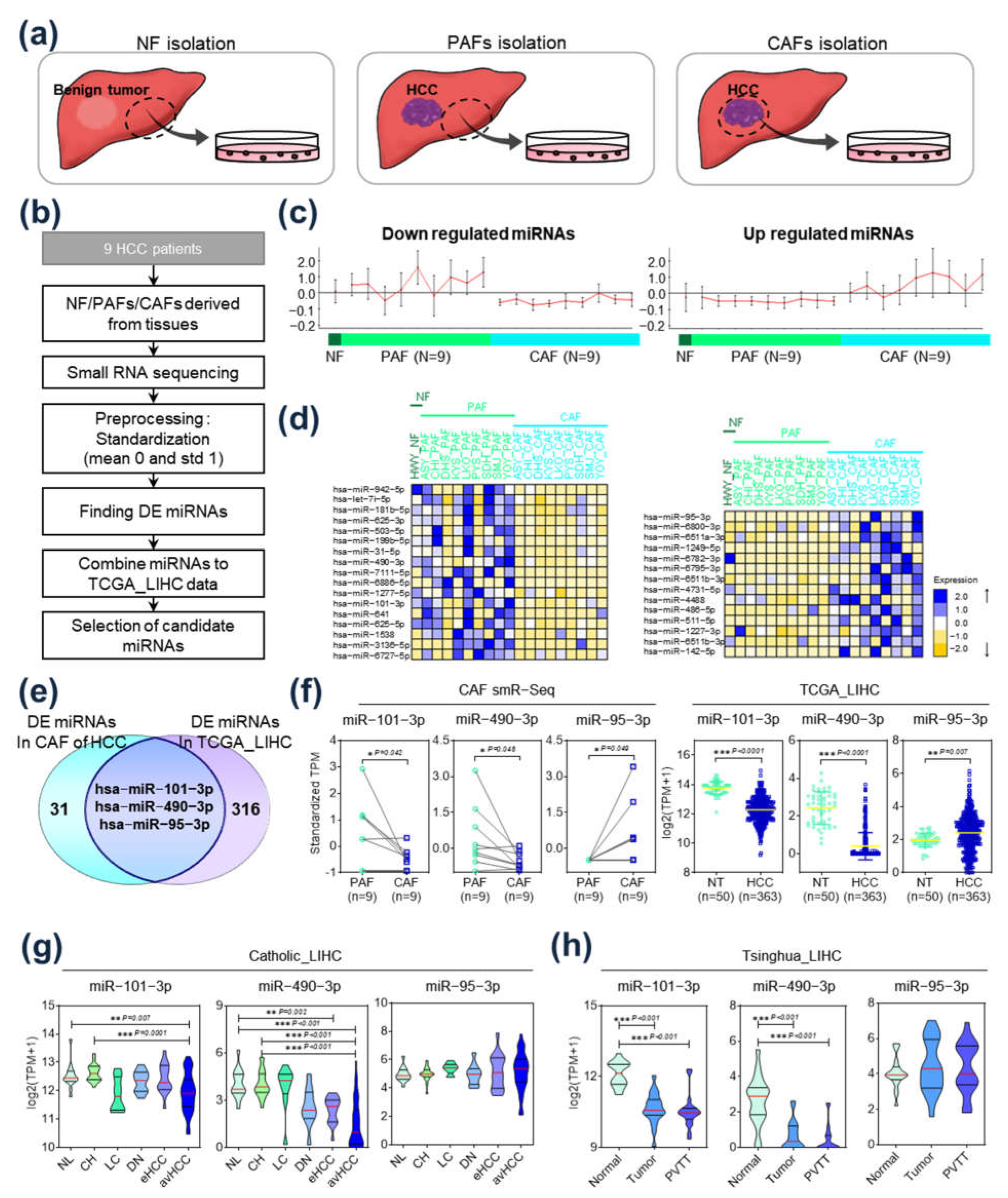
2.1. Identification of Differentially Expressed miRs in HCC-CAFs
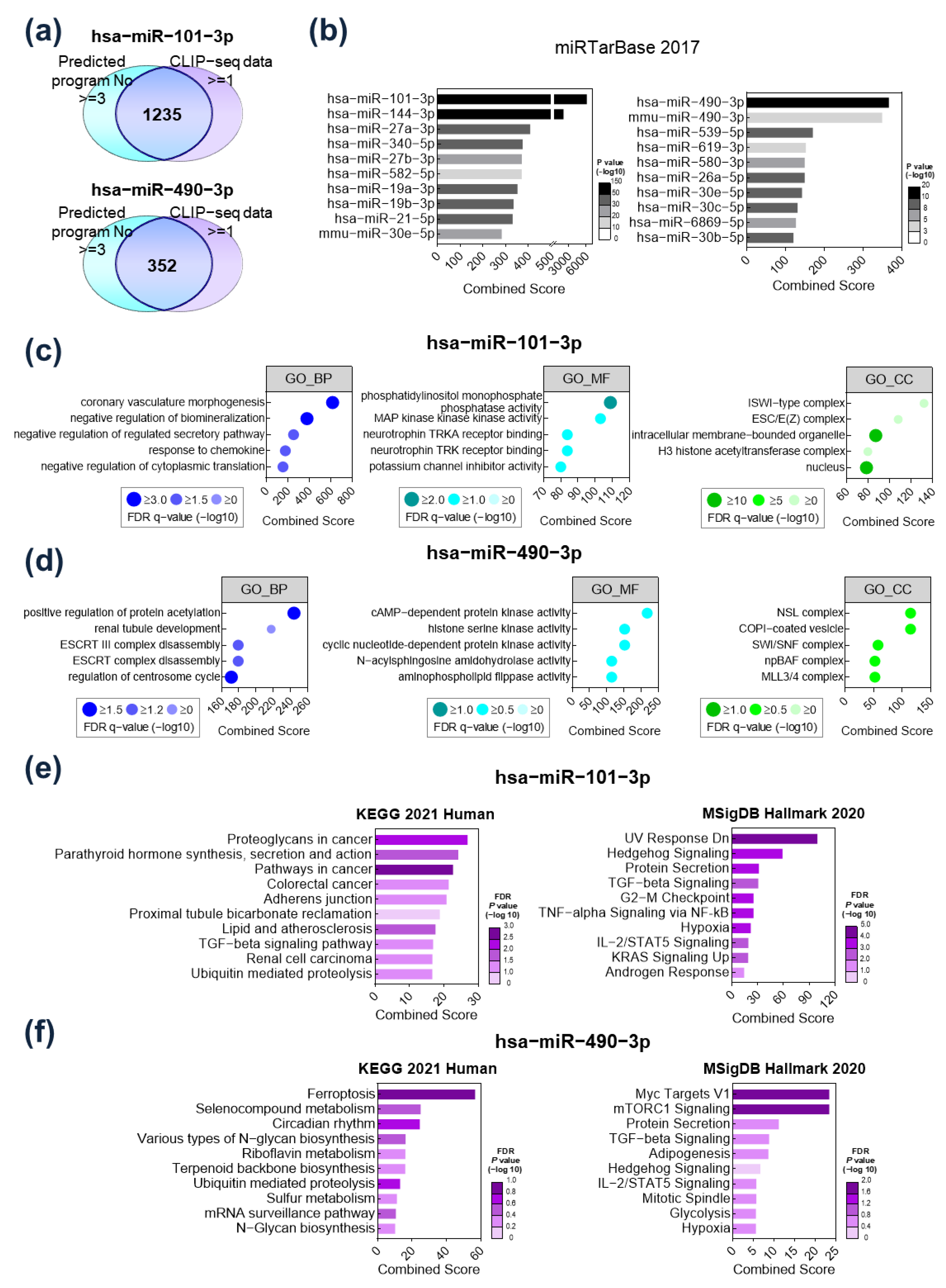
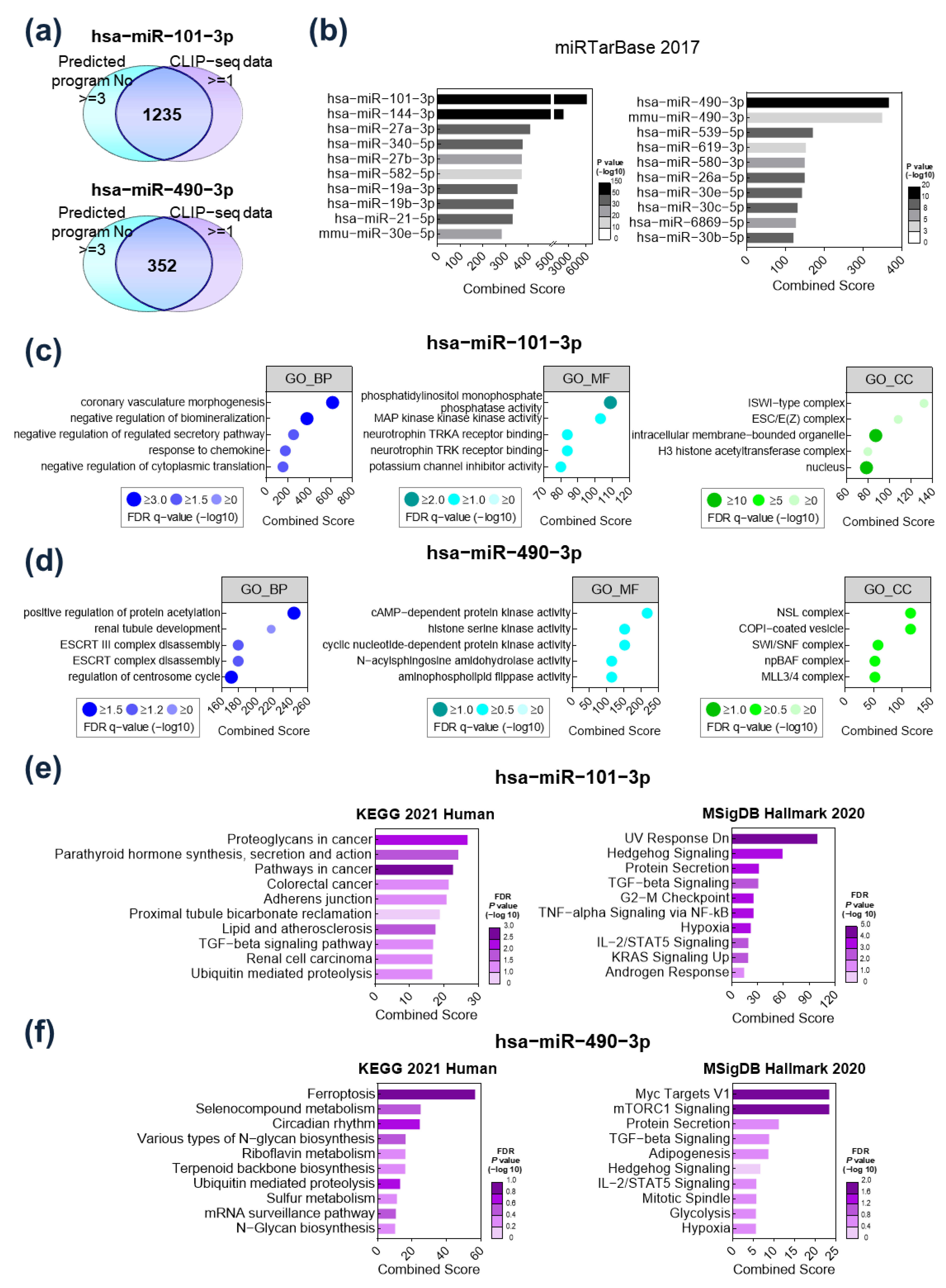
2.2. Functional Analysis of Target Gene Signatures Regulated by miR-101-3P and miR-490-3p
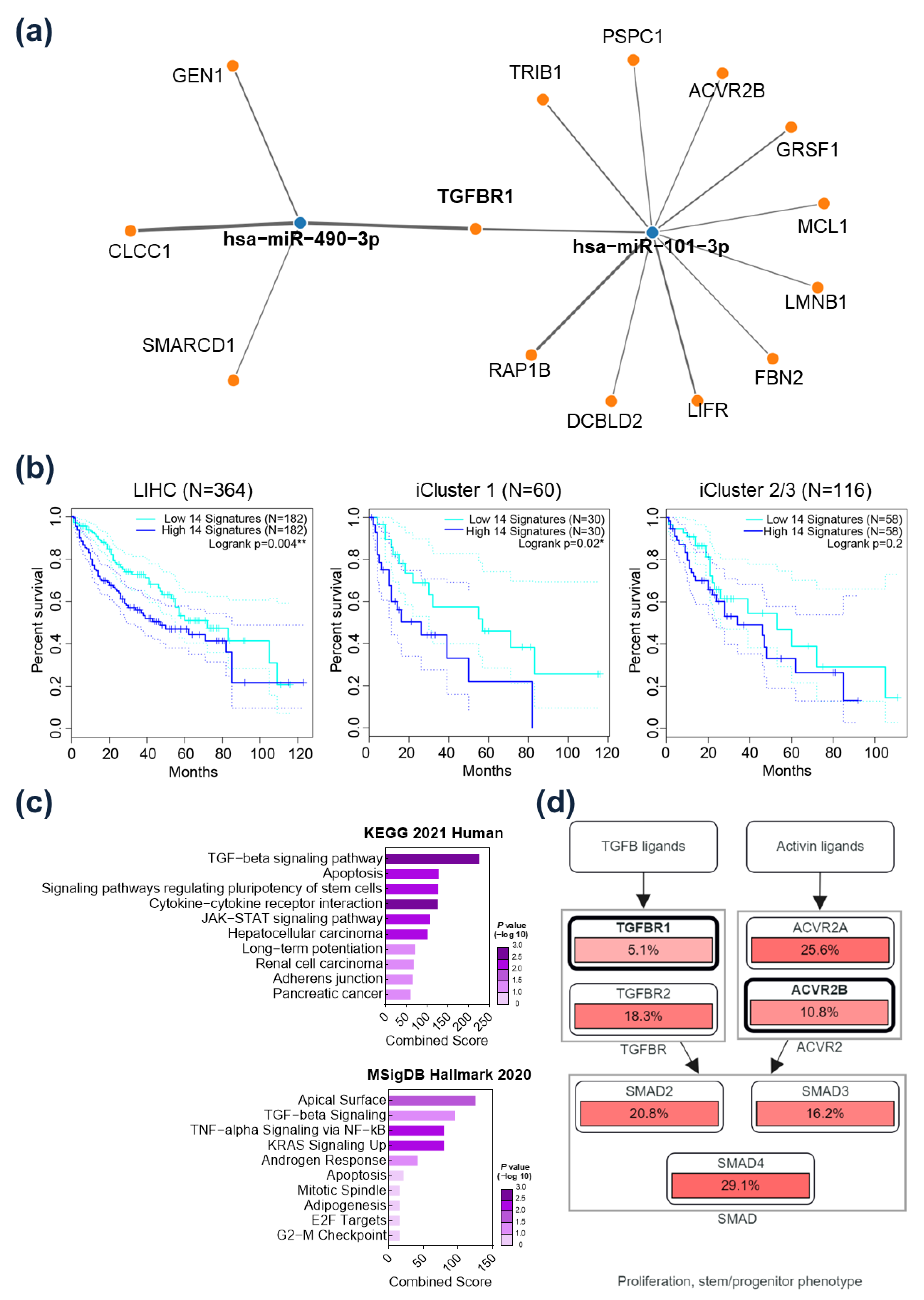
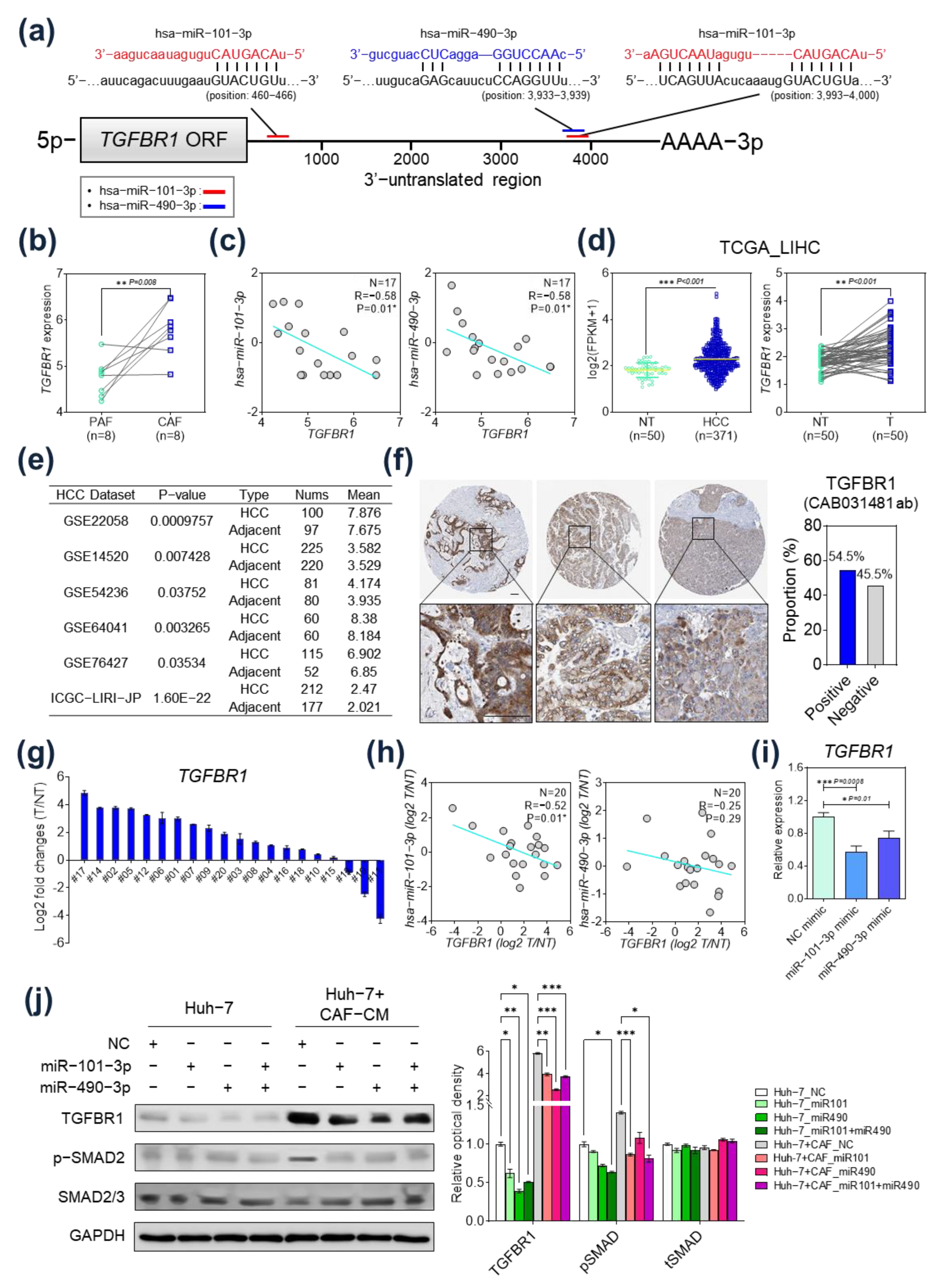
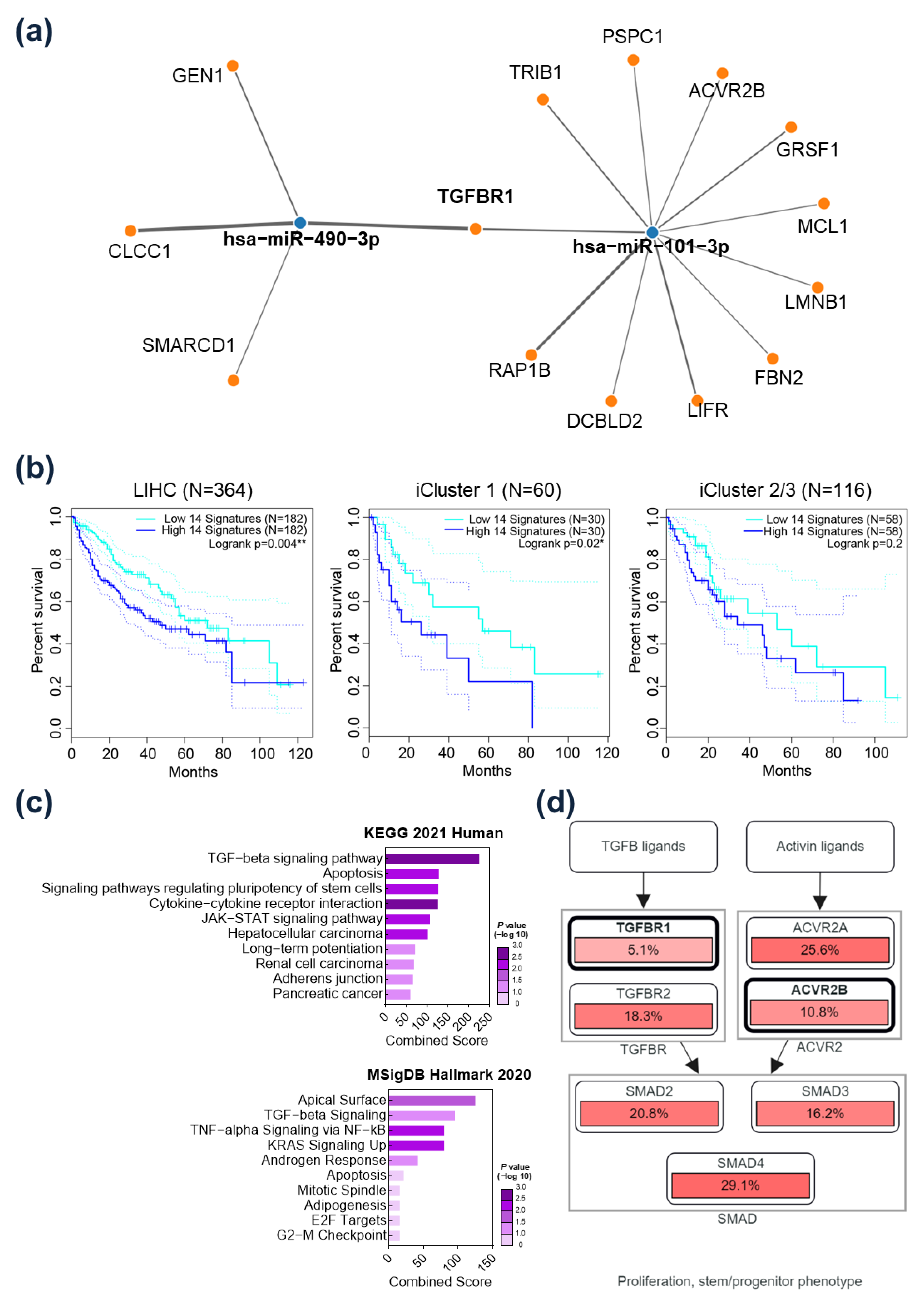
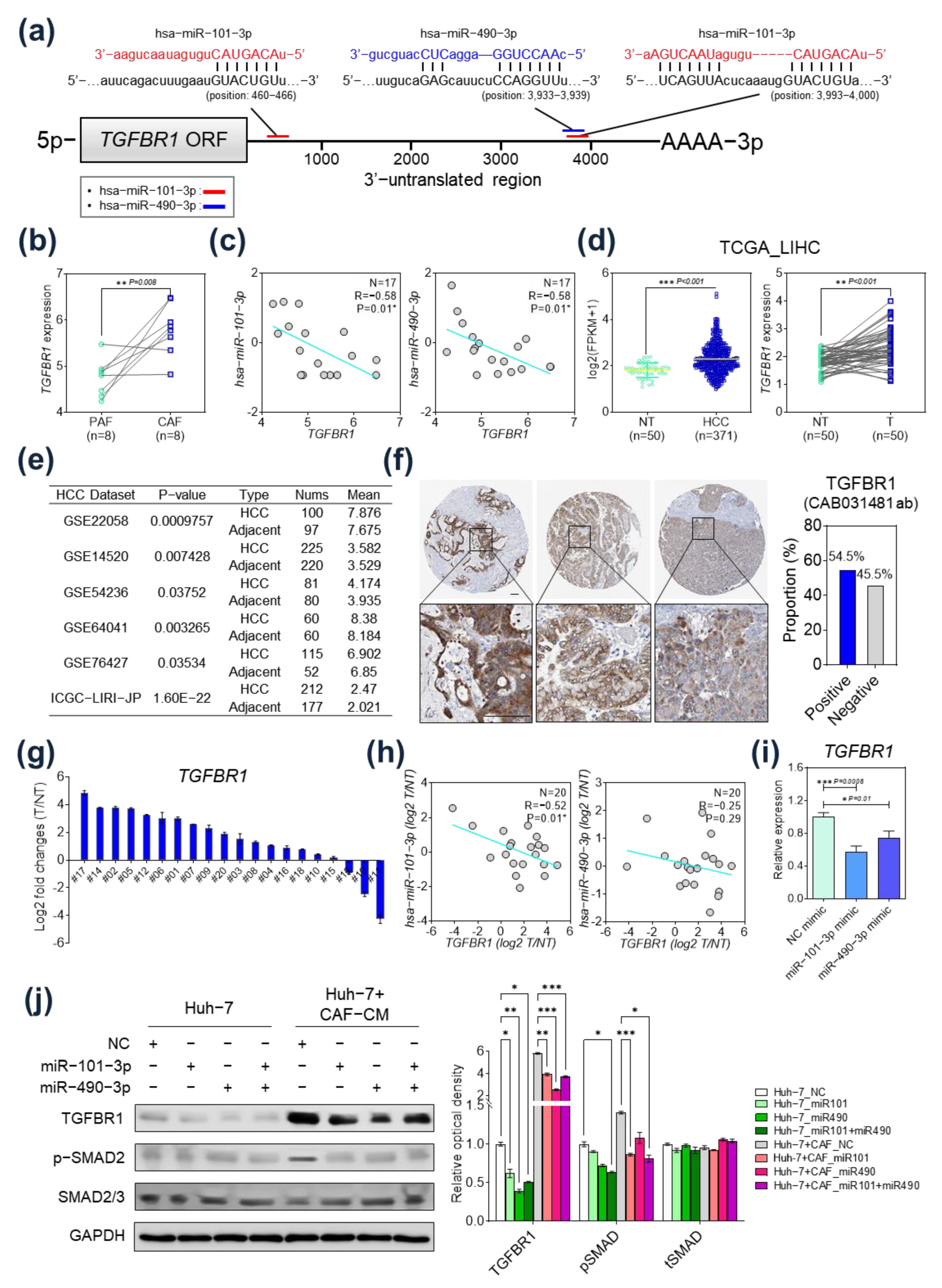
2.3. TGFBR1 Was Identified as a Common Target Gene of miR-101-3p and miR-490-3p
2.4. TGFBR1 Expression Was Negatively Regulated by hsa-miR-101-3p and hsa-miR-490-3p
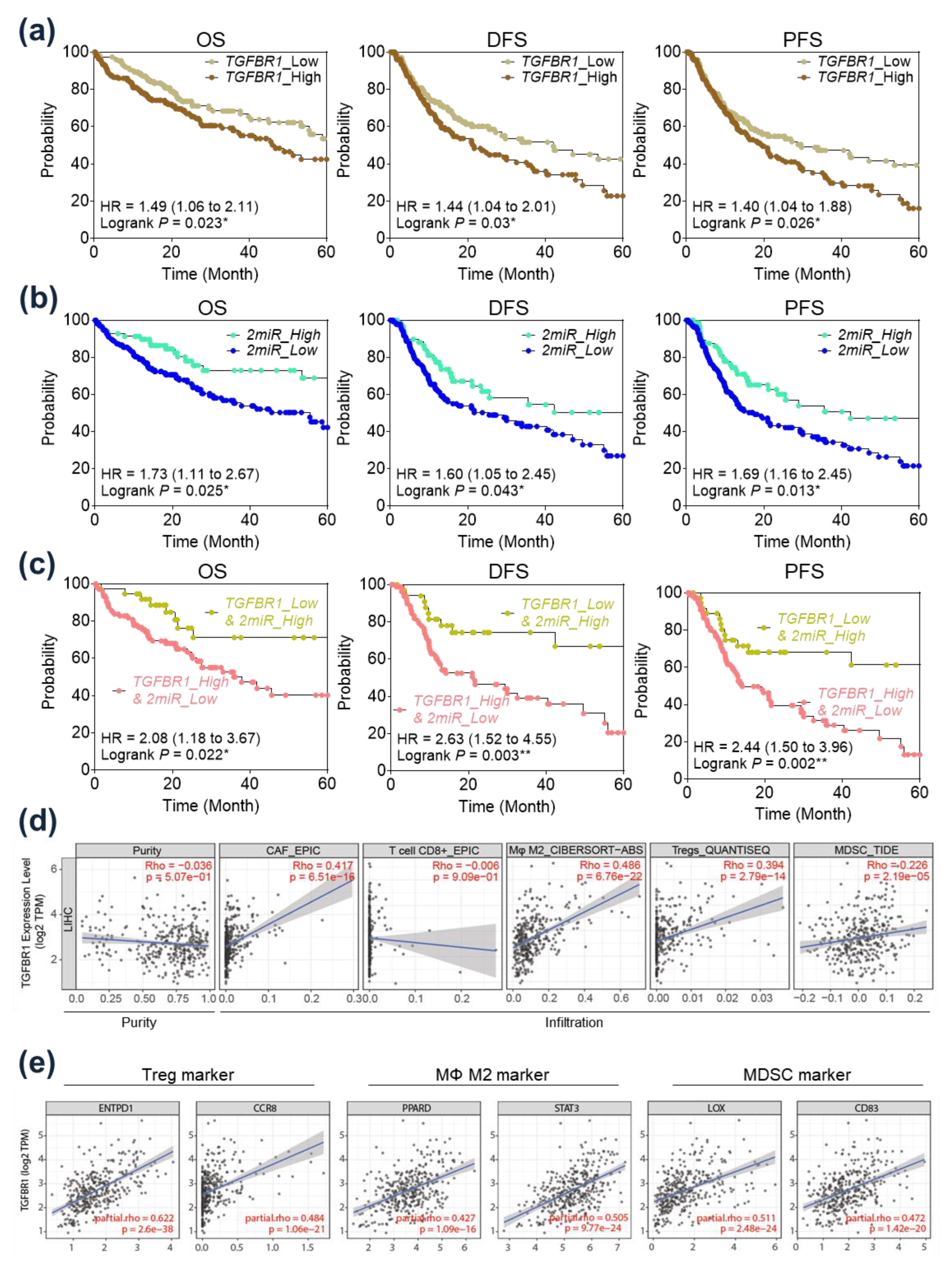
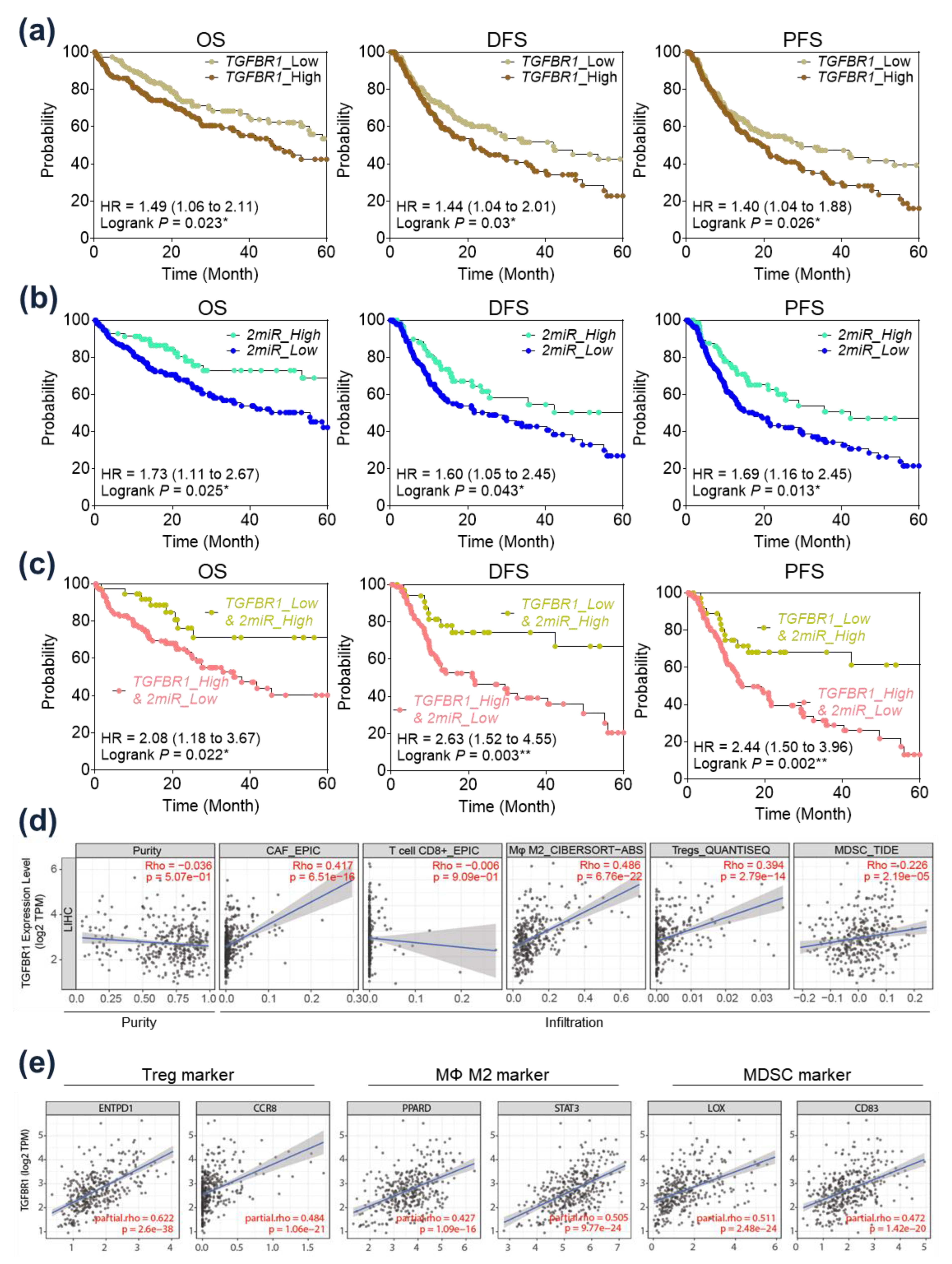
2.5. Prognostic Implication of TGFBR1 in HCC Patients
2.6. Immunological Implication of TGFBR1 in HCC Patients
3. Discussion
4. Materials and Methods
4.1. Isolation of CAFs, PAFs, and NFs from Surgically Resected Liver Tissue
4.2. Small-RNA-Sequencing
4.3. Gene Expression Profiling Using Public Omics Databases
