Abstract
Soybean-seed development is controlled in multiple ways, as in many known regulating genes. Here, we identify a novel gene, Novel Seed Size (NSS), involved in seed development, by analyzing a T-DNA mutant (S006). The S006 mutant is a random mutant of the GmFTL4pro:GUS transgenic line, with phenotypes with small and brown seed coats. An analysis of the metabolomics and transcriptome combined with RT-qPCR in the S006 seeds revealed that the brown coat may result from the increased expression of chalcone synthase 7/8 genes, while the down-regulated expression of NSS leads to small seed size. The seed phenotypes and a microscopic observation of the seed-coat integument cells in a CRISPR/Cas9-edited mutant nss1 confirmed that the NSS gene conferred small phenotypes of the S006 seeds. As mentioned in an annotation on the Phytozome website, NSS encodes a potential DNA helicase RuvA subunit, and no such genes were previously reported to be involved in seed development. Therefore, we identify a novel gene in a new pathway controlling seed development in soybeans.
1. Introduction
Soybean (Glycine max (L.) Merr.) is one of the most important multiuse crops, providing plant proteins and edible oils for humans and animals [1]. With the continuous growth in the population, the global demand for soybean is also increasing rapidly. Thus, the improvement of soybean yield is urgently needed. The yield-component factors of soybean include plant architecture, the number of seeds per pod, and seed weight/size [2]. Seed size not only determines yield but also influences plant fitness and adaption [3]. Therefore, the identification of genes controlling seed size and the exploration of the relevant mechanism are among the focuses of soybean research.
Until now, several genes controlling seed size have been isolated and characterized in soybean [4]. The BIG SEEDS1 (BS1) gene, encoding a plant-specific transcription regulator, has been reported to control seed size and weight through a regulatory module that targets primary cell proliferation in soybean [5]. In addition, GmKIX8-1, a negative regulator for cell proliferation, is also involved in the control of seed size [6]. Cytochrome-P450 family members have been confirmed to regulate organ size and development [7]. Furthermore, GmCYP78A10, GmCYP78A57, GmCYP78A70, and GmCYP78A72 all demonstrate positive control over seed size in soybean [8,9]. A putative soybean invertase inhibitor, GmCIF1, participates in controlling seed maturation through specifically depressing CWI (cell-wall invertase) activities [10]. A novel PSK-encoding gene, GmPSKγ1, increases seed size by encouraging cell expansion [11]. A SPINDLY-like gene, GmSSS1, positively regulates soybean-seed size by influencing cell expansion and cell division [12]. In addition, some regulatory genes with natural allelic variations, which relate to seed size and quality, were reported recently [4]. A phosphatase 2C-1 (PP2C-1) allele contributes to increasing seed weight/size by enhancing integument-cell size and activating seed-trait-related genes [13]. In addition, GmSWEET10a and GmSWEET10b, which contribute to sugar allocation from seed coat to embryo, determine the oil and protein contents and seed size in soybean [14,15,16]. Furthermore, GmGA3ox1, which encodes gibberellin 3β-hydroxylase, positively regulates soybean-seed weight [17]. A semi-dominant locus, ST1 (Seed Thickness 1), affects seed thickness and oil content [18], and GmPDAT is responsible for the seed size and oil content in soybean [19]. The discovery of more genes related to seed size will accelerate soybean breeding for yield improvement [4].
Here, we identified a T-DNA insertion mutant, S006, which has brown seed coats and small seeds. Metabolomes revealed that flavonoids accumulated in the S006 seeds, and a transcriptome analysis showed that the up-regulated expression of CHS7/8 might result in the pigmentation of seed coats in the S006 mutant. The resequencing data showed that T-DNA is inserted into chromosome 8 of the soybean genome, which affects the expression levels of flanking genes, including a down-regulated gene, Glyma.08G309000. The CRISPR/Cas9-edited mutants of the Glyma.08G309000 gene demonstrated similar phenotypes of small seeds to the S006 mutant seeds. Therefore, Glyma.08G309000 was named as Novel Seed Size (NSS). Altogether, we identified a novel gene controlling seed development in soybean.
