
Inhibition of Succinate Dehydrogenase by Pesticides (SDHIs) and Energy Metabolism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. SDH and Cellular Energy Metabolism
Comparison between SDH/Complex II and the Other Mitochondrial Respiratory Complexes
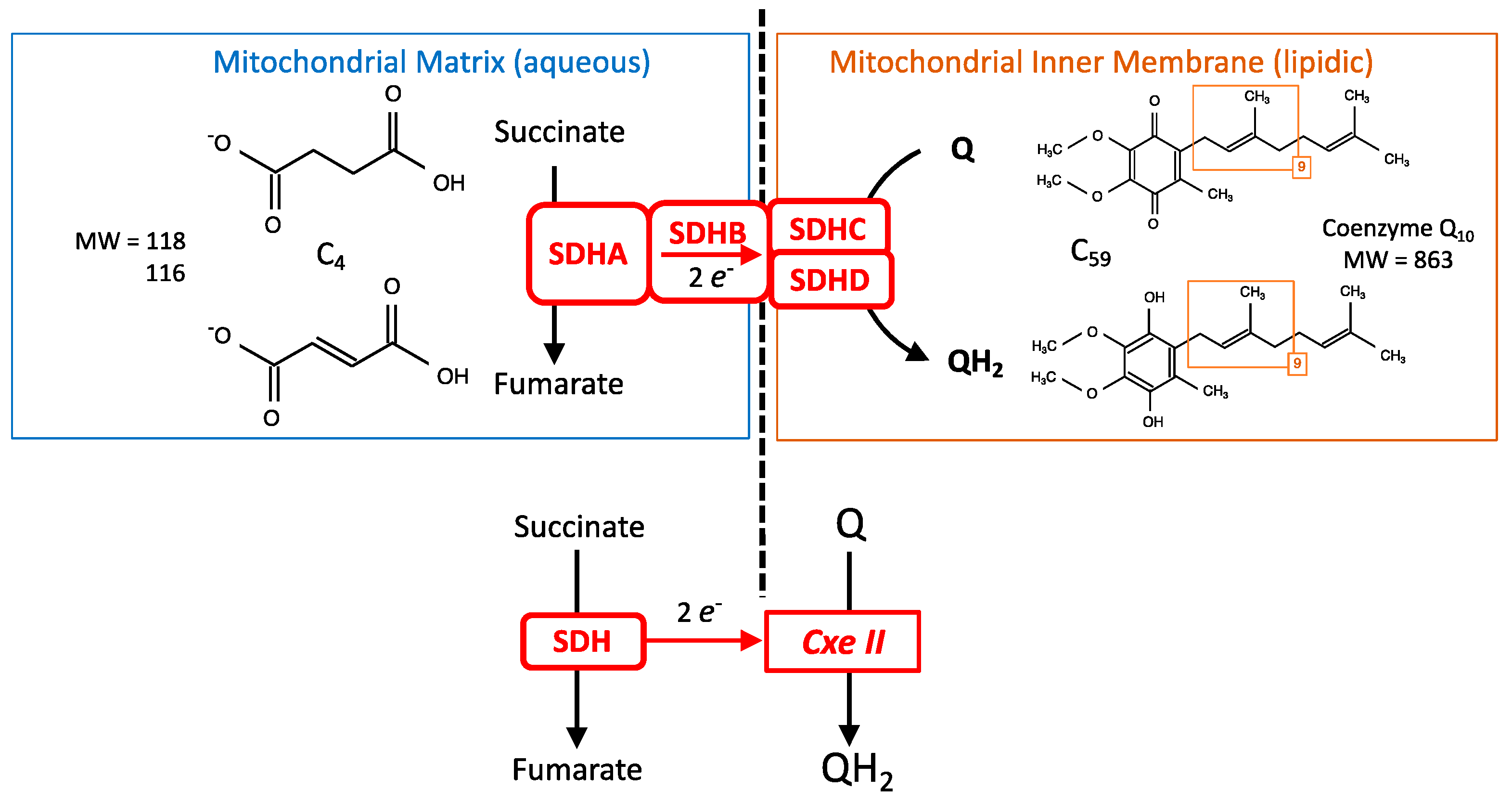
- Respiratory complexes are very large heteromeric assemblies (more than one million dalton) inserted in the mitochondrial inner membrane. This means a complex/long assembly process. A consequence is a long lifetime that allows integration and binding of inhibitors with time. The large majority of the subunits constituting these respiratory complexes are encoded by the nuclear genome, but in addition complexes I, III, IV and V contain a minority of subunits coded by the mitochondrial genome. In contrast, all subunits of complex II are coded by the nuclear genome.