4.4. Prediction of miR Target Genes
4.5. Gene Ontology Analysis and Molecular Pathway Mining
4.6. Network Analysis between the Hub-Target Genes and CAF-Related miRs
4.7. Survival Analyses
4.8. TGFBR1 Protein Expression in Human HCC tissues
4.9. Cell Culture, Collection of CAF-CM, and miR Mimic Transfection
4.10. RNA Isolation and Quantitative Reverse-Transcription Polymerase Chain Reaction Analysis
4.11. Western Blot Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi Rad, H.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O’Byrne, K.; Rezaei, N.; Kulasinghe, A. Understanding the tumor microenvironment for effective immunotherapy. Med. Res. Rev. 2021, 41, 1474–1498. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or tumor suppressor? The duplicity of microRNAs in cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Barrera, L.N.; Ridley, P.M.; Bermejo-Rodriguez, C.; Costello, E.; Perez-Mancera, P.A. The role of microRNAs in the modulation of cancer-associated fibroblasts activity during pancreatic cancer pathogenesis. J. Physiol. Biochem. 2022, 79, 193–204. [Google Scholar] [CrossRef]
- Savardashtaki, A.; Shabaninejad, Z.; Movahedpour, A.; Sahebnasagh, R.; Mirzaei, H.; Hamblin, M.R. miRNAs derived from cancer-associated fibroblasts in colorectal cancer. Epigenomics 2019, 11, 1627–1645. [Google Scholar] [CrossRef]
- Cancer Gemone Atlas Research Network. Electronic address: Sheeler@bcm.edu; Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aprelikova, O.; Green, J.E. MicroRNA regulation in cancer-associated fibroblasts. Cancer Immunol. Immunother. 2012, 61, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoepp, M.; Strose, A.J.; Haier, J. Dysregulation of miRNA expression in cancer associated fibroblasts (CAFs) and its consequences on the tumor microenvironment. Cancers 2017, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Chen, M.; Cao, L.; Hu, Y.; Li, X.; Zhang, Q.; Ren, Y.; Wu, X.; Meng, Z.; Xu, K. Cancer-associated fibroblasts promote migration and invasion of non-small cell lung cancer cells via miR-101-3p mediated VEGFA secretion and AKT/eNOS pathway. Front. Cell Dev. Biol. 2021, 9, 764151. [Google Scholar] [CrossRef]
