
Long-Term SMN- and Ncald-ASO Combinatorial Therapy in SMA Mice and NCALD-ASO Treatment in hiPSC-Derived Motor Neurons Show Protective Effects

 , ,
, ,
Abstract

1. Introduction
2. Results
2.1. Re-Injection of Ncald-ASO Significantly Reduces NCALD in Brain and Spinal Cord
2.2. Long-Term Combinatorial Treatment with Ncald- and SMN-ASOs Ameliorates Electrophysiological Defects and NMJ Denervation in SMA Mice
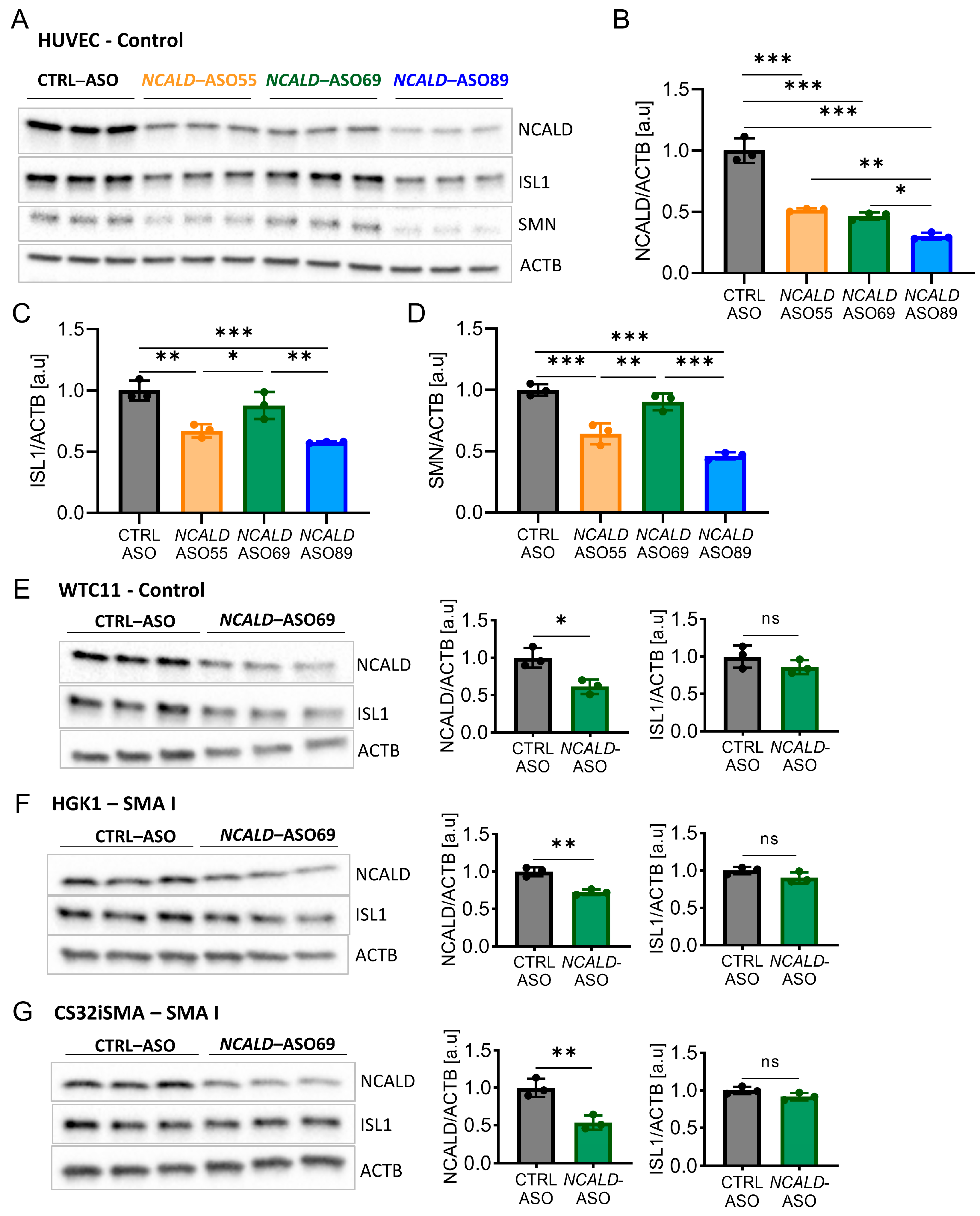
2.3. Testing of Human NCALD-ASOs in MNs Derived from SMA and Control hiPSCs
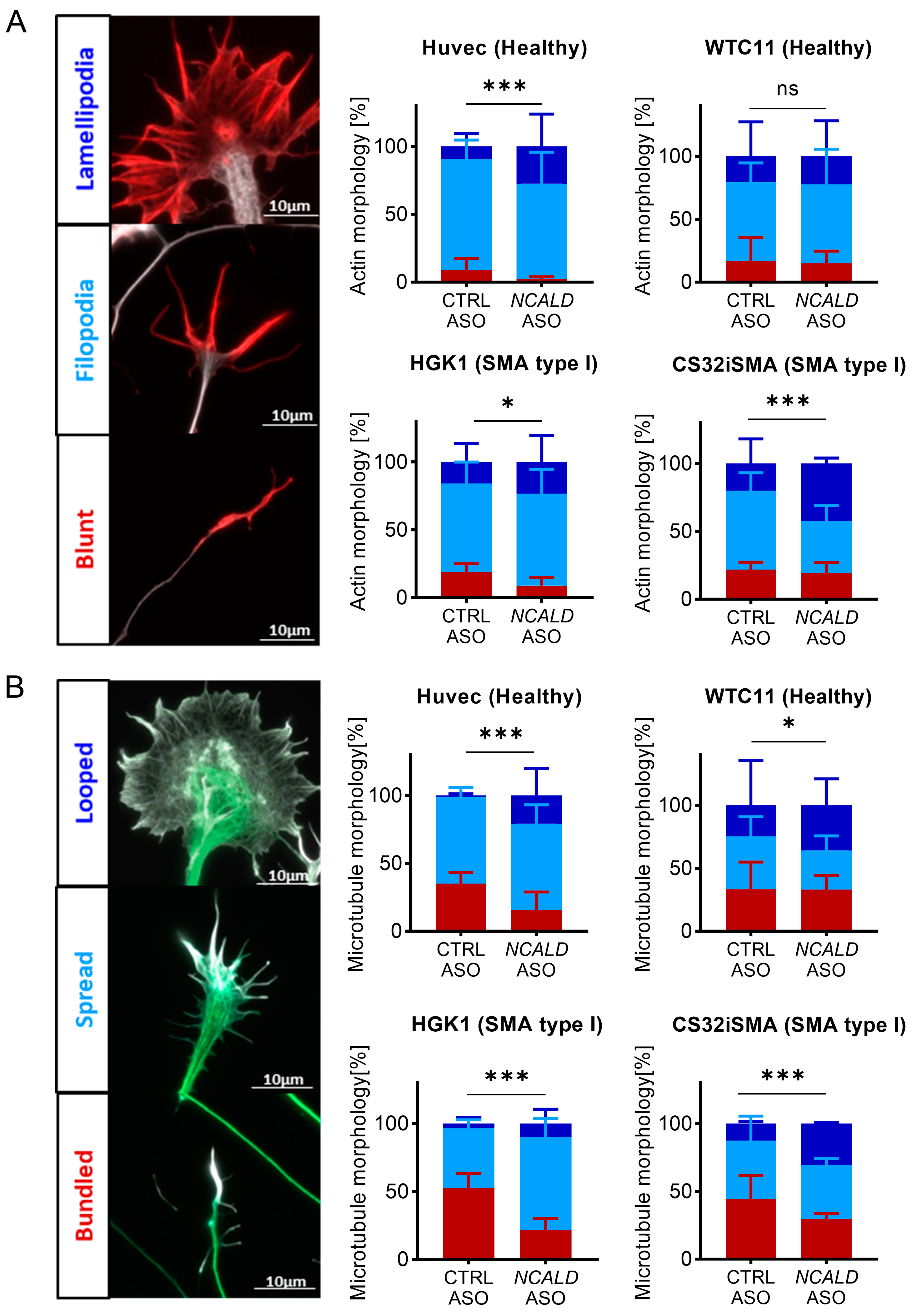
2.4. Treatment with NCALD-ASO69 Influences Growth Cone Morphology
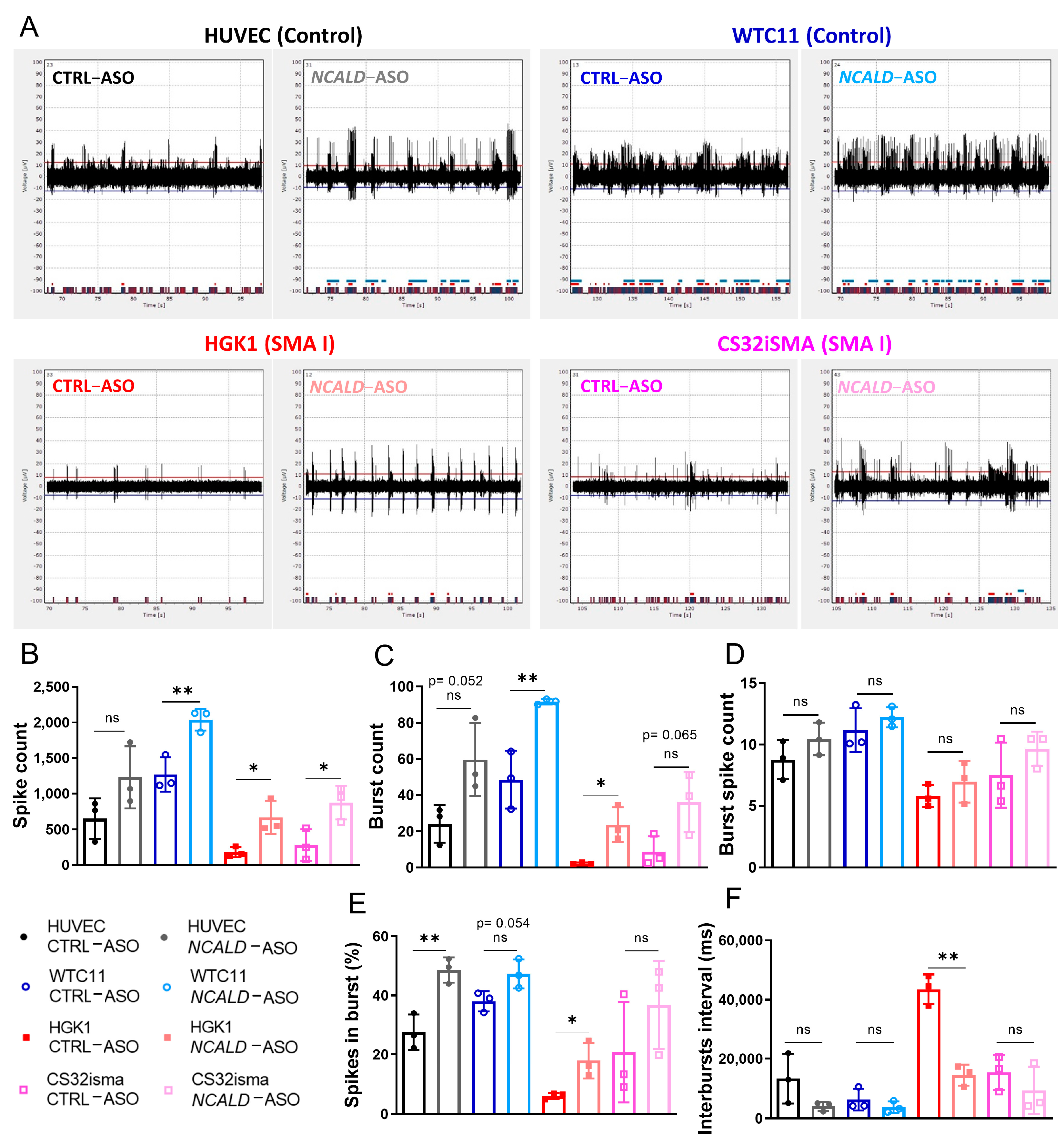
2.5. Treatment with NCALD-ASO69 Increases Neuronal Activity
3. Discussion
3.1. Ncald-ASO Re-Injection Prolongs Amelioration of Electrophysiological Defects and NMJ Pathology in SMA Mice
3.2. NCALD-ASO69 Treatment Improves Cytoskeleton Dynamics and Neuronal Activity in hiPSC-Derived MNs
4. Materials and Methods
4.1. Mouse Model and Genotyping
4.2. Antisense Oligonucleotides (ASOs)
4.3. ASOs Injection In Vivo
4.4. Experimental Design
4.5. Western Blot
4.6. Compound Muscle Action Potential and Motor Unit Number Estimation
4.7. Analysis NMJ from the Transversus Abdominis (TVA)
4.8. Muscle Fiber Analysis
4.9. hiPSC Maintenance and Differentiation into MNs
4.10. NCALD-ASOs Treatment in hiPSC-Derived MNs
4.11. hiPSCs and MNs Immunohistochemistry
4.12. Multielectrode Array
4.13. Image Acquisition and Analysis
4.14. Image Acquisition and Analysis
4.15. Statistics
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in Type 1 Spinal Muscular Atrophy. N. Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Darras, B.T.; Masson, R.; Mazurkiewicz-Beldzinska, M.; Rose, K.; Xiong, H.; Zanoteli, E.; Baranello, G.; Bruno, C.; Vlodavets, D.; Wang, Y.; et al. Risdiplam-Treated Infants with Type 1 Spinal Muscular Atrophy versus Historical Controls. N. Engl. J. Med. 2021, 385, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Wirth, B. Spinal Muscular Atrophy: In the Challenge Lies a Solution. Trends Neurosci. 2021, 44, 306–322. [Google Scholar] [CrossRef]