2. Results
2.1. Phenotypes of S006 Mutant
In previous studies, we created a binary expression vector, which carried a GmFTL4 (Glyma.16G044100) promoter driving the GUS reporter gene to elucidate the expression specificity of GmFTL4 (Figure S1), and obtained five independent stable transgenic soybean lines by Agrobacterium-mediated transformation. Unexpectedly, we found a unique line with brown seed coats and small seeds (named S006), which was different from the other transformed lines, which had normal yellow seed coats. Compared with wild-type plants (TL), seeds of the S006 mutant exhibited significantly lower seed width (Figure 1a,d), which resulted in a decrease in 100-seed weight in this mutant (Figure 1c). Consistent with these results, the cotyledon area of the S006 also decreased compared with the wild-type plants (Figure 1f). However, the plant height, number of pods per plant and number of seeds per plant showed no significant difference between the S006 mutant and the wild type (Figure S2).
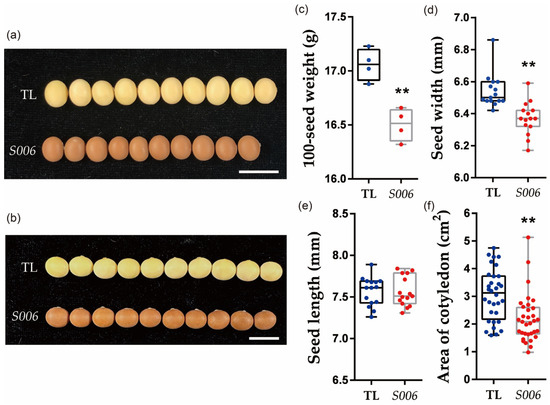
Figure 1.
Comparative analysis of phenotypes between S006 mutant and wild-type seeds. (a) Image of seeds showing decreases in width in S006 mutant (10 seeds are shown in each case); (b) image of seeds showing no significant changes in length in S006 mutant; (c) 100-seed weight was significantly decreased in S006 mutant; (d) seed width significantly decreased in S006 mutant.; (e) there was no significant difference in seed length between S006 mutant and wild type; (f) cotyledon area significantly decreased in S006 mutant. Asterisks indicate a significant difference according to Student’s t-test (**, p < 0.01).
Because the seed size was greatly affected by the integument size, which is determined by cell proliferation and cell expansion [13,20], the areas of the outer-integument cells in the seeds of the wild-type and S006 plants were compared. We found that the area of the outer-integument cells in the seeds of the S006 mutant was significantly decreased compared with that of the wild type (Figure 2a–c).

Figure 2.
Comparison of seed-integument cells between S006 and wild-type plants. (a) Image of integument cells in wild type; (b) image of integument cells in S006 mutant; (c) statistical analysis of cell area of S006 and wild-type seeds. Asterisks indicate a significant difference according to Student’s t-test (**, p < 0.01).
2.2. Metabolite Profiling of S006 Mutant and Wild-Type Seeds
To further explore the changes in metabolism in the S006 mutant seeds, differential metabolites were analyzed among the seed coats of the S006 (SZP) versus the TL (TZP) and the cotyledons of the S006 (SZZ) versus the TL (TZZ). In total, 255 significant diverse metabolites were identified among the SZP versus the TZP, comprising 159 up-regulated and 96 down-regulated metabolites (Figure S3a). Most of these up-regulated metabolites were flavonoids, while the down-regulated metabolites were phenolic acids (Table S1). Furthermore, a total of 268 significant diverse metabolites were identified among the SZZ versus the TZZ, with 202 up-regulated and 66 down-regulated metabolites (Figure S3b). Compared with the SZP versus the TZP, the up-regulated metabolites of the SZZ versus the TZZ included lipids, organic acids, amino acids and derivatives, in addition to flavonoids (Table S2). The heat maps of the differential metabolites among these two combinations also clearly indicated the similar variations (Figure 3a,b).
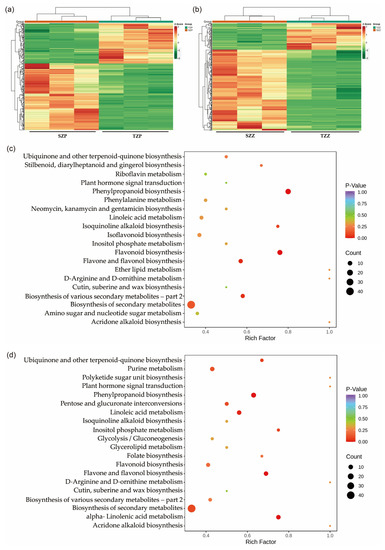
Figure 3.
Differentially accumulated metabolites among seed coats and cotyledons of S006 vs. TL. (a) A heat map of differential metabolites among SZP vs. TZP; (b) a heat map of differential metabolites among SZZ vs. TZZ; (c) KEGG-pathway-enrichment analysis of differential metabolites among SZP vs. TZP; (d) KEGG-pathway-enrichment analysis of differential metabolites among SZZ vs. TZZ.