- Zhao, L.; Sun, Y.; Hou, Y.; Peng, Q.; Wang, L.; Luo, H.; Tang, X.; Zeng, Z.; Liu, M. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int. J. Biochem. Cell Biol. 2012, 44, 2051–2059. [Google Scholar] [CrossRef]
- Yang, J.; Lu, Y.; Lin, Y.Y.; Zheng, Z.Y.; Fang, J.H.; He, S.; Zhuang, S.M. Vascular mimicry formation is promoted by paracrine TGF-beta and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer Lett. 2016, 383, 18–27. [Google Scholar] [CrossRef]
- Chen, S.; Chen, X.; Xiu, Y.L.; Sun, K.X.; Zhao, Y. MicroRNA-490-3P targets CDK1 and inhibits ovarian epithelial carcinoma tumorigenesis and progression. Cancer Lett. 2015, 362, 122–130. [Google Scholar] [CrossRef]
- Xu, X.; Chen, R.; Li, Z.; Huang, N.; Wu, X.; Li, S.; Li, Y.; Wu, S. MicroRNA-490-3p inhibits colorectal cancer metastasis by targeting TGFbetaR1. BMC Cancer 2015, 15, 1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, W.; Choi, S.K.; Kim, D.; Kim, H.G.; Park, J.K.; Han, J.E.; Cho, G.J.; Yun, S.; Yu, W.; Han, S.H.; et al. ZNF507 affects TGF-beta signaling via TGFBR1 and MAP3K8 activation in the progression of prostate cancer to an aggressive state. J. Exp. Clin. Cancer Res. 2021, 40, 291. [Google Scholar] [CrossRef] [PubMed]
- Moore-Smith, L.; Pasche, B. TGFBR1 signaling and breast cancer. J. Mammary Gland Biol. Neoplasia 2011, 16, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Pasche, B.; Pennison, M.J.; Jimenez, H.; Wang, M. TGFBR1 and cancer susceptibility. Trans. Am. Clin. Climatol. Assoc. 2014, 125, 300–312. [Google Scholar] [PubMed]
- Rosman, D.S.; Phukan, S.; Huang, C.C.; Pasche, B. TGFBR1*6A enhances the migration and invasion of MCF-7 breast cancer cells through RhoA activation. Cancer Res. 2008, 68, 1319–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, S.; Shetty, K.; Yu, H.; Wong, L.; Rao, S.; Jogunoori, W.; Amdur, R.; Li, S.; Latham, P.; Nguyen, B.N. TGF-β