
Approaches for sRNA Analysis of Human RNA-Seq Data: Comparison, Benchmarking
 , , , , , and
, , , , , and
Abstract
1. Introduction
1.1. sRNA Expression Methods
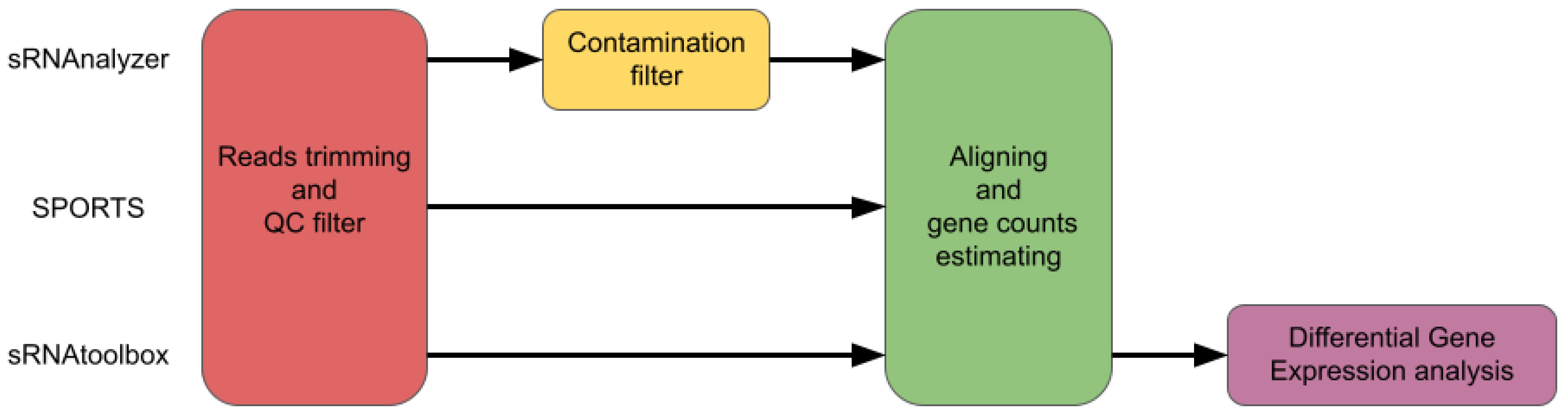
1.2. Alignment-Based Tools
2. Results
2.1. Input Data
2.2. Trimming
2.3. Genome or Transcriptome Aligning
2.4. Assigning


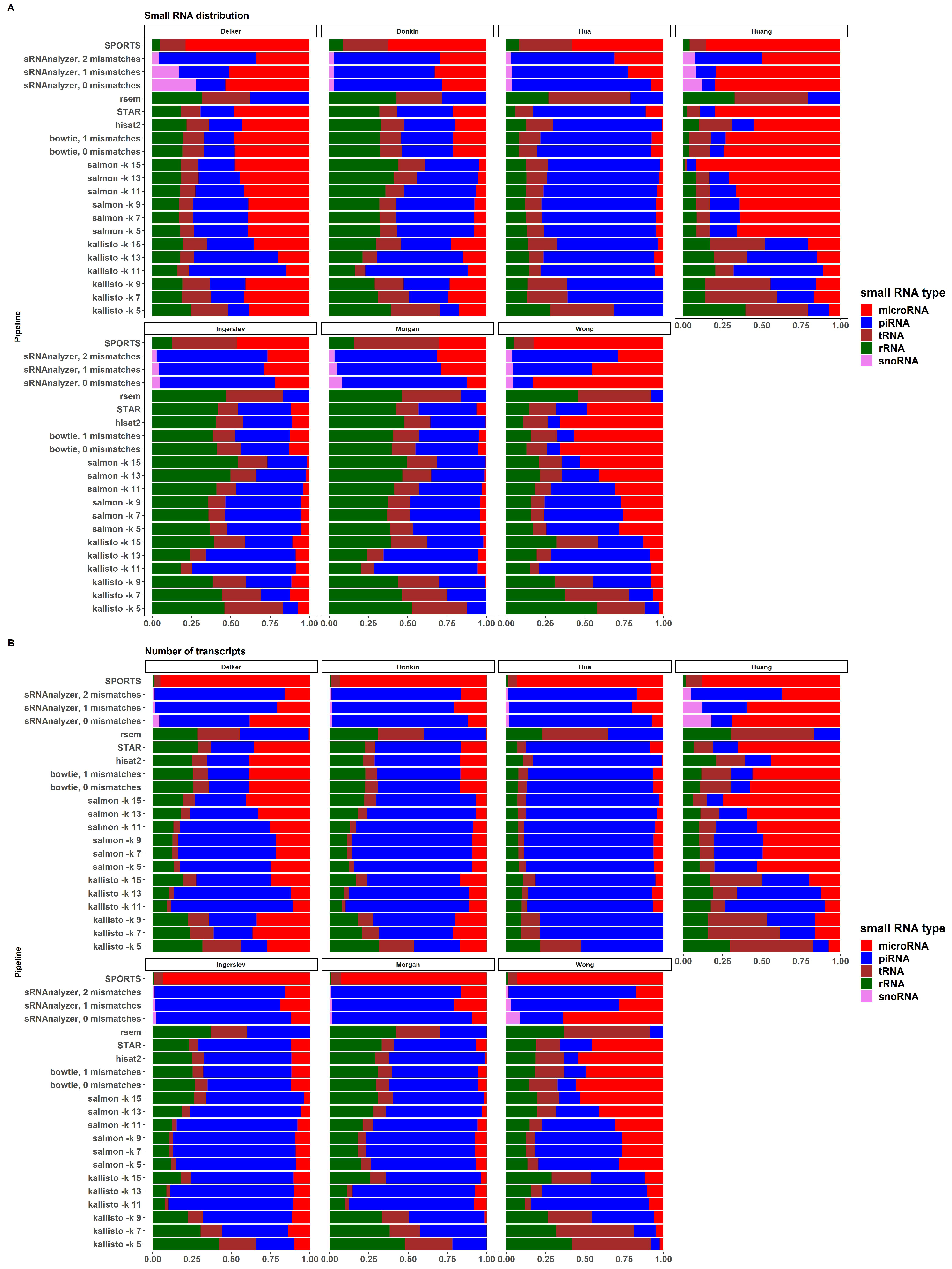
2.5. sRNA Biotypes Distribution

2.6. Filtering
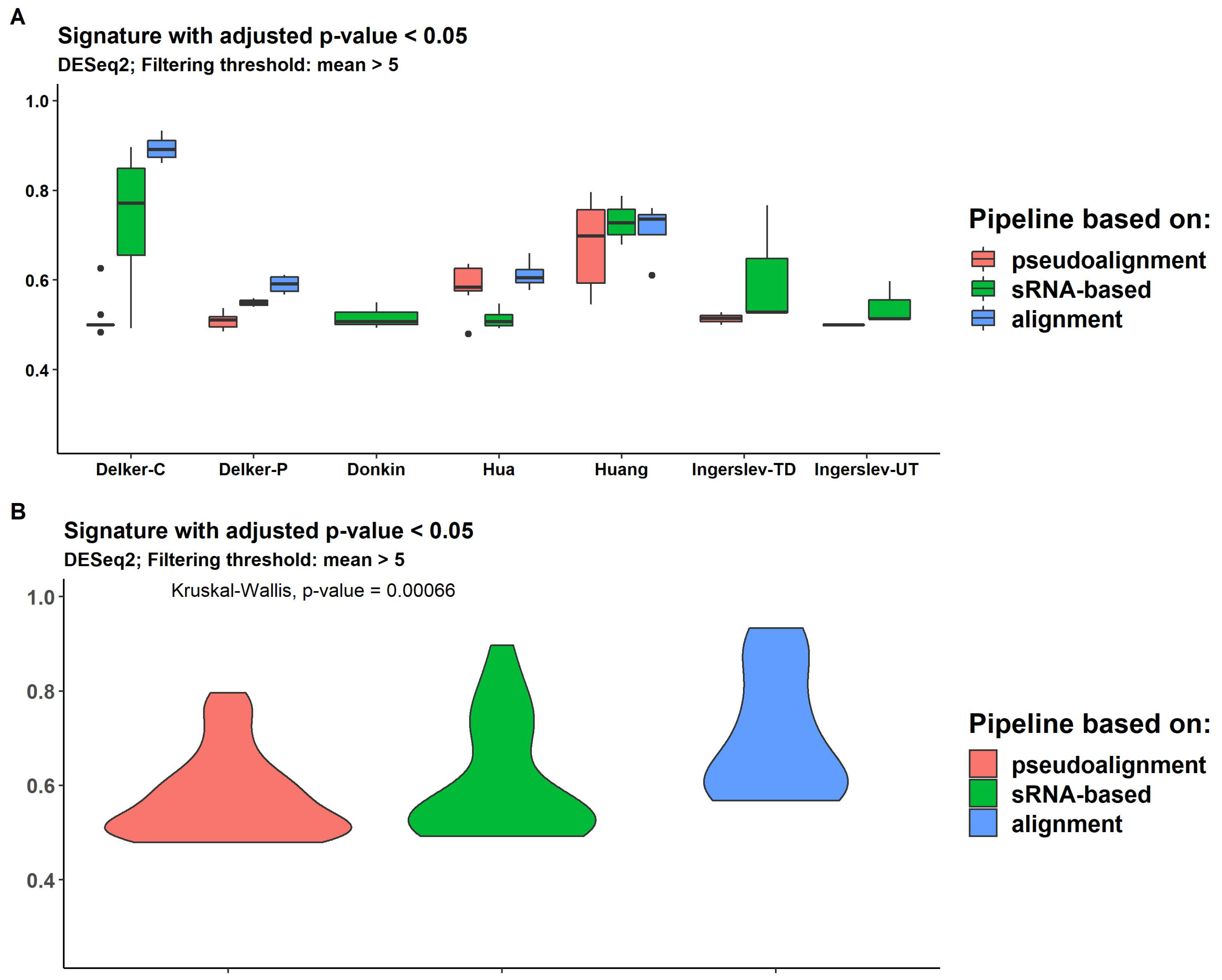
2.7. Differential Expression
2.8. Expression Signature Quality Estimation
2.9. Transfer RNA Fragments Analysis

2.10. sRNA Tools Performance
2.11. Overall Performance and Recommendation