To further investigate the biological processes involved, the differential metabolites were assigned to KEGG pathways. As shown in Figure 3c, the differential metabolites of the SZP versus the TZP were significantly involved in flavonoid, flavone, flavonol, and phenylpropanoid biosynthesis. In addition, the differential metabolites of the SZZ versus the TZZ were involved in the biosynthesis of ubiquinone and other terpenoid quinones, as well as linoleic acid and alpha-linolenic-acid metabolism (Figure 3d). Seed-coat color is affected by many factors, among which the most important is the flavonoids [21,22]. Based on the above results, we speculated that the pigmentation of the S006 seed coat was caused by the accumulation of flavonoids.
2.3. Up-Regulated Expressions of CHS-Related Genes Induced the Pigmentation of Seed Coat in S006 Mutant
To elucidate the mechanisms underlying the changes in color and cell area of the seed coat in the S006 mutant, the transcriptomes of the seed coat of the wild type and the S006 mutant were compared by RNA-seq analysis. In particular, the expressions of 2225 genes (foldchange ≥ 1.5, p < 0.05, Table S3) were significantly changed in the S006 mutant, including 824 up-regulated genes and 1401 down-regulated genes (Figure 4a,b). This group of significantly changed genes contains a cluster of bHLH transcription factors (Figure S4), indicating that the phenotypic changes in the S006 mutant may be correlated with the bHLH transcription factors. To further assess the biological functions of the differentially expressed genes (DEGs), we performed gene ontology (GO) and top-10-KEGG analyses. The results showed that the DEGs in the S006 mutant were mainly enriched in three pathways: photosynthesis, porphyrin and chlorophyll metabolism, and flavonoid biosynthesis (Figure 4c). Next, we selected some genes related to flavonoid biosynthesis for the RT-qPCR analysis to confirm the RNA-seq data. These genes included CHS1 (Glyma.08G109400), CHS2 (Glyma.05G153200), CHS3 (Glyma.08G109300), CHS4 (Glyma.08G110700), CHS5 (Glyma.08G109200), CHS6 (Glyma.09G075200), CHS7/8 (Glyma.01G228700 and Glyma.11G011500), and CHS9 (Glyma.08G109500). The RT-qPCR results were in agreement with the RNA-seq data (Figure 4d). Combined with previous research results [23,24], we assumed that the up-regulated transcription of CHS7/8 resulted in the accumulation of flavonoids in the seed coat, thus pigmenting the seed coat in the S006 mutant.
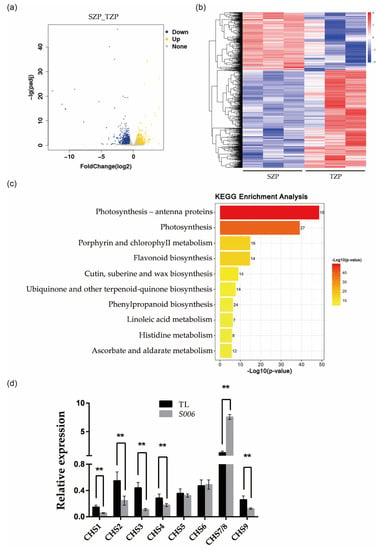
Figure 4.
Transcriptome analysis of S006 and wild type. (a) A volcano plot of DEGs between S006 and wild type; (b) a heat map of DEGs between S006 and wild type; (c) the ten KEGG pathways that were the most enriched in DEGs; (d) the RT-qPCR verified the expression of nine CHS genes. Asterisks indicate a significant difference according to Student’s t-test (**, p < 0.01).
2.4. T-DNA Insertion in Genome Caused the Expression Changes in Flanking Genes
Because the phenotypes of the S006 mutant were different from those of other transformed lines, we speculated that its phenotype variation may be related to the insertion of T-DNA. To investigate the molecular mechanism for these variations, the T-DNA insertion site was identified firstly by resequencing. The results showed that T-DNA inserted in 42,785,587 bp of chromosome 8 in soybean genome (Figure S5). Next, the expression levels of 10 genes around upstream and downstream of this insertion site were analyzed. The results of the RT-qPCR showed that the Glyma.08G308900 (Gm89) was up-regulated and the Glyma.08G309000 (Gm90) was down-regulated (Figure 5a). Therefore, these two genes were employed as candidate genes for further study. As shown by the data below, Gm90 may be related to seed development; thus, it was renamed as Novel Seed Size (NSS).