receptors 1 and 2 are functional biomarkers that stratify risk of hepatocellular cancer (HCC). Artificial intelligence based validation at three centers. Cancer Res. 2021, 81, 2544. [Google Scholar] [CrossRef]
- Wang, J.; Xiang, H.; Lu, Y.; Wu, T. Role and clinical significance of TGFbeta1 and TGFbetaR1 in malignant tumors. Int. J. Mol. Med. 2021, 47, 55. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Tauriello, D.V.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Gonzalez-Sanchez, E.; Vaquero, J.; Fernandez-Barrena, M.G.; Lasarte, J.J.; Avila, M.A.; Sarobe, P.; Reig, M.; Calvo, M.; Fabregat, I. The TGF-β pathway: A pharmacological target in hepatocellular carcinoma? Cancers 2021, 13, 3248. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, Z.; Xu, X. Molecular subtyping of hepatocellular carcinoma: A step toward precision medicine. Cancer Commun. 2020, 40, 681–693. [Google Scholar] [CrossRef]
- Rhee, H.; Kim, H.; Park, Y.N. Clinico-radio-pathological and molecular features of hepatocellular carcinomas with keratin 19 expression. Liver Cancer 2020, 9, 663–681. [Google Scholar] [CrossRef] [PubMed]
- Vasievich, E.A.; Huang, L. The suppressive tumor microenvironment: A challenge in cancer immunotherapy. Mol. Pharm. 2011, 8, 635–641. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Dong, C.; Jiang, K.; Xu, Z.; Li, R.; Guo, K.; Shao, S.; Wang, L. Heterogeneity of cancer-associated fibroblasts and roles in the progression, prognosis, and therapy of hepatocellular carcinoma. J. Hematol. Oncol. 2019, 12, 101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eun, J.W.; Ahn, H.R.; Baek, G.O.; Yoon, M.G.; Son, J.A.; Weon, J.H.; Yoon, J.H.; Kim, H.S.; Han, J.E.; Kim, S.S.; et al. Aberrantly Expressed MicroRNAs in Cancer-Associated Fibroblasts and Their Target Oncogenic Signatures in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 4272. https://doi.org/10.3390/ijms24054272