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu. Rev. Genom. Hum. Genet. 2020, 21, 231–261. [Google Scholar] [CrossRef]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Meyer, O.H.; Simonds, A.K.; Schroth, M.K.; Graham, R.J.; Kirschner, J.; Iannaccone, S.T.; Crawford, T.O.; Woods, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 2018, 28, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Princivalle, A.; Forti, F.; Lizier, C.; Zeviani, M. Expression of the SMN gene, the spinal muscular atrophy determining gene, in the mammalian central nervous system. Hum. Mol. Genet. 1997, 6, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Coovert, D.D.; Le, T.T.; McAndrew, P.E.; Strasswimmer, J.; Crawford, T.O.; Mendell, J.R.; Coulson, S.E.; Androphy, E.J.; Prior, T.W.; Burghes, A.H. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997, 6, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Zilio, E.; Piano, V.; Wirth, B. Mitochondrial Dysfunction in Spinal Muscular Atrophy. Int. J. Mol. Sci. 2022, 23, 10878. [Google Scholar] [CrossRef]
- Cuartas, J.; Gangwani, L. R-loop Mediated DNA Damage and Impaired DNA Repair in Spinal Muscular Atrophy. Front. Cell Neurosci. 2022, 16, 826608. [Google Scholar] [CrossRef]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Groen, E.J.N.; Perenthaler, E.; Courtney, N.L.; Jordan, C.Y.; Shorrock, H.K.; van der Hoorn, D.; Huang, Y.T.; Murray, L.M.; Viero, G.; Gillingwater, T.H. Temporal and tissue-specific variability of SMN protein levels in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2018, 27, 2851–2862. [Google Scholar] [CrossRef]
- Kariya, S.; Obis, T.; Garone, C.; Akay, T.; Sera, F.; Iwata, S.; Homma, S.; Monani, U.R. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J. Clin. Investig. 2014, 124, 785–800. [Google Scholar] [CrossRef]