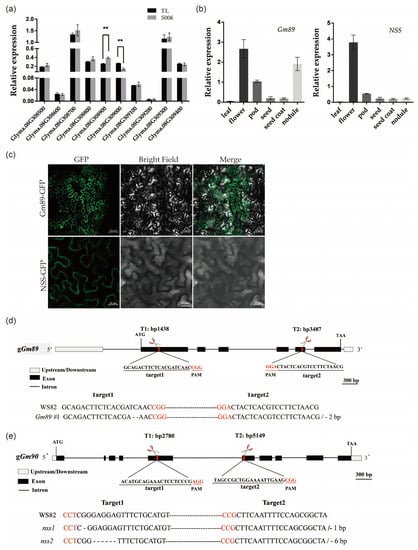
Figure 5.
Expression patterns, protein subcellular localization, and mutant constructions of Gm89 and NSS genes. (a) Flanking-gene expressions of T-DNA insertion site in S006 mutant. Asterisks indicate a significant difference according to Student’s t-test (**, p < 0.01); (b) expression patterns of Gm89 and NSS. (c) subcellular localization of Gm89 and NSS proteins. Bars for upper row = 50 μm, bars for lower row = 20 μm; (d) identification of Gm89 mutant. The red letters represent the PAM site; (e) identification of nss mutants. The red letters represent the PAM site.
2.5. Expression Patterns, Protein Subcellular Localizations and Mutant Constructions of Gm89 and NSS
To study the specific expression profiles of the Gm89 and NSS genes in different organs, we examined the transcript accumulation in the soybean organs by RT-qPCR. The results showed that the flowers accumulated the highest amount of Gm89 and NSS transcripts. Furthermore, the nodules had high levels of Gm89 transcripts. In addition, Gm89 and NSS transcripts were also detected in the pods, seeds, and seed coats (Figure 5b). The data implied wide roles of Gm89 and NSS in soybean development.
To visualize the subcellular localization of the Gm89 and NSS proteins, fusion genes of Gm89:GFP and NSS:GFP driven by a 35S promoter were constructed individually and then infiltrated into Nicotiana benthamiana leaves mediated by Agrobacterium tumefaciens. The GFP fluorescence signals in the cytoplasm (Figure 5c) indicated that two genes may function in the cytoplasm.
Next, we constructed mutants of Gm89 and NSS in the WS82 background by normal CRISPR/Cas9 technology to unveil their respective biological functions. The PCR amplifications were performed to screen mutant lines of the Gm89 and NSS. In particular, Gm89#1 lost 2 bp at target 1, leading to the early termination of the translation (Figure 5d). We also obtained two nss mutants. The nss1 mutant was terminated early due to the loss of 1 bp at target 1, whereas nss2 only missed two amino acids at target 1 (Figure 5e). Next, the Gm89#1, nss1, and nss2 mutants were used for a phenotypic analysis.
2.6. NSS Is Involved in Regulating Seed Size in Soybean
In order to further clarify the functions of the Gm89 and NSS genes in the S006 phenotypes, the agronomic traits of the transformed plants were investigated. The results showed that the 100-seed weight of nss1 was significantly lower than that of WS82, while the 100-seed weight of Gm89#1 and nss2 was not significantly different from that of the wild type (Figure 6a,b). The integument cells of the mutant seeds were further observed by confocal microscopy, and the cell area of nss1 was significantly reduced compared with the wild type, indicating that NSS-gene mutation contributed to S006-seed-size phenotypes. The cell area of Gm89#1 and nss2 was not significantly different from that of WS82, which was consistent with the phenotypes of seed size, suggesting that neither the Gm89 gene nor the two-amino-acid deletion in nss2 produced seed-size phenotypes.
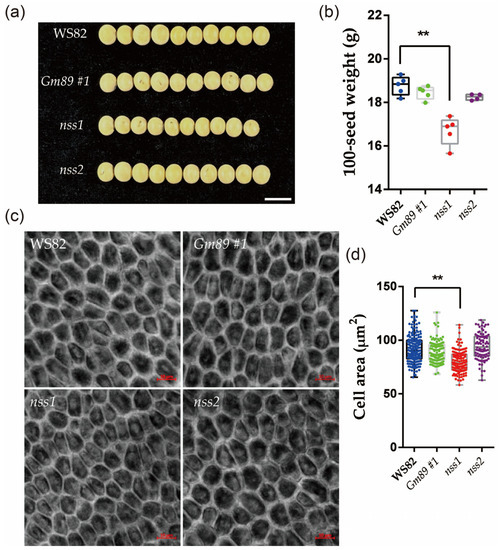
Figure 6.