3. Discussion
- Trimming with the lower length bound = 15 and the upper-length bound = Read length − 40% of Adapter length;
- Mapping on a reference genome with bowtie aligner, with one mismatch allowed (-v 1 parameter);
- Filtering by mean threshold > 5;
- DESeq2 for DE analysis with adjusted p-value < 0.05.
4. Materials and Methods
4.1. Data Sources
4.2. Preprocessing of RNA-seq Data
- 45 nt
- Read length − X × Adapter lengthwhere X = 10%, 20%, 30%, ..., 100%
- Read length − X ntwhere X = 3, 6, 9, ..., 30 nt
- Read length × (1 − Xwhere X = 0.05, 0.1, 0.15, ..., 0.5
4.3. Processing of Small RNA-seq Data
4.3.1. sRNAs
4.3.2. tRNA Fragments
4.3.3. sRNA Tools
4.4. Filtering Thresholds
- Min filtering: expression value for a transcript in all samples is higher than the threshold (N)min(counts) > (N);
- Mean filtering: mean expression value for a transcript in all samples is higher than the threshold (N)mean(counts) > N;
- Median filtering: median expression value for a transcript in all samples is higher than the threshold (N)median(counts) > N.
4.5. Differential Expression
- p-value < 0.05;
- Adjusted p-value < 0.05.
- Adjusted p-value threshold (preferable);
- Combination of p-value and absolute fold change thresholds.