- Lauria, F.; Bernabo, P.; Tebaldi, T.; Groen, E.J.N.; Perenthaler, E.; Maniscalco, F.; Rossi, A.; Donzel, D.; Clamer, M.; Marchioretto, M.; et al. SMN-primed ribosomes modulate the translation of transcripts related to spinal muscular atrophy. Nat. Cell Biol. 2020, 22, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Vill, K.; Schwartz, O.; Blaschek, A.; Glaser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Roschinger, W.; Becker, M.; Czibere, L.; et al. Newborn screening for spinal muscular atrophy in Germany: Clinical results after 2 years. Orphanet. J. Rare Dis. 2021, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Farrar, M.A.; Muntoni, F.; Saito, K.; Mendell, J.R.; Servais, L.; McMillan, H.J.; Finkel, R.S.; Swoboda, K.J.; Kwon, J.M.; et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: The Phase III SPR1NT trial. Nat. Med. 2022, 28, 1381–1389. [Google Scholar] [CrossRef]
- Strauss, K.A.; Farrar, M.A.; Muntoni, F.; Saito, K.; Mendell, J.R.; Servais, L.; McMillan, H.J.; Finkel, R.S.; Swoboda, K.J.; Kwon, J.M.; et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: The Phase III SPR1NT trial. Nat. Med. 2022, 28, 1390–1397. [Google Scholar] [CrossRef]
- Dangouloff, T.; Hiligsmann, M.; Deconinck, N.; D’Amico, A.; Seferian, A.M.; Boemer, F.; Servais, L. Financial cost and quality of life of patients with spinal muscular atrophy identified by symptoms or newborn screening. Dev. Med. Child. Neurol. 2023, 65, 67–77. [Google Scholar] [CrossRef]
- Oprea, G.E.; Krober, S.; McWhorter, M.L.; Rossoll, W.; Muller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Riessland, M.; Kaczmarek, A.; Schneider, S.; Swoboda, K.J.; Lohr, H.; Bradler, C.; Grysko, V.; Dimitriadi, M.; Hosseinibarkooie, S.; Torres-Benito, L.; et al. Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am. J. Hum. Genet. 2017, 100, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Torres-Benito, L.; Schneider, S.; Rombo, R.; Ling, K.K.; Grysko, V.; Upadhyay, A.; Kononenko, N.L.; Rigo, F.; Bennett, C.F.; Wirth, B. NCALD Antisense Oligonucleotide Therapy in Addition to Nusinersen further Ameliorates Spinal Muscular Atrophy in Mice. Am. J. Hum. Genet. 2019, 105, 221–230. [Google Scholar] [CrossRef]
- Hosseinibarkooie, S.; Peters, M.; Torres-Benito, L.; Rastetter, R.H.; Hupperich, K.; Hoffmann, A.; Mendoza-Ferreira, N.; Kaczmarek, A.; Janzen, E.; Milbradt, J.; et al. The Power of Human Protective Modifiers: PLS3 and CORO1C Unravel Impaired Endocytosis in Spinal Muscular Atrophy and Rescue SMA Phenotype. Am. J. Hum. Genet. 2016, 99, 647–665. [Google Scholar] [CrossRef] [PubMed]