Eun JW, Ahn HR, Baek GO, Yoon MG, Son JA, Weon JH, Yoon JH, Kim HS, Han JE, Kim SS, et al. Aberrantly Expressed MicroRNAs in Cancer-Associated Fibroblasts and Their Target Oncogenic Signatures in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2023; 24(5):4272. https://doi.org/10.3390/ijms24054272
Chicago/Turabian StyleEun, Jung Woo, Hye Ri Ahn, Geum Ok Baek, Moon Gyeong Yoon, Ju A Son, Ji Hyang Weon, Jung Hwan Yoon, Hyung Seok Kim, Ji Eun Han, Soon Sun Kim, and et al. 2023. "Aberrantly Expressed MicroRNAs in Cancer-Associated Fibroblasts and Their Target Oncogenic Signatures in Hepatocellular Carcinoma" International Journal of Molecular Sciences 24, no. 5: 4272. https://doi.org/10.3390/ijms24054272
APA StyleEun, J. W., Ahn, H. R., Baek, G. O., Yoon, M. G., Son, J. A., Weon, J. H., Yoon, J. H., Kim, H. S., Han, J. E., Kim, S. S., Cheong, J. Y., Kim, B.-w., & Cho, H. J. (2023). Aberrantly Expressed MicroRNAs in Cancer-Associated Fibroblasts and Their Target Oncogenic Signatures in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 24(5), 4272. https://doi.org/10.3390/ijms24054272