Comparative analysis of phenotypes between mutants and wild-type seeds. (a) The seed size decreased in width in nss1 mutant (10 seeds are shown in each line); (b) the 100-seed weight was significantly decreased in the nss1 mutant; (c) the integument cells in mutants and wild-type seeds. Bars = 10 μm; (d) the cell area of mutants and wild-type seeds. Asterisks indicate a significant difference according to Student’s t-test (**, p < 0.01).
Because the seeds of the nss1 mutant showed small sizes, lower 100-seed weights and reduced areas of integument cells (Figure 6), it is suggested that the NSS gene is involved in controlling seed size. A sequence analysis provided by Phytozome showed that NSS encoded a functional unknown protein, which contained a peptidase-c1 domain as a potential DNA helicase RuvA subunit and may be involved in apoptosis. In previous studies [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19], some genes encoding transcription regulators or enzymes were demonstrated to control seed size. However, no such genes or pathways have been reported to be related to seed development. From these results, we concluded that NSS is a novel gene involved in regulating seed size.
3. Discussion
Flavonoids are the most important components that affect seed-coat color. They mainly include anthocyanins, proanthocyanidins, flavonoids, isoflavones, and xanthones [25]. The metabonomic analysis of the S006 mutant showed that more than 200 different metabolites were detected in the seed coat, among which flavonoids and phenolic acids accounted for the largest proportion. The KEGG pathway was significantly enriched in the synthesis of phenylpropanoids and flavonoids (Figure 3c), and the content of dihydrokaempferol was significantly increased, indicating that the anthocyanin formation was significantly increased in the S006 mutant, resulting in the pigmentation of the seed coat. In addition, many flavonoids were also among the differential metabolites in the cotyledons, further indicating that the flavonoid metabolism in the seeds changed significantly in the S006 mutant.
The endogenous RNA interference (RNAi) of chalcone synthase (CHS) genes inhibits pigmentation of seed coats in soybean [26]. It was found that CHS genes formed CHS1-CHS3-CHS4 + 5.87 kb + CHS1-CHS3-CHS4 inverted tandem repeat structure (GmIRCHS) on the chromosome [27]. The CHS3 is a pseudogene due to partial truncation and forms an inverted repeat sequence (IR), which produces a dsRNA region and induces the post-transcriptional gene silencing of the CHS genes [27,28]. The pigmentation in seed coats is related to the release of CHS7/8 genes from the silencing effect [24,29]. In addition, although CHS genes are expressed in both the seed coat and the cotyledon, the RNAi of CHS genes occurs specifically in the seed coat [26]. The results of our transcriptome analysis showed that the pigmentation of the S006 mutant seed coat may have been due to the change in the expression levels of the genes related to the flavonoid-synthesis pathway (Figure 4c). Through the RT-qPCR experiments, it was verified that CHS1, CHS2, CHS3, CHS4, and CHS9 were significantly down-regulated and that CHS7/8 were significantly up-regulated in the S006 mutant (Figure 4d). Consistent with previous research results, the up-regulated expression of CHS7/8 may be the main reason for the pigmentation of the S006 seed coat. However, not only the nss mutant but also the Gm89#1 mutant appeared to have a normal seed-coat color, suggesting these two genes did not participate in the pigmentation of the S006 mutant’s seed coat.
No DEGs of known genes related to cell division or expansion were found in the transcriptome analysis, despite the reduction in the outer-integument-cell area in the S006 mutant. The expression levels of Gm89 and NSS were confirmed to have changed in the S006 mutant; however, only the nss1, and not the Gm89 mutant seeds showed small size, lower 100-seed weight, and reduced area of integument cells (Figure 6), suggesting that the NSS gene is a novel gene controlling seed development. The NSS gene encodes a functional unknown protein, which contains a peptidase-c1 domain as a potential DNA helicase RuvA subunit and may be involved in apoptosis. There are no reports showing genes with similar functions participating in seed development [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19]. Since the nss1 mutant exhibited small integument cells, the NSS gene may be related to cell expansion. Regarding the encouragement of cell expansion by GmPSKγ1 and GmSSS1 [11,12], it would be of interest to investigate the relationship between them and their functions in cell expansion in seed coats.