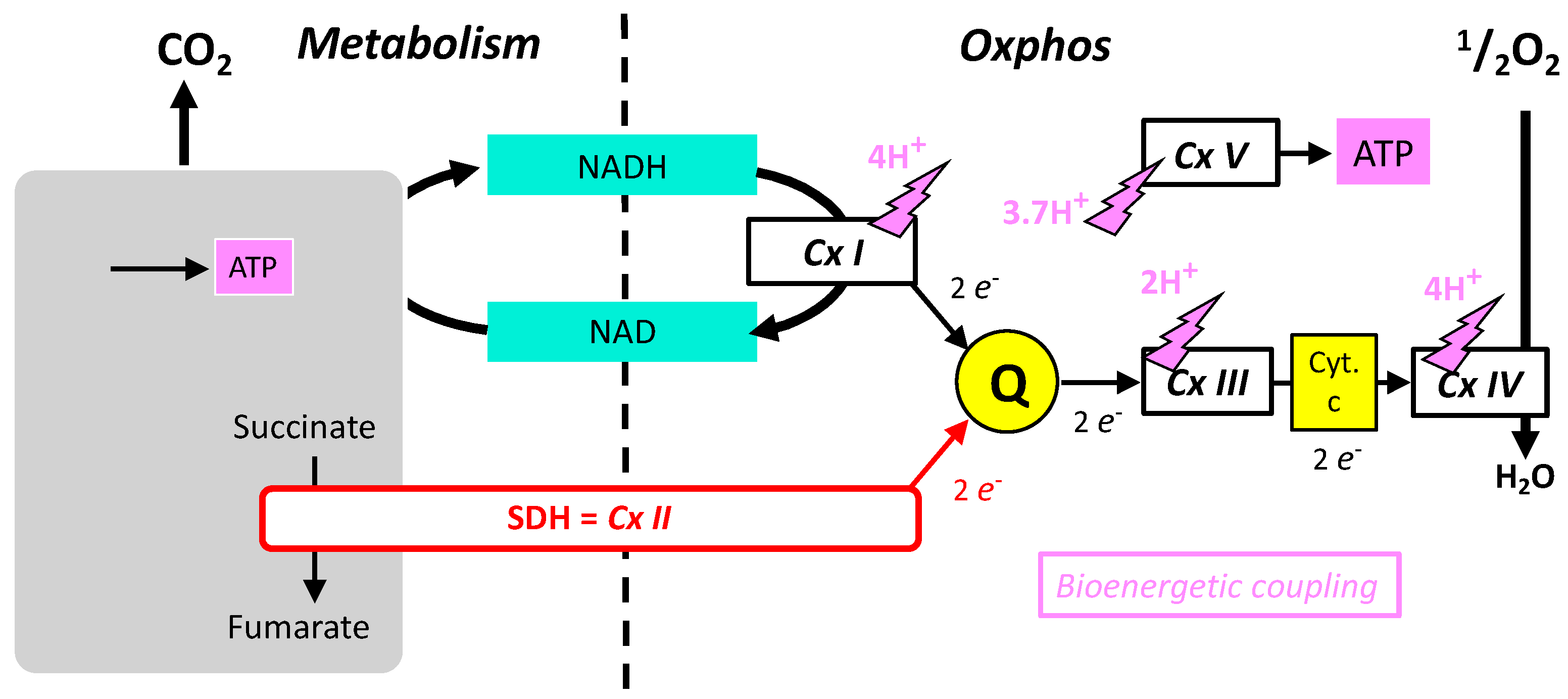
- The oxidative phosphorylation mechanism follows Mitchell’s chemiosmotic theory, according to which proton gradient and movement explain the bioenergetic coupling (Figure 2). Accordingly, complexes I, III, IV, and V associate proton movement to another reaction: redox reactions to proton pumping in complexes I, III, and IV, or phosphorylation of ADP into ATP to proton reentry in complex V. In contrast, complex II does not catalyze proton movement and is uniquely a redox enzyme.