- Wolff, L.; Strathmann, E.A.; Muller, I.; Mahlich, D.; Veltman, C.; Niehoff, A.; Wirth, B. Plastin 3 in health and disease: A matter of balance. Cell Mol. Life Sci. 2021, 78, 5275–5301. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadi, M.; Derdowski, A.; Kalloo, G.; Maginnis, M.S.; O’Hern, P.; Bliska, B.; Sorkac, A.; Nguyen, K.C.; Cook, S.J.; Poulogiannis, G.; et al. Decreased function of survival motor neuron protein impairs endocytic pathways. Proc. Natl. Acad. Sci. USA 2016, 113, E4377–E4386. [Google Scholar] [CrossRef] [PubMed]
- Kaifer, K.A.; Villalon, E.; Osman, E.Y.; Glascock, J.J.; Arnold, L.L.; Cornelison, D.D.W.; Lorson, C.L. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2017, 2, e89970. [Google Scholar] [CrossRef] [PubMed]
- Alrafiah, A.; Karyka, E.; Coldicott, I.; Iremonger, K.; Lewis, K.E.; Ning, K.; Azzouz, M. Plastin 3 Promotes Motor Neuron Axonal Growth and Extends Survival in a Mouse Model of Spinal Muscular Atrophy. Mol. Ther. Methods Clin. Dev. 2018, 9, 81–89. [Google Scholar] [CrossRef]
- Walsh, M.B.; Janzen, E.; Wingrove, E.; Hosseinibarkooie, S.; Muela, N.R.; Davidow, L.; Dimitriadi, M.; Norabuena, E.M.; Rubin, L.L.; Wirth, B.; et al. Genetic modifiers ameliorate endocytic and neuromuscular defects in a model of spinal muscular atrophy. BMC Biol. 2020, 18, 127. [Google Scholar] [CrossRef]
- Muinos-Buhl, A.; Rombo, R.; Janzen, E.; Ling, K.K.; Hupperich, K.; Rigo, F.; Bennett, C.F.; Wirth, B. Combinatorial ASO-mediated therapy with low dose SMN and the protective modifier Chp1 is not sufficient to ameliorate SMA pathology hallmarks. Neurobiol. Dis. 2022, 171, 105795. [Google Scholar] [CrossRef]
- Janzen, E.; Mendoza-Ferreira, N.; Hosseinibarkooie, S.; Schneider, S.; Hupperich, K.; Tschanz, T.; Grysko, V.; Riessland, M.; Hammerschmidt, M.; Rigo, F.; et al. CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain 2018, 141, 2343–2361. [Google Scholar] [CrossRef]