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
The S006 homozygous mutants were generated via Agrobacterium-mediated transformation in the G. max (L.) Mer. cultivar Tianlong1 (TL), and TL wild-type plants were used as controls. The Gm89#1, nss1, and nss2 genetically modified lines were generated in Williams 82 (WS82) background mediated by Agrobacterium tumefaciens, and WS82 used as control. All soybean plants were grown in a growth room under short-day conditions (8-hour-light/16-hour-dark cycle) at 25 ± 1 °C with light illumination. Nicotiana benthamiana plants were grown in a growth room at 22 ± 1 °C with a 16-hour-light/8-hour-dark cycle. The light conditions were from a LED light source (GreenPower LED top lighting, PhilipsHorticulture LED).
4.2. Vector Construction and Soybean Transformation
The 8.9 kb from start coden ATG of GmFTL4 promoter was amplified from soybean-cultivar TL1 genome and then inserted into the Fu79-GUS vector [30] to fuse with GUS reporter gene. The binary vector, Fu39-2:GmFTL4pro:GUS, was constructed through LR reaction (Invitrogen, Carlsbad, CA, USA) and transformed into TL1 following the cotyledonary node method [31].
The network-based tool CRISPR-P (http://crispr.hzau.edu.cn/cgi-bin/CRISPR2/CRISPR, accessed on 21 December 2020) was used to design sgRNAs for Gm89 and Gm90 (NSS) genes [32]. For soybean-genome editing, two entry vectors that individually expressed the Cas9 protein [33] and sgRNAs were constructed, as in our previous report [34]. Next, the genome-editing vectors were introduced into Agrobacterium tumefaciens. Soybean transformations were completed by Wuhan Edgene Bio-tech CO., LT (Edgene, Wuhan, Hubei, China).
4.3. RNA-Extraction and -Expression Analysis
Total RNA of seeds or other tissues were extracted using an EasyPure RNA Kit (TransGen Biotech, Beijing, China). About 1 μg of total RNA was used in reverse transcription (Tiangen, China) for SYBR detection of RT-qPCR products. The ChamQTM SYBR qPCR Master Mix (High ROX Premixed) (Vazyme, Nanjing, Jiangsu, China) was used to detect the expression levels of genes. In the experiment, GmUKN2 (Glyma.06G04180) was used as the internal reference gene [35] for the detection of gene expressions. All primers are listed in Supplemental Table S4.
4.4. Subcellular Localization
The Gm89:GFP and NSS:GFP fusion genes driven by the Cauliflower mosaic virus 35S promoter were transiently expressed in the leaves of 2- to 4-week-old N. benthamiana leaves. The fluorescence signals were visualized by Zeiss LSM700 confocal laser scanning microscope.
4.5. Metabolite Detection and Data Analysis
Seed coats and cotyledons from mature seeds of S006 and TL1 were collected and stored in refrigerator at −80 °C. We used MetWare (Wuhan, China) to perform metabolite identification and quantification according to their standard procedures.
4.6. Transcriptome Analysis
In R5 stage, seed coats of S006 and TL1 were collected and stored in refrigerator at −80 °C. Total RNA extraction, sequencing with an Illumina HiSeq instrument and data analysis were performed by Annoroad Gene Technology (Beijing, China).
5. Conclusions
We identified a novel gene participating in seed development from a random T-DNA mutant, whose mutation resulted in small integument cells, small seeds, and lower 100-seed weights.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24044189/s1.