4.6. Expression Signature Quality Evaluation
4.7. Stages and Metrics of Benchmarking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| bowtie -v 1 | bowtie with one mismatch allowed |
| bowtie -v 0 | bowtie with no mismatches allowed |
| CLI | command-line-based interfaces |
| DE | Differential expression |
| GEO | Gene Expression Omnibus |
| GO | gene ontology |
| GUI | graphic-based user interface |
| H-score | Hobotnica score |
| ITAS | Integrated Transcript Annotation of Small RNA |
| KEGG | Kyoto encyclopedia of genes and genomes |
| lncRNA | long non-coding RNA |
| miRNA | microRNA |
| NGS | next generation sequencing |
| piRNA | piwi-interacting RNA |
| snoRNA | small nucleolar RNA |
| sRNA | small non-coding RNA |
| rsRNA | small rRNA-derived RNA |
| tsRNA | tRNA-derived small RNA |
References
- Storz, G. An expanding universe of noncoding RNAs. Science 2002, 296, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Peng, J.; Yi, C. The epitranscriptome of small non-coding RNAs. Non-Coding RNA Res. 2021, 6, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Penner-Goeke, S.; Binder, E.B. Epigenetics and depression. Dialogues Clin. Neurosci. 2022, 21, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.; Silva, J.V.; Howl, J.; Santos, M.A.; Fardilha, M. All you need to know about sperm RNAs. Hum. Reprod. Update 2022, 28, 67–91. [Google Scholar] [CrossRef]
- Krawetz, S.A.; Kruger, A.; Lalancette, C.; Tagett, R.; Anton, E.; Draghici, S.; Diamond, M.P. A survey of small RNAs in human sperm. Hum. Reprod. 2011, 26, 3401–3412. [Google Scholar] [CrossRef]
- Oluwayiose, O.A.; Houle, E.; Whitcomb, B.W.; Suvorov, A.; Rahil, T.; Sites, C.K.; Krawetz, S.A.; Visconti, P.; Pilsner, J.R. Altered non-coding RNA profiles of seminal plasma extracellular vesicles of men with poor semen quality undergoing in vitro fertilization treatment. Andrology 2022. [Google Scholar] [CrossRef]
- Marcho, C.; Oluwayiose, O.A.; Pilsner, J.R. The preconception environment and sperm epigenetics. Andrology 2020, 8, 924–942. [Google Scholar] [CrossRef]
- Kotsyfakis, M.; Patelarou, E. MicroRNAs as biomarkers of harmful environmental and occupational exposures: A systematic review. Biomarkers 2019, 24, 623–630. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Rassoulzadegan, M.; Tuorto, F.; Chen, Q. Sperm RNA code programmes the metabolic health of offspring. Nat. Rev. Endocrinol. 2019, 15, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Cecere, G. Small RNAs in epigenetic inheritance: From mechanisms to trait transmission. Febs. Lett. 2021, 595, 2953–2977. [Google Scholar] [CrossRef] [PubMed]
- Micheel, J.; Safrastyan, A.; Wollny, D. Advances in Non-Coding RNA Sequencing. Non-Coding RNA 2021, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Benesova, S.; Kubista, M.; Valihrach, L. Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis. Diagnostics 2021, 11, 964. [Google Scholar] [CrossRef]
- Zytnicki, M.; Gaspin, C. srnaMapper: An optimal mapping tool for sRNA-Seq reads. BMC Bioinform. 2022, 23, 495. [Google Scholar] [CrossRef]
- Roovers, E.F.; Rosenkranz, D.; Mahdipour, M.; Han, C.T.; He, N.; de Sousa Lopes, S.M.C.; van der Westerlaken, L.A.; Zischler, H.; Butter, F.; Roelen, B.A.; et al. Piwi proteins and piRNAs in mammalian oocytes and early embryos. Cell Rep. 2015, 10, 2069–2082. [Google Scholar] [CrossRef]
- Han, B.W.; Wang, W.; Zamore, P.D.; Weng, Z. piPipes: A set of pipelines for piRNA and transposon analysis via small RNA-seq, RNA-seq, degradome-and CAGE-seq, ChIP-seq and genomic DNA sequencing. Bioinformatics 2015, 31, 593–595. [Google Scholar] [CrossRef]