- Lewelt, A.; Krosschell, K.J.; Scott, C.; Sakonju, A.; Kissel, J.T.; Crawford, T.O.; Acsadi, G.; D’Anjou, G.; Elsheikh, B.; Reyna, S.P.; et al. Compound muscle action potential and motor function in children with spinal muscular atrophy. Muscle Nerve 2010, 42, 703–708. [Google Scholar] [CrossRef]
- Arnold, W.D.; Porensky, P.N.; McGovern, V.L.; Iyer, C.C.; Duque, S.; Li, X.; Meyer, K.; Schmelzer, L.; Kaspar, B.K.; Kolb, S.J.; et al. Electrophysiological Biomarkers in Spinal Muscular Atrophy: Preclinical Proof of Concept. Ann. Clin. Transl. Neurol. 2014, 1, 34–44. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Ruiz, S.; Yi, F.; Herrerias, A.; Batchelder, E.M.; Izpisua Belmonte, J.C. Rapid and highly efficient generation of induced pluripotent stem cells from human umbilical vein endothelial cells. PLoS ONE 2011, 6, e19743. [Google Scholar] [CrossRef] [PubMed]
- Garbes, L.; Heesen, L.; Holker, I.; Bauer, T.; Schreml, J.; Zimmermann, K.; Thoenes, M.; Walter, M.; Dimos, J.; Peitz, M.; et al. VPA response in SMA is suppressed by the fatty acid translocase CD36. Hum. Mol. Genet. 2013, 22, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Rossoll, W.; Jablonka, S.; Andreassi, C.; Kroning, A.K.; Karle, K.; Monani, U.R.; Sendtner, M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 2003, 163, 801–812. [Google Scholar] [CrossRef]
- Moradi, M.; Sivadasan, R.; Saal, L.; Luningschror, P.; Dombert, B.; Rathod, R.J.; Dieterich, D.C.; Blum, R.; Sendtner, M. Differential roles of alpha-, beta-, and gamma-actin in axon growth and collateral branch formation in motoneurons. J. Cell Biol. 2017, 216, 793–814. [Google Scholar] [CrossRef]
- Iino, S.; Kobayashi, S.; Hidaka, H. Neurocalcin-immunopositive nerve terminals in the muscle spindle, Golgi tendon organ and motor endplate. Brain Res. 1998, 808, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Ivings, L.; Pennington, S.R.; Jenkins, R.; Weiss, J.L.; Burgoyne, R.D. Identification of Ca2+-dependent binding partners for the neuronal calcium sensor protein neurocalcin delta: Interaction with actin, clathrin and tubulin. Biochem. J. 2002, 363, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Khazaei, M.R.; Girouard, M.P.; Alchini, R.; Ong Tone, S.; Shimada, T.; Bechstedt, S.; Cowan, M.; Guillet, D.; Wiseman, P.W.; Brouhard, G.; et al. Collapsin response mediator protein 4 regulates growth cone dynamics through the actin and microtubule cytoskeleton. J. Biol. Chem. 2014, 289, 30133–30143. [Google Scholar] [CrossRef]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urban, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the brain: Endocannabinoids shape neuronal connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Van Alstyne, M.; Tattoli, I.; Delestree, N.; Recinos, Y.; Workman, E.; Shihabuddin, L.S.; Zhang, C.; Mentis, G.Z.; Pellizzoni, L. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat. Neurosci. 2021, 24, 930–940. [Google Scholar] [CrossRef]
- Arnold, W.D.; Sheth, K.A.; Wier, C.G.; Kissel, J.T.; Burghes, A.H.; Kolb, S.J. Electrophysiological Motor Unit Number Estimation (MUNE) Measuring Compound Muscle Action Potential (CMAP) in Mouse Hindlimb Muscles. J. Vis. Exp. 2015, 103, e52899. [Google Scholar] [CrossRef]
- Boyd, P.J.; Gillingwater, T.H. Axonal and Neuromuscular Junction Pathology in Spinal Muscular Atrophy. In Spinal Musculat Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 133–151. [Google Scholar]
- Woschitz, V.; Mei, I.; Hedlund, E.; Murray, L.M. Mouse models of SMA show divergent patterns of neuronal vulnerability and resilience. Skelet. Muscle 2022, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Gromova, A.; La Spada, A.R. Harmony Lost: Cell-Cell Communication at the Neuromuscular Junction in Motor Neuron Disease. Trends Neurosci. 2020, 43, 709–724. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Tzartos, S.; Evoli, A.; Palace, J.; Burns, T.M.; Verschuuren, J. Myasthenia gravis. Nat. Rev. Dis. Primers 2019, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Simard, L.R. Survival motor neuron (SMN) protein: Role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum. Mol. Genet. 2002, 11, 1605–1614. [Google Scholar] [CrossRef]