Author Contributions
X.Z., Y.F. and X.F. conceived and supervised the research project. M.Z., R.D. and P.H. performed the research. X.Z., Y.F. and M.L. performed data analysis. X.Z. and Y.F. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (grant no.: 32071925), the National Key R&D Project (grant no.: 2021YFF1001202) from the Ministry of Science and Technology of China, the Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences, and the Guizhou University cultivation project (Guida cultivation (2020) no. 37).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lu, X.; Li, Q.T.; Xiong, Q.; Li, W.; Bi, Y.D.; Lai, Y.C.; Liu, X.L.; Man, W.Q.; Zhang, W.K.; Ma, B. The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 2016, 86, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, S.; Wang, Z.; Yuan, Y.; Zhang, Z.; Liang, Q.; Yang, X.; Duan, Z.; Liu, Y.; Kong, F. Progress in soybean functional genomics over the past decade. Plant Biotechnol. J. 2022, 20, 256–282. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, R.; Li, Y. Molecular networks of seed size control in plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, M.; Zhang, Z.; Liang, S.; Fan, L.; Yang, X.; Yuan, Y.; Pan, Y.; Zhou, G.; Liu, S. Natural allelic variation of GmST05 controlling seed size and quality in soybean. Plant Biotechnol. J. 2022, 20, 1807–1818. [Google Scholar] [CrossRef]
- Ge, L.; Yu, J.; Wang, H.; Luth, D.; Bai, G.; Wang, K.; Chen, R. Increasing seed size and quality by manipulating BIG SEEDS1 in legume species. Proc. Natl. Acad. Sci. USA 2016, 113, 12414–12419. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.X.; Paddock, K.J.; Zhang, Z.; Stacey, M.G. GmKIX8-1 regulates organ size in soybean and is the causative gene for the major seed weight QTL qSw17-1. New Phytol. 2021, 229, 920–934. [Google Scholar] [CrossRef]
- Du, J.; Wang, S.; He, C.; Zhou, B.; Ruan, Y.-L.; Shou, H. Identification of regulatory networks and hub genes controlling soybean seed set and size using RNA sequencing analysis. J. Exp. Bot. 2017, 68, 1955–1972. [Google Scholar] [CrossRef]
- Zhao, B.; Dai, A.; Wei, H.; Yang, S.; Wang, B.; Jiang, N.; Feng, X. Arabidopsis KLU homologue GmCYP78A72 regulates seed size in soybean. Plant Mol. Biol. 2016, 90, 33–47. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Zhang, H.; Sun, G.; Zhang, W.; Qiu, L. Evolution and association analysis of GmCYP78A10 gene with seed size/weight and pod number in soybean. Mol. Biol. Rep. 2015, 42, 489–496. [Google Scholar] [CrossRef]
- Tang, X.; Su, T.; Han, M.; Wei, L.; Wang, W.; Yu, Z.; Xue, Y.; Wei, H.; Du, Y.; Greiner, S. Suppression of extracellular invertase inhibitor gene expression improves seed weight in soybean (Glycine max). J. Exp. Bot. 2017, 68, 469–482. [Google Scholar] [CrossRef]
- Yu, L.; Liu, Y.; Zeng, S.; Yan, J.; Wang, E.; Luo, L. Expression of a novel PSK-encoding gene from soybean improves seed growth and yield in transgenic plants. Planta 2019, 249, 1239–1250. [Google Scholar] [CrossRef]
- Zhu, W.; Yang, C.; Yong, B.; Wang, Y.; Li, B.; Gu, Y.; Wei, S.; An, Z.; Sun, W.; Qiu, L. An enhancing effect attributed to a nonsynonymous mutation in SOYBEAN SEED SIZE 1, a SPINDLY-like gene, is exploited in soybean domestication and improvement. New Phytol. 2022, 236, 1375–1392. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xiong, Q.; Cheng, T.; Li, Q.-T.; Liu, X.-L.; Bi, Y.-D.; Li, W.; Zhang, W.-K.; Ma, B.; Lai, Y.-C. A PP2C-1 allele underlying a quantitative trait locus enhances soybean 100-seed weight. Mol. Plant 2017, 10, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, S.; Wang, J.; Yokosho, K.; Zhou, B.; Yu, Y.-C.; Liu, Z.; Frommer, W.B.; Ma, J.F.; Chen, L.-Q. Simultaneous changes in seed size, oil content and protein content driven by selection of SWEET homologues during soybean domestication. Natl. Sci. Rev. 2020, 7, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Yang, S.; Zhang, K.; He, J.; Wu, C.; Ren, Y.; Gai, J.; Li, Y. Natural variation and selection in GmSWEET39 affect soybean seed oil content. New Phytol. 2020, 225, 1651–1666. [Google Scholar] [CrossRef]
- Zhang, H.; Goettel, W.; Song, Q.; Jiang, H.; Hu, Z.; Wang, M.L.; An, Y.-q.C. Selection of GmSWEET39 for oil and protein improvement in soybean. PLoS Genetics 2020, 16, e1009114. [Google Scholar] [CrossRef]