- Ray, R.; Pandey, P. piRNA analysis framework from small RNA-Seq data by a novel cluster prediction tool-PILFER. Genomics 2018, 110, 355–365. [Google Scholar] [CrossRef]
- Jung, I.; Park, J.C.; Kim, S. piClust: A density based piRNA clustering algorithm. Comput. Biol. Chem. 2014, 50, 60–67. [Google Scholar] [CrossRef]
- Rosenkranz, D.; Zischler, H. proTRAC-a software for probabilistic piRNA cluster detection, visualization and analysis. BMC Bioinform. 2012, 13, 5. [Google Scholar] [CrossRef]
- Hackenberg, M.; Rodríguez-Ezpeleta, N.; Aransay, A.M. miRanalyzer: An update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res. 2011, 39, W132–W138. [Google Scholar] [CrossRef] [PubMed]
- Stocks, M.B.; Mohorianu, I.; Beckers, M.; Paicu, C.; Moxon, S.; Thody, J.; Dalmay, T.; Moulton, V. The UEA sRNA Workbench (version 4.4): A comprehensive suite of tools for analyzing miRNAs and sRNAs. Bioinformatics 2018, 34, 3382–3384. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Chen, W.X.; Mei, S.Q.; Yang, Y.D.; Yang, J.H.; Qu, L.H.; Zheng, L.L. tsRFun: A comprehensive platform for decoding human tsRNA expression, functions and prognostic value by high-throughput small RNA-Seq and CLIP-Seq data. Nucleic Acids Res. 2022, 50, D421–D431. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Puerta, E.; Lebrón, R.; Rueda, A.; Gómez-Martín, C.; Giannoukakos, S.; Jaspez, D.; Medina, J.M.; Zubkovic, A.; Jurak, I.; Fromm, B.; et al. sRNAbench and sRNAtoolbox 2019: Intuitive fast small RNA profiling and differential expression. Nucleic Acids Res. 2019, 47, W530–W535. [Google Scholar] [CrossRef]
- Wu, X.; Kim, T.K.; Baxter, D.; Scherler, K.; Gordon, A.; Fong, O.; Etheridge, A.; Galas, D.J.; Wang, K. sRNAnalyzer—A flexible and customizable small RNA sequencing data analysis pipeline. Nucleic Acids Res. 2017, 45, 12140–12151. [Google Scholar] [CrossRef]
- Shi, J.; Ko, E.A.; Sanders, K.M.; Chen, Q.; Zhou, T. SPORTS1. 0: A tool for annotating and profiling non-coding RNAs optimized for rRNA-and tRNA-derived small RNAs. Genom. Proteom. Bioinform. 2018, 16, 144–151. [Google Scholar] [CrossRef]
- Pogorelcnik, R.; Vaury, C.; Pouchin, P.; Jensen, S.; Brasset, E. sRNAPipe: A Galaxy-based pipeline for bioinformatic in-depth exploration of small RNAseq data. Mob. DNA 2018, 9, 25. [Google Scholar] [CrossRef]
- Panero, R.; Rinaldi, A.; Memoli, D.; Nassa, G.; Ravo, M.; Rizzo, F.; Tarallo, R.; Milanesi, L.; Weisz, A.; Giurato, G. iSmaRT: A toolkit for a comprehensive analysis of small RNA-Seq data. Bioinformatics 2017, 33, 938–940. [Google Scholar] [CrossRef]
- Rahman, R.U.; Gautam, A.; Bethune, J.; Sattar, A.; Fiosins, M.; Magruder, D.S.; Capece, V.; Shomroni, O.; Bonn, S. Oasis 2: Improved online analysis of small RNA-seq data. BMC Bioinform. 2018, 19, 54. [Google Scholar] [CrossRef]
- Stupnikov, A.; Bezuglov, V.; Skakov, I.; Shtratnikova, V.; Pilsner, J.R.; Suvorov, A.; Sergeyev, O. ITAS: Integrated Transcript Annotation for Small RNA. Non-Coding RNA 2022, 8, 30. [Google Scholar] [CrossRef]
- Quek, C.; Jung, C.h.; Bellingham, S.A.; Lonie, A.; Hill, A.F. iSRAP–a one-touch research tool for rapid profiling of small RNA-seq data. J. Extracell. Vesicles 2015, 4, 29454. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, S.; La Ferlita, A.; Carapezza, G.; Alaimo, S.; Isacchi, A.; Ferro, A.; Pulvirenti, A.; Bosotti, R. A benchmarking of pipelines for detecting ncRNAs from RNA-Seq data. Brief. Bioinform. 2019, 21, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Bruno, V.M.; Rasko, D.A.; Cuomo, C.A.; Muñoz, J.F.; Livny, J.; Shetty, A.C.; Mahurkar, A.; Dunning Hotopp, J.C. Best practices on the differential expression analysis of multi-species RNA-seq. Genome Biol. 2021, 22, 21. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Davis, J.; Li, X.; Smith, K.S.; Battle, A.; Montgomery, S.B. High-resolution transcriptome analysis with long-read RNA sequencing. PLoS ONE 2014, 9, e108095. [Google Scholar] [CrossRef] [PubMed]