- Nolle, A.; Zeug, A.; van Bergeijk, J.; Tonges, L.; Gerhard, R.; Brinkmann, H.; Al Rayes, S.; Hensel, N.; Schill, Y.; Apkhazava, D.; et al. The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum. Mol. Genet. 2011, 20, 4865–4878. [Google Scholar] [CrossRef]
- Hensel, N.; Claus, P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neuroscientist 2018, 24, 54–72. [Google Scholar] [CrossRef]
- Monani, U.R.; Coovert, D.D.; Burghes, A.H. Animal models of spinal muscular atrophy. Hum. Mol. Genet. 2000, 9, 2451–2457. [Google Scholar] [CrossRef]
- Fletcher, E.V.; Simon, C.M.; Pagiazitis, J.G.; Chalif, J.I.; Vukojicic, A.; Drobac, E.; Wang, X.; Mentis, G.Z. Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nat. Neurosci. 2017, 20, 905–916. [Google Scholar] [CrossRef]
- Tharaneetharan, A.; Cole, M.; Norman, B.; Romero, N.C.; Wooltorton, J.R.A.; Harrington, M.A.; Sun, J. Functional Abnormalities of Cerebellum and Motor Cortex in Spinal Muscular Atrophy Mice. Neuroscience 2021, 452, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.M.; Blanco-Redondo, B.; Buettner, J.M.; Pagiazitis, J.G.; Fletcher, E.V.; Sime Longang, J.K.; Mentis, G.Z. Chronic Pharmacological Increase of Neuronal Activity Improves Sensory-Motor Dysfunction in Spinal Muscular Atrophy Mice. J. Neurosci. 2021, 41, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, S.; Beck, M.; Lechner, B.D.; Mayer, C.; Sendtner, M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J. Cell Biol. 2007, 179, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, B.; Krober, S.; Torres-Benito, L.; Borgmann, A.; Peters, M.; Hosseini Barkooie, S.M.; Tejero, R.; Jakubik, M.; Schreml, J.; Milbradt, J.; et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 2013, 22, 1328–1347. [Google Scholar] [CrossRef] [PubMed]
- Glascock, J.J.; Osman, E.Y.; Coady, T.H.; Rose, F.F.; Shababi, M.; Lorson, C.L. Delivery of therapeutic agents through intracerebroventricular (ICV) and intravenous (IV) injection in mice. J. Vis. Exp. 2011, 56, e2968. [Google Scholar] [CrossRef]
- DeVos, S.L.; Miller, T.M. Direct intraventricular delivery of drugs to the rodent central nervous system. J. Vis. Exp. 2013, 75, e50326. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovas, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef]