- Huang, F.; Tian, Z.; Yu, D. Downregulation of a gibberellin 3b-hydroxylase enhances photosynthesis and increases seed yield in soybean. New Phytol. 2022, 235, 502–517. [Google Scholar]
- Li, J.; Zhang, Y.; Ma, R.; Huang, W.; Hou, J.; Fang, C.; Wang, L.; Yuan, Z.; Sun, Q.; Dong, X. Identification of ST1 reveals a selection involving hitchhiking of seed morphology and oil content during soybean domestication. Plant Biotechnol. J. 2022, 20, 1110–1121. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Zhang, Y.-W.; Han, X.; Zuo, J.-F.; Zhang, Z.; Shang, H.; Song, Q.; Zhang, Y.-M. An evolutionary population structure model reveals pleiotropic effects of GmPDAT for traits related to seed size and oil content in soybean. J. Exp. Bot. 2020, 71, 6988–7002. [Google Scholar] [CrossRef]
- Wu, X.; Cai, X.; Zhang, B.; Wu, S.; Wang, R.; Li, N.; Li, Y.; Sun, Y.; Tang, W. ERECTA regulates seed size independently of its intracellular domain via MAPK-DA1-UBP15 signaling. Plant Cell 2022, 34, 3773–3789. [Google Scholar] [CrossRef]
- Lepiniec, L.; Debeaujon, I.; Routaboul, J.-M.; Baudry, A.; Pourcel, L.; Nesi, N.; Caboche, M. Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 2006, 57, 405–430. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, J.H.; Zabala, G.; Varala, K.; Hudson, M.; Vodkin, L.O. Endogenous, tissue-specific short interfering RNAs silence the chalcone synthase gene family in Glycine max seed coats. Plant Cell 2009, 21, 3063–3077. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, J.H.; Clough, S.J.; Chan, W.-C.; Vodkin, L.O. Tissue-specific gene silencing mediated by a naturally occurring chalcone synthase gene cluster in Glycine max. Plant Cell 2004, 16, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Yong, H.; Kan, J.; Jin, C. Recent advances in flavonoid-grafted polysaccharides: Synthesis, structural characterization, bioactivities and potential applications. Int. J. Biol. Macromol. 2018, 116, 1011–1025. [Google Scholar] [CrossRef]
- Kurauchi, T.; Kasai, A.; Tougou, M.; Senda, M. Endogenous RNA interference of chalcone synthase genes in soybean: Formation of double-stranded RNA of GmIRCHS transcripts and structure of the 5′ and 3′ ends of short interfering RNAs. J. Plant Physiol. 2011, 168, 1264–1270. [Google Scholar] [CrossRef]
- Kasai, A.; Kasai, K.; Yumoto, S.; Senda, M. Structural features of GmIRCHS, candidate of the I gene inhibiting seed coat pigmentation in soybean: Implications for inducing endogenous RNA silencing of chalcone synthase genes. Plant Mol. Biol. 2007, 64, 467–479. [Google Scholar] [CrossRef]
- Senda, M.; Nishimura, S.; Kasai, A.; Yumoto, S.; Takada, Y.; Tanaka, Y.; Ohnishi, S.; Kuroda, T. Comparative analysis of the inverted repeat of a chalcone synthase pseudogene between yellow soybean and seed coat pigmented mutants. Breed. Sci. 2013, 63, 384–392. [Google Scholar] [CrossRef]
- Senda, M.; Jumonji, A.; Yumoto, S.; Ishikawa, R.; Harada, T.; Niizeki, M.; Akada, S. Analysis of the duplicated CHS1 gene related to the suppression of the seed coat pigmentation in yellow soybeans. Theor. Appl. Genet. 2002, 104, 1086–1091. [Google Scholar] [CrossRef]
- Wang, X.; Fan, C.; Zhang, X.; Zhu, J.; Fu, Y.-F. BioVector, a flexible system for gene specific-expression in plants. BMC Plant Biol. 2013, 13, 1–10. [Google Scholar] [CrossRef]
- Paz, M.M.; Martinez, J.C.; Kalvig, A.B.; Fonger, T.M.; Wang, K. Improved cotyledonary node method using an alternative explant derived from mature seed for efficient Agrobacterium-mediated soybean transformation. Plant Cell Rep. 2006, 25, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Lu, L.; Liu, H.Y.; Li, S.; Xing, F.; Chen, L.L. CRISPR-P: A web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol. Plant 2014, 7, 1494–1496. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hu, Z.; Chen, R.; Jiang, Q.; Song, G.; Zhang, H.; Xi, Y. Targeted mutagenesis in soybean using the CRISPR-Cas9 system. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Cheng, Z.; Zhang, X.-M.; Huang, P.; Fan, C.; Yu, G.; Chen, F.; Xu, K.; Chen, Q.; Miao, Y. Spatial divergence of PHR-PHT1 modules maintains phosphorus homeostasis in soybean nodules. Plant Physiol. 2020, 184, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Fan, C.; Li, H.; Zhang, Q.; Fu, Y.-F. Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Mol. Biol. 2009, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).