- Complexes I, II, III, and V could operate in the opposite direction depending on forces, which for complexes I, III, and V include the proton gradient (membrane potential). The proton gradient has no direct effect on complex II (SDH) that (in absence of inhibitors) is uniquely sensitive to the ratio of succinate/fumarate and to the reduction/oxidation state of quinone.
- Complexes I, III, and IV of the MRC accept electrons from electron shuttles (NAD, quinone, cytochrome c) and, therefore, the link with metabolic redox reactions is indirect. In contrast, the link between carbon metabolism and electron supply to the respiratory chain is direct and relies on electron transfer within the SDH itself.
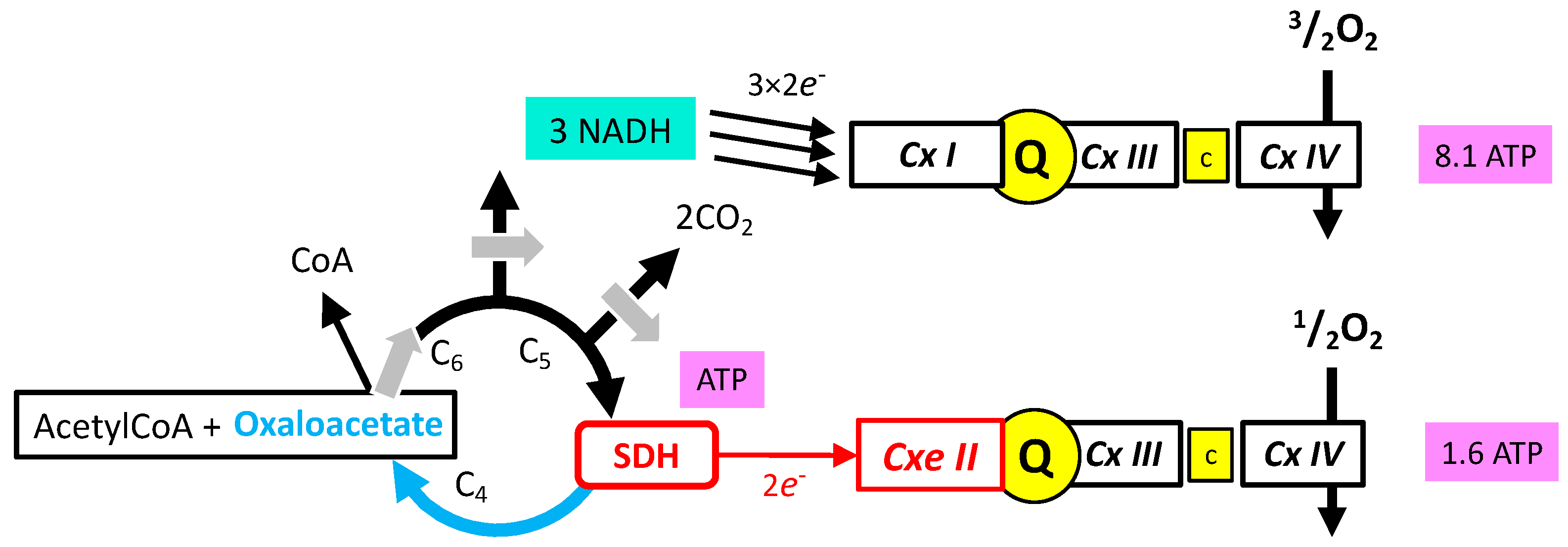
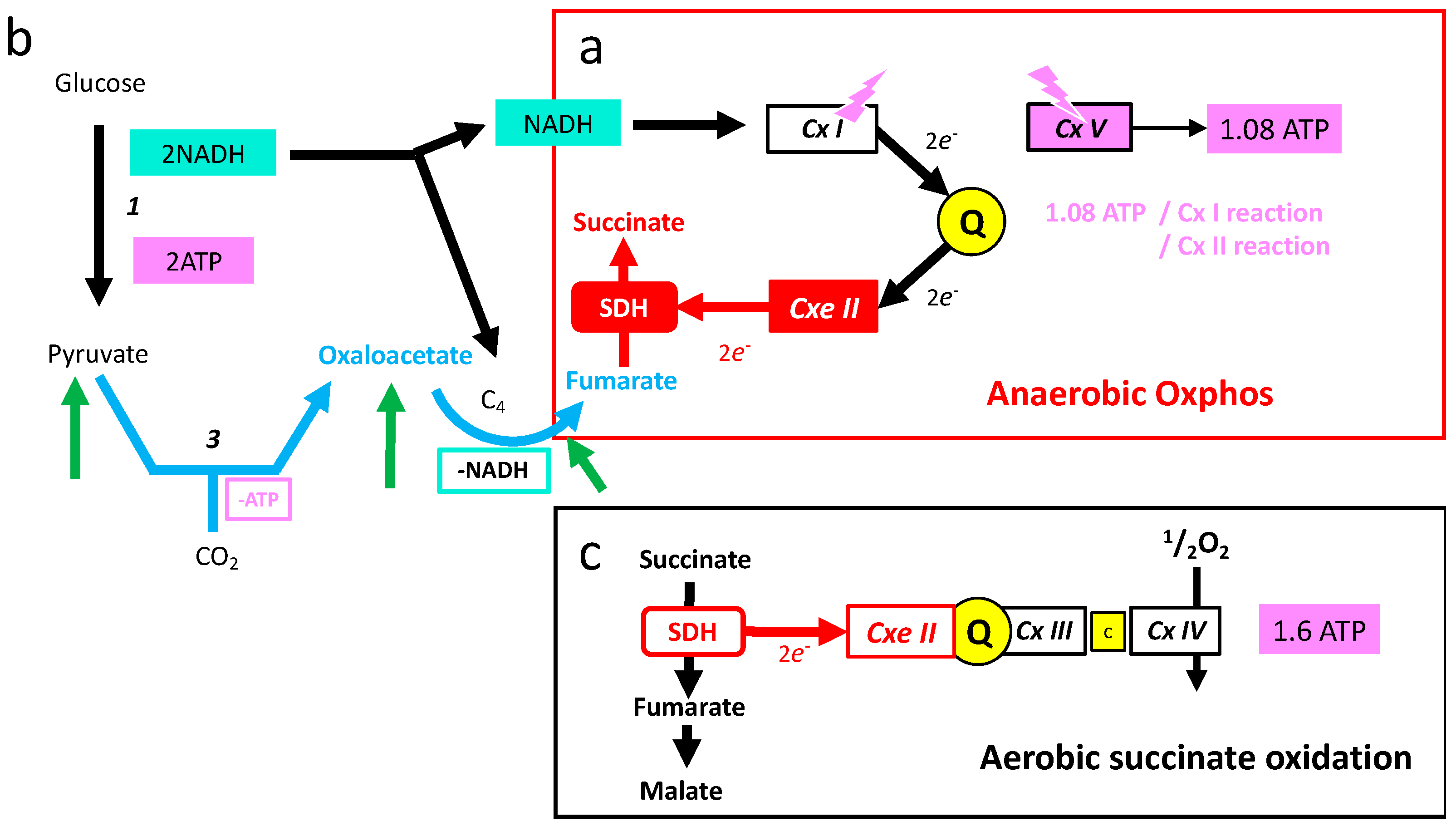
- Oxidation of glucose and of fatty acids loads the largest part of the electrons on the NAD/NADH shuttle and complex I is, therefore, the main entry for electrons in the respiratory chain. However, complex II and the electron-transferring flavoprotein complex reoxidizing FADH2 from beta oxidation of fatty acids and a few others yield electrons directly to quinone with two consequences: (i) the yield in ATP of the electron transfer to oxygen is lower (1.6 ATP) per two electrons (oxygen atom) instead of 2.7 with complex I, and, (ii) quinone appears to be the point of convergence for the entry of electrons, with only one exit (complex III) and, consequently, competition between electron donors could take place, notably between complex I and II.
3. The Succinate Dehydrogenase or Mitochondrial Complex II
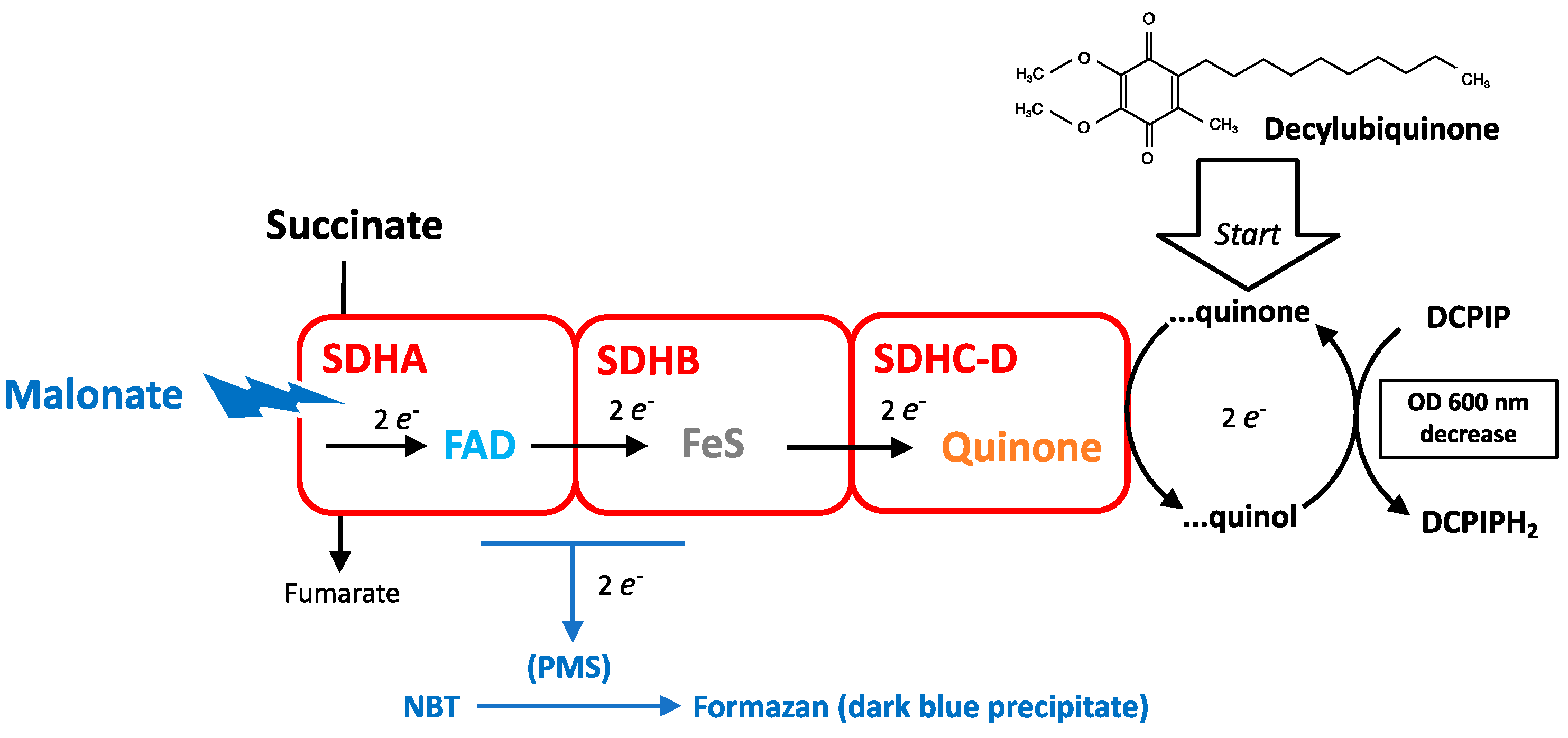
3.1. Measurement of SDH Activity
3.2. SDH Affinity for Succinate and Maximal Enzymatic Activity
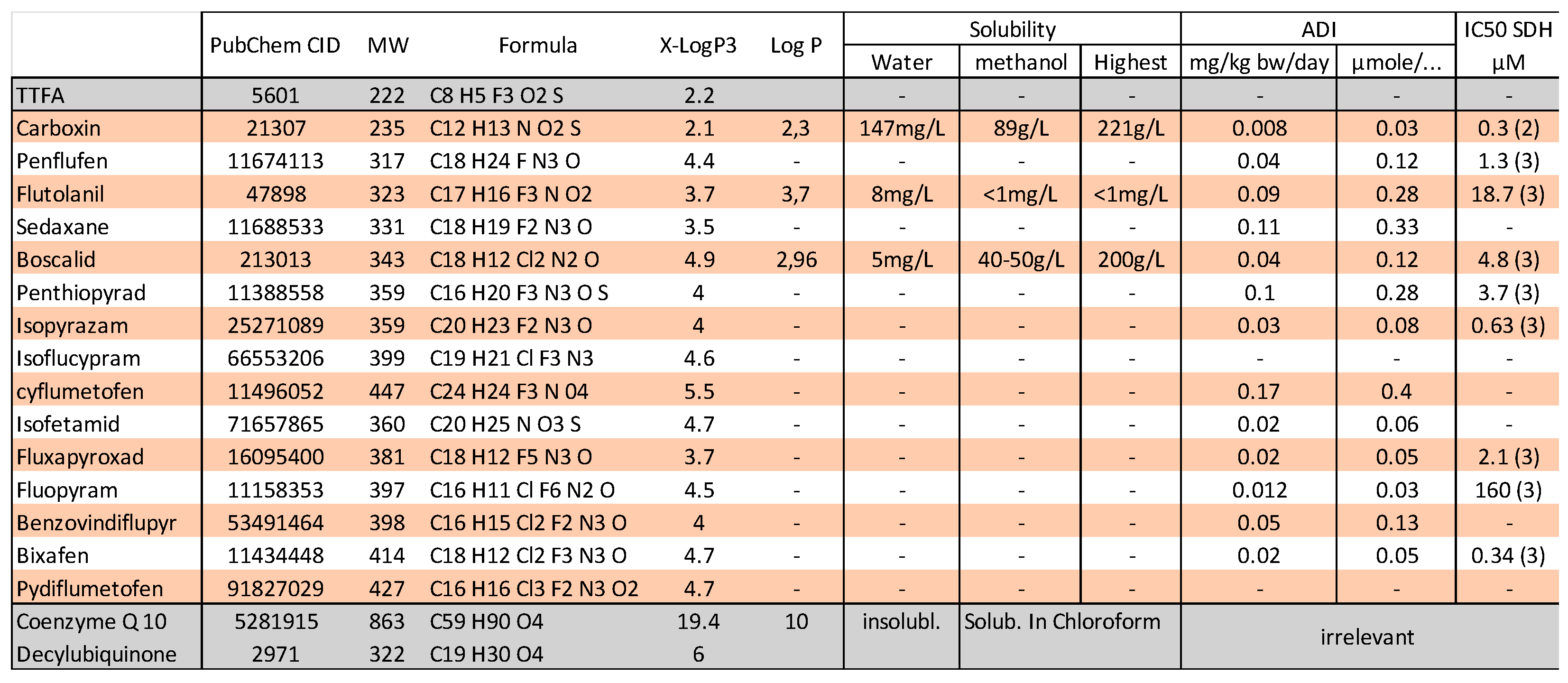
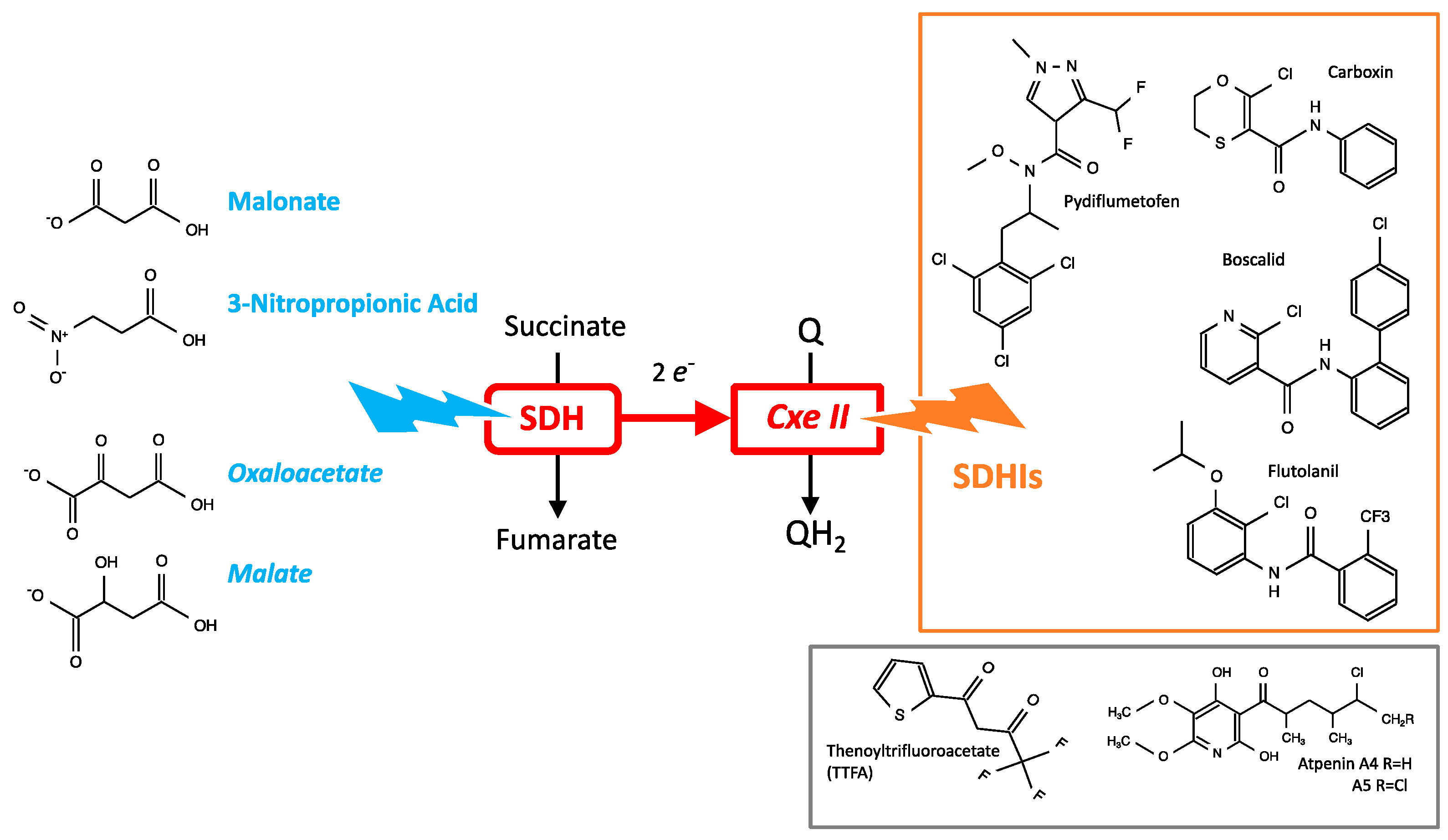
3.3. Chemical Inhibitors of SDH