- Delle Vedove, A.; Natarajan, J.; Zanni, G.; Eckenweiler, M.; Muinos-Buhl, A.; Storbeck, M.; Guillen Boixet, J.; Barresi, S.; Pizzi, S.; Holker, I.; et al. CAPRIN1(P512L) causes aberrant protein aggregation and associates with early-onset ataxia. Cell. Mol. Life Sci. 2022, 79, 526. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Phenotype | SMN1/SMN2 Copies | Sex | Age Sampling | Reprogrammed |
|---|---|---|---|---|---|
| HUVEC | Healthy control | 2/2 | male | fetal | Retrovirus |
| WTC11 | Healthy control | 2/2 | male | 30 years | Episomal plasmid |
| HGK1 | SMA I | 0/2 | female | 6 months | Retrovirus |
| CS32iSMA | SMA I | 0/2 | male | 7 months | Episomal plasmid |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muiños-Bühl, A.; Rombo, R.; Ling, K.K.; Zilio, E.; Rigo, F.; Bennett, C.F.; Wirth, B. Long-Term SMN- and Ncald-ASO Combinatorial Therapy in SMA Mice and NCALD-ASO Treatment in hiPSC-Derived Motor Neurons Show Protective Effects. Int. J. Mol. Sci. 2023, 24, 4198. https://doi.org/10.3390/ijms24044198
Muiños-Bühl A, Rombo R, Ling KK, Zilio E, Rigo F, Bennett CF, Wirth B. Long-Term SMN- and Ncald-ASO Combinatorial Therapy in SMA Mice and NCALD-ASO Treatment in hiPSC-Derived Motor Neurons Show Protective Effects. International Journal of Molecular Sciences. 2023; 24(4):4198. https://doi.org/10.3390/ijms24044198
Chicago/Turabian StyleMuiños-Bühl, Anixa, Roman Rombo, Karen K. Ling, Eleonora Zilio, Frank Rigo, C. Frank Bennett, and Brunhilde Wirth. 2023. "Long-Term SMN- and Ncald-ASO Combinatorial Therapy in SMA Mice and NCALD-ASO Treatment in hiPSC-Derived Motor Neurons Show Protective Effects" International Journal of Molecular Sciences 24, no. 4: 4198. https://doi.org/10.3390/ijms24044198
APA StyleMuiños-Bühl, A., Rombo, R., Ling, K. K., Zilio, E., Rigo, F., Bennett, C. F., & Wirth, B. (2023). Long-Term SMN- and Ncald-ASO Combinatorial Therapy in SMA Mice and NCALD-ASO Treatment in hiPSC-Derived Motor Neurons Show Protective Effects. International Journal of Molecular Sciences, 24(4), 4198. https://doi.org/10.3390/ijms24044198