- Stupnikov, A.; Glazko, G.V.; Emmert-Streib, F. Effects of subsampling on characteristics of RNA-seq data from triple-negative breast cancer patients. Chin. J. Cancer 2015, 34, 36. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Delorenzi, M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinform. 2013, 14, 91. [Google Scholar] [CrossRef]
- Rapaport, F.; Khanin, R.; Liang, Y.; Pirun, M.; Krek, A.; Zumbo, P.; Mason, C.E.; Socci, N.D.; Betel, D. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013, 14, 3158. [Google Scholar] [CrossRef]
- Assefa, A.T.; De Paepe, K.; Everaert, C.; Mestdagh, P.; Thas, O.; Vandesompele, J. Differential gene expression analysis tools exhibit substandard performance for long non-coding RNA-sequencing data. Genome Biol. 2018, 19, 96. [Google Scholar] [CrossRef]
- Stupnikov, A.; McInerney, C.; Savage, K.; McIntosh, S.; Emmert-Streib, F.; Kennedy, R.; Salto-Tellez, M.; Prise, K.; McArt, D. Robustness of differential gene expression analysis of RNA-seq. Comput. Struct. Biotechnol. J. 2021, 19, 3470–3481. [Google Scholar] [CrossRef]
- Wong, R.K.; MacMahon, M.; Woodside, J.V.; Simpson, D.A. A comparison of RNA extraction and sequencing protocols for detection of small RNAs in plasma. BMC Genom. 2019, 20, 446. [Google Scholar] [CrossRef]
- Huang, G.; Cao, M.; Huang, Z.; Xiang, Y.; Liu, J.; Wang, Y.; Wang, J.; Yang, W. Small RNA-sequencing identified the potential roles of neuron differentiation and MAPK signaling pathway in dilated cardiomyopathy. Biomed. Pharmacother. 2019, 114, 108826. [Google Scholar] [CrossRef]
- Kanth, P.; Hazel, M.W.; Boucher, K.M.; Yang, Z.; Wang, L.; Bronner, M.P.; Boylan, K.E.; Burt, R.W.; Westover, M.; Neklason, D.W.; et al. Small RNA sequencing of sessile serrated polyps identifies microRNA profile associated with colon cancer. Genes Chromosom. Cancer 2019, 58, 23–33. [Google Scholar] [CrossRef]
- Morgan, C.P.; Shetty, A.C.; Chan, J.C.; Berger, D.S.; Ament, S.A.; Epperson, C.N.; Bale, T.L. Repeated sampling facilitates within- and between-subject modeling of the human sperm transcriptome to identify dynamic and stress-responsive sncRNAs. Sci. Rep. 2020, 10, 17498. [Google Scholar] [CrossRef] [PubMed]
- Hua, M.; Liu, W.; Chen, Y.; Zhang, F.; Xu, B.; Liu, S.; Chen, G.; Shi, H.; Wu, L. Identification of small non-coding RNAs as sperm quality biomarkers for in vitro fertilization. Cell Discov. 2019, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.R.; Jørgensen, N.; Kristiansen, V.B.; et al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell Metab. 2016, 23, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Ingerslev, L.R.; Donkin, I.; Fabre, O.; Versteyhe, S.; Mechta, M.; Pattamaprapanont, P.; Mortensen, B.; Krarup, N.T.; Barrès, R. Endurance training remodels sperm-borne small RNA expression and methylation at neurological gene hotspots. Clin. Epigenet. 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software (accessed on 11 August 2022).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010; Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 11 August 2022).
- Available online: https://international.neb.com/faqs/2017/07/17/how-should-my-nebnext-small-rna-library-be-trimmed (accessed on 11 August 2022).
- Available online: https://support.illumina.com/bulletins/2016/12/what-sequences-do-i-use-for-adapter-trimming.html (accessed on 11 August 2022).
- Available online: https://perkinelmer-appliedgenomics.com/wp-content/uploads/marketing/NEXTFLEX/miRNA/NEXTflex_Small_RNA_v3_Trimming_Instructions.pdf (accessed on 11 August 2022).
- Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26/ (accessed on 11 August 2022).
- Stupnikov, A.; Sizykh, A.; Budkina, A.; Favorov, A.; Afsari, B.; Wheelan, S.; Marchionni, L.; Medvedeva, Y. Hobotnica: Exploring molecular signature quality [version 2; peer review: 2 approved]. F1000Research 2022, 10, 1260. [Google Scholar] [CrossRef]
- Lamb, J. The Connectivity Map: A new tool for biomedical research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef]
- Musa, A.; Ghoraie, L.S.; Zhang, S.D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Brief. Bioinform. 2018, 19, 506–523. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. goseq: Gene Ontology testing for RNA-seq datasets. R Bioconductor 2012, 8, 1–25. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J.; et al. The UCSC genome browser database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Year | Last Update | Status | Interface | Type | RNA Classes | Output |
|---|---|---|---|---|---|---|---|
| DSAP | 2010 | 2010 | not supported | GUI | map and | ncRNA (Rfam), miRNA | DE |
| for Solexa only | remove | transcripts | |||||
| Oasis 2 | 2018 | 2018 | not supported | online | map and | miRNA, piRNA, | transcripts |
| remove | small nucleolar RNA (snoRNA) | counts | |||||
| iSmaRT | 2017 | 2017 | supported | GUI | map and | miRNA, piRNA | DE |
| from ftp-server | remove | transcripts | |||||
| sRNAPipe | 2018 | 2021 | supported | GUI | map and | miRNA, piRNA, | transcripts |
| Galaxy-based | remove | rRNA, tRNAs, | counts | ||||
| transposable elements | |||||||
| iSRAP | 2015 | 2016 | supported | CLI | map and | miRNA, piRNA, snoRNA | DE |
| remove | transcripts | ||||||
| miARma-seq | 2016 | 2019 | not supported | CLI | map and | miRNA, snoRNA | DE |
| remove | transcripts | ||||||
| tsRFun | 2022 | 2022 | supported | online | map and | tsRNA | DE |
| remove | transcripts | ||||||
| piPipes | 2015 | 2016 | supported | CLI | map | piRNA | transcripts |
| counts | |||||||
| PILFER | 2018 | 2018 | supported | CLI | map | piRNA | transcripts |
| counts | |||||||
| miRanalyzer | 2011 | 2014 | not supported | online | map and | miRNA | DE |
| remove | transcripts | ||||||
| sRNA workbench | 2018 | 2018 | not supported | CLI | map | miRNA | alignment |
| sRNAtoolbox | 2022 | 2022 | supported | online | map and | miRNA, tRNA, | DE |
| remove | ncRNA, cDNA | transcripts | |||||
| sRNAnalyzer | 2017 | 2017 | supported | CLI | map and | miRNA, piRNA, tRNA, | transcripts |
| remove | snoRNA | counts | |||||
| SPORTS | 2018 | 2021 | supported | CLI | map and | miRNA, piRNA, rRNAs, | transcripts |
| remove | tRNAs, tRNA fragments | counts |
| Stage | Pipeline Command | Justification |
|---|---|---|
| Trimming | Read length: lower bound-15 and | Retain sufficient and the same number of reads after |
| upper bound-Read length − 40% of adapter length | trimming for downstream analyses for all datasets. | |
| Aligning | bowtie aligner with 1 mismatch allowed | The high alignment rate and H-score for all datasets. |
| Assigning | ITAS [30] | Optimized annotation for small RNA. |
| Filtering | mean count > 5 | Sufficient number of transcripts for the |
| downstream analysis and higher H-score. | ||
| DE analysis | DESeq2 | Sufficient number of significant transcripts and |
| high H-score. |
| Dataset | Biological Object and Contrast | Alignment Rate | Assignment * Rate | Number of Filtered Transcripts ** | Number of Findings *** | H-Score |
|---|---|---|---|---|---|---|
| ”Hua“ | Sperm | 0.94 | 0.29 | 901 | 71 | 0.66 |
| ”Donkin“ | Sperm | 0.97 | 0.07 | 966 | 0 | - |
| ”Ingerslev“ | Sperm untrained contrast | 0.9 | 0.06 | 1236 | 0 | - |
| detrained contrast | 0 | - | ||||
| “Morgan” | Sperm | 0.89 | 0.1 | - | - | - |
| “Delker” | colon cancer polyps contrast | 0.94 | 0.55 | 1142 | 21 | 0.6 |
| controls contrast | 5 | 0.86 | ||||
| “Huang” | Blood | 0.94 | 0.48 | 498 | 17 | 0.73 |
| “Wong” | Blood plasma | 0.68 | 0.38 | - | - | - |
| GEO ID | Cite | Object | Total Samples Number (Contrast Groups) | Raw Reads Length | Library Kit |
|---|---|---|---|---|---|
| GSE118125 | |||||
| https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE118125 | |||||
| (accessed on 11 August 2022) | [58] | Blood plasma | 30 | 76 | NEXTflex |
| CleanTag, Qiaseq | |||||
| GSE117841 | |||||
| https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117841 | |||||
| (accessed on 11 August 2022) | |||||
| [59] | Blood | 20 (10 vs. 10) | 50 | Truseq | |
| GSE118504 | |||||
| https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE118504 | |||||
| (accessed on 11 August 2022) | |||||
| [60] | Colon Cancer | 108 (16 vs. 14 and 15 vs. 15) | 50 | NEBNext, Truseq | |
| GSE159155 | |||||
| https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE159155 | |||||
| (accessed on 11 August 2022) | |||||
| [61] | Sperm | 98 | 50 | Truseq | |
| GSE110190 | |||||
| https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE110190 | |||||
| (accessed on 11 August 2022) | |||||
| [62] | Sperm | 87 (64 vs. 23) | 150 | Illumina | |
| GSE74426 | |||||
| https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74426 | |||||
| (accessed on 11 August 2022) | |||||
| [63] | Sperm | 23 (13 vs. 10) | 42 | NEBNext | |
| GSE109478 | |||||
| https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109478 | |||||
| (accessed on 11 August 2022) | |||||
| [64] | Sperm | 24 (9 vs. 9 and 9 vs. 6) | 51 | NEBNext |
| Stage | Metrics |
|---|---|
| Input data | Biological object, lab kit, read length, adapter length, reads quality |
| Trimming | Part of reads-processed trimming, length threshold |
| Aligning (for genome-alignment-based pipelines) | Alignment rate |
| Assigning | Assignment rate, distribution by sRNA type |
| Filtering | Number of transcripts after trimming |
| DE | Number of significant transcripts |
| Expression signature quality evaluation | H-score |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezuglov, V.; Stupnikov, A.; Skakov, I.; Shtratnikova, V.; Pilsner, J.R.; Suvorov, A.; Sergeyev, O. Approaches for sRNA Analysis of Human RNA-Seq Data: Comparison, Benchmarking. Int. J. Mol. Sci. 2023, 24, 4195. https://doi.org/10.3390/ijms24044195
Bezuglov V, Stupnikov A, Skakov I, Shtratnikova V, Pilsner JR, Suvorov A, Sergeyev O. Approaches for sRNA Analysis of Human RNA-Seq Data: Comparison, Benchmarking. International Journal of Molecular Sciences. 2023; 24(4):4195. https://doi.org/10.3390/ijms24044195
Chicago/Turabian StyleBezuglov, Vitalik, Alexey Stupnikov, Ivan Skakov, Victoria Shtratnikova, J. Richard Pilsner, Alexander Suvorov, and Oleg Sergeyev. 2023. "Approaches for sRNA Analysis of Human RNA-Seq Data: Comparison, Benchmarking" International Journal of Molecular Sciences 24, no. 4: 4195. https://doi.org/10.3390/ijms24044195
APA StyleBezuglov, V., Stupnikov, A., Skakov, I., Shtratnikova, V., Pilsner, J. R., Suvorov, A., & Sergeyev, O. (2023). Approaches for sRNA Analysis of Human RNA-Seq Data: Comparison, Benchmarking. International Journal of Molecular Sciences, 24(4), 4195. https://doi.org/10.3390/ijms24044195