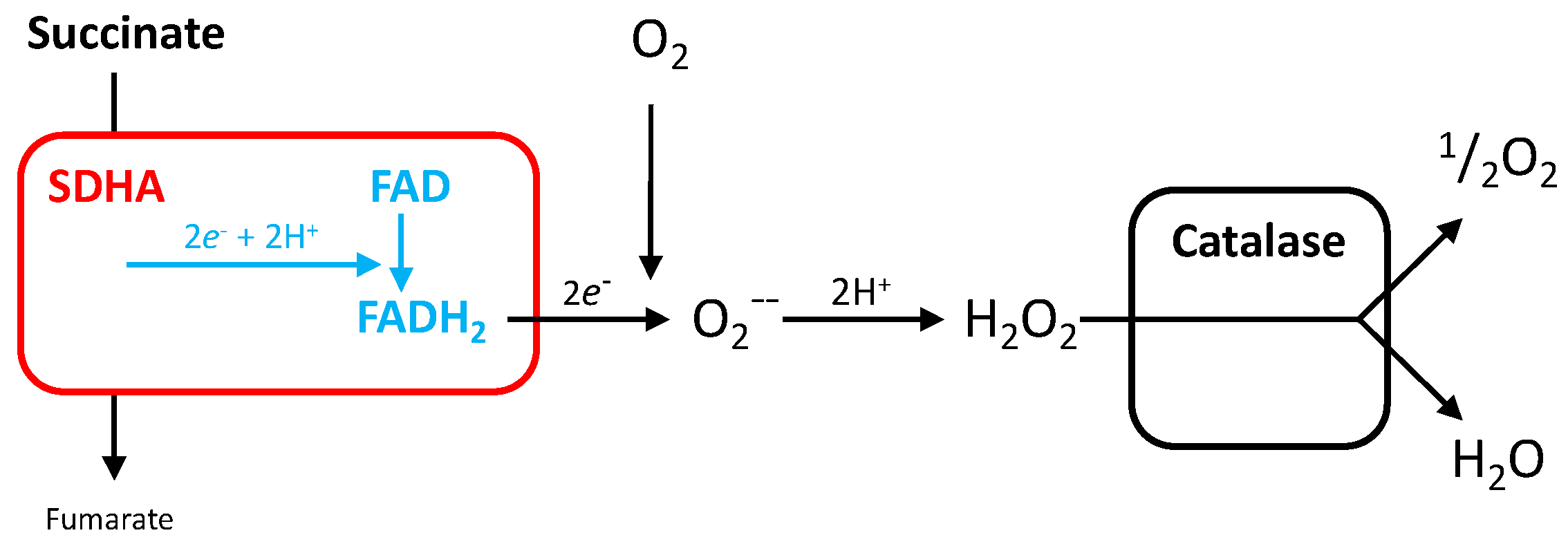
3.4. Reactive Oxygen Species Production by SDH
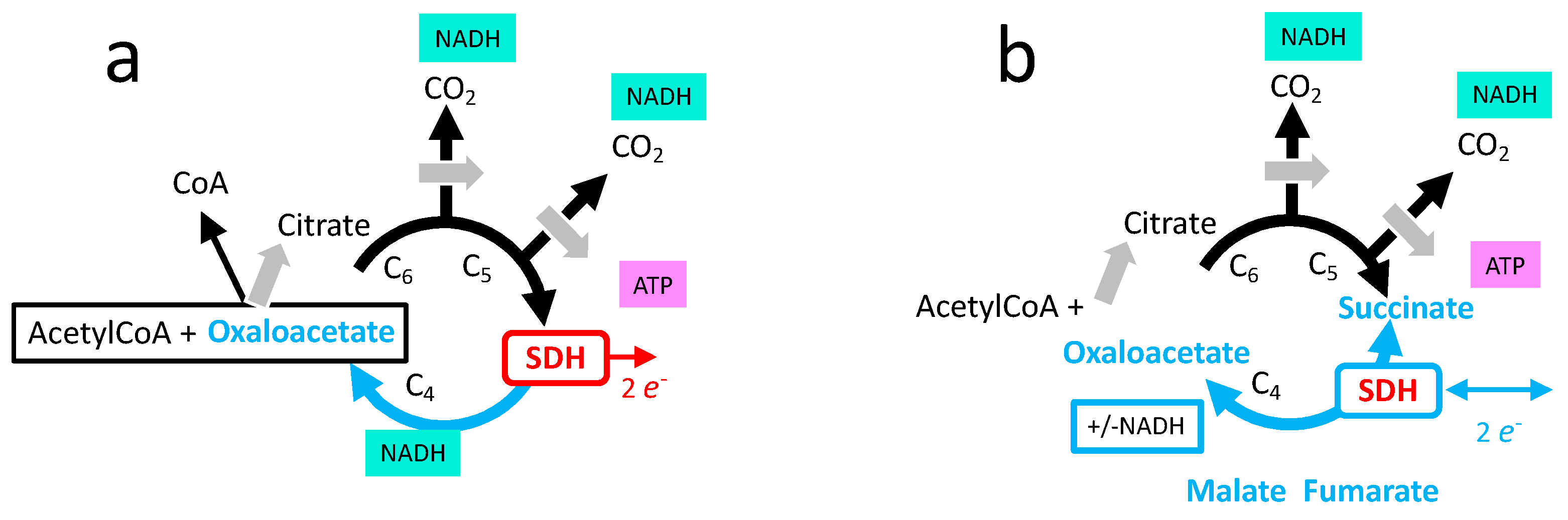
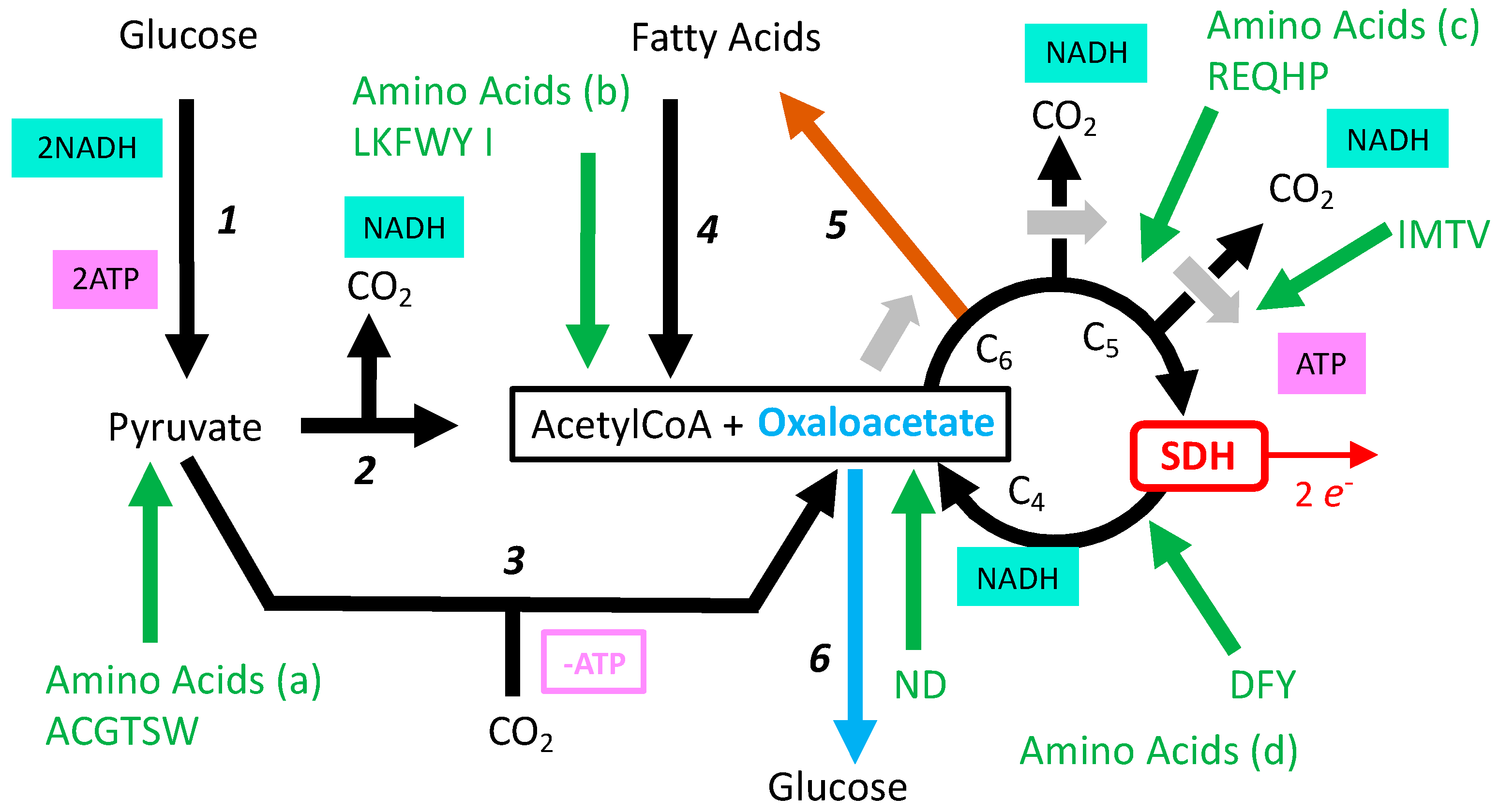
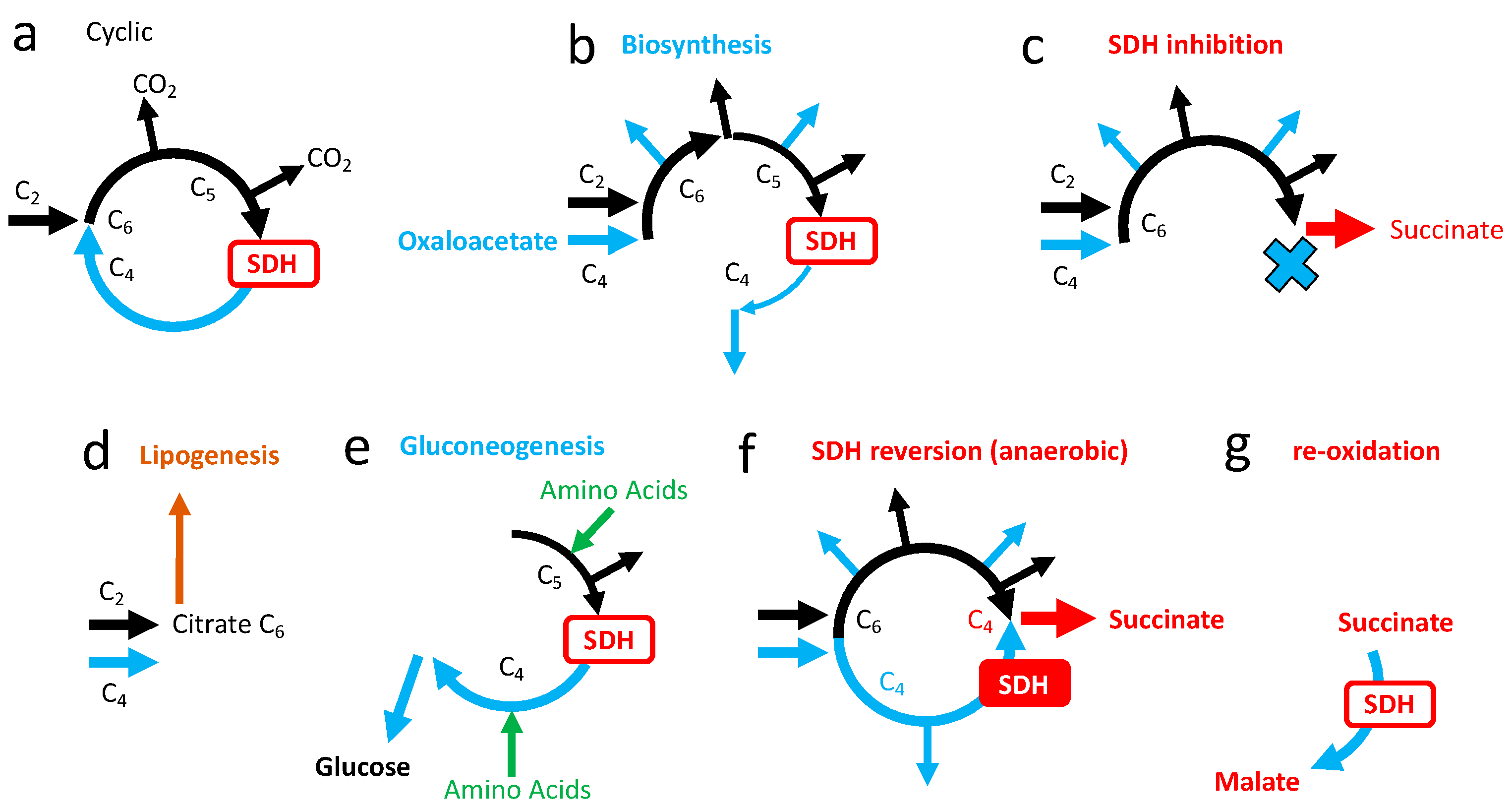
3.5. Role of SDH in the Tricarboxylic Cycle
4. Loss of SDH Activity
4.1. SDH Negative Tumors
4.2. Inhibition of SDH in Experimental Models
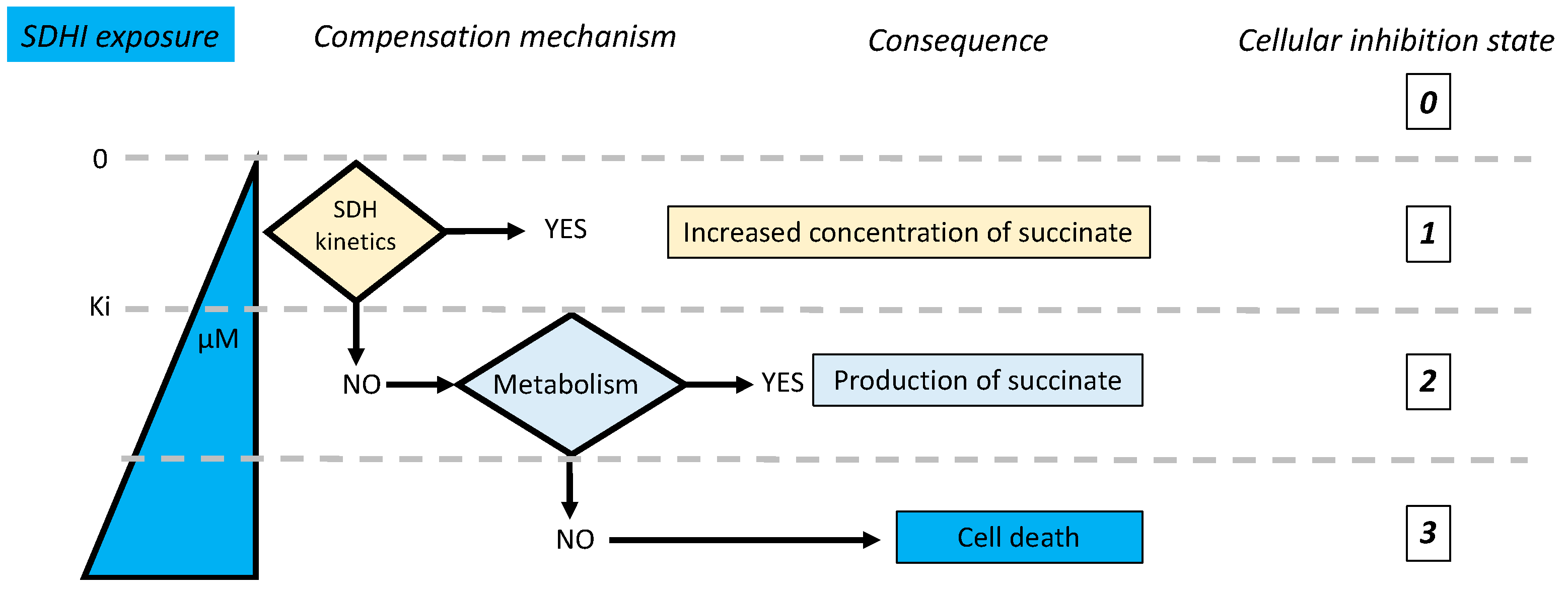
5. Consequences of SDH Inhibition
- Is inhibition complete or partial?
- If inhibition is partial, could the remaining enzyme activity be sufficient to ensure normal reaction rate?
- Are there salvage pathways that would compensate for or alleviate the consequences of inhibition of the target enzyme?
5.1. Salvage Pathways to SDH Inhibition
5.1.1. Succinate Accumulation
5.1.2. Succinate Cycle
5.1.3. Electron Escape from SDHA
5.2. Kinetic Compensation by SDH
5.3. Adaptive Responses to SDH Inhibition
- Full aerobic oxidation is not always/everywhere pertinent and we should consider other metabolic schemes.
- Is a steady state increase in succinate concentration without consequences?
- The interaction between SDHIs and other active substances is to be considered.
- Finally, are the experimental conditions of exposure to SDHIs appropriate to evaluate their possible impact, and particularly with regard to differences between animal models and humans?
6. Complex II and the Hypoxic Challenge
6.1. The Hypoxic Challenge
6.2. Lactic Fermentation and Oxygen Debt: Later or Elsewhere
6.3. The Mitochondrial Respiratory Chain in Anaerobic Mode
6.4. High Requirement for Electron Flux in Complexes I and II under Anaerobic Mode
6.5. Succinate Is a Signal
7. Metabolic Events
7.1. Mild Inhibition of SDH and Lipid Synthesis
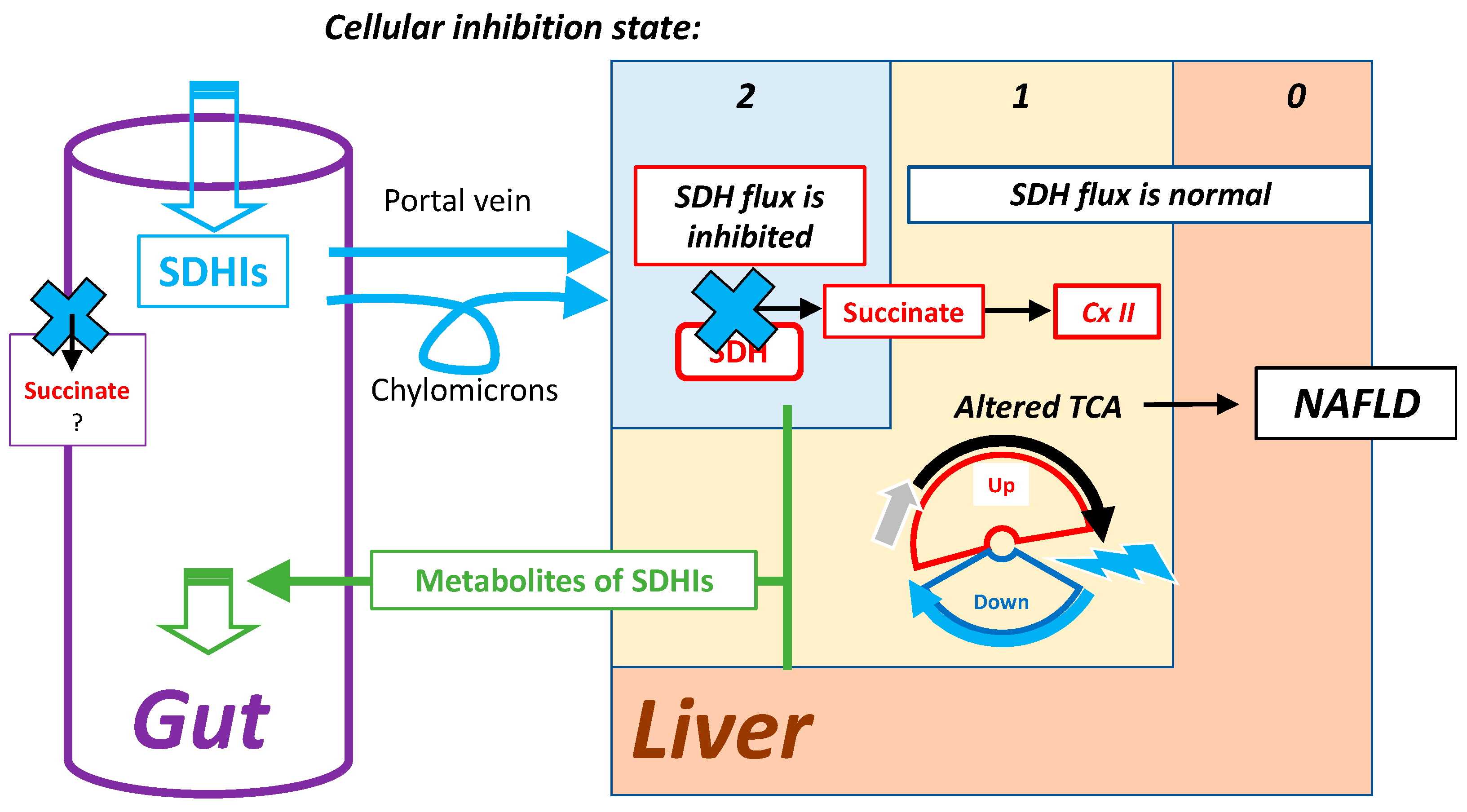
7.2. Highest Exposure to Oral SDHI Gut and Liver
- Its cumulative nature needs a long time to develop.
- It appears unlikely that inquiries about effects related to metabolic diseases were required in the process of evaluation of these active substances.
- Whenever hepatic carcinogenesis is recorded in rodents, it might be considered as specific to rodents and irrelevant to human exposure. In this respect, comparison with transgenic animals that, equipped with SDH, mutated to acquire resistance to a given SDHI would help to dissociate effects caused by exposure to the xenobiotic (SDHI) from that resulting from SDH inhibition.
- Unless the effect would be extremely potent, it would not develop spontaneously, but would require diet manipulation (high fat, high sucrose diet), and rodents are relatively resistant to it.
7.3. Metabolic Issues with Animal Models
8. Convergent Interferences
8.1. Quinone
8.2. Mitochondrial Respiration
9. Assimilation of SDHIs and Diet
10. Conclusions
10.1. To Understand How Cells/Organisms Withstand SDH Inhibition
- There is a significant “reserve” in SDH activity that could be mobilized under conditions of partial inhibition. This results in a threshold effect and, for example, existing animal models exposed to the SDH inhibitor NPA require a 30% to 50% inhibition of the SDH activity to result into detectable lesions.
- In contrast with NPA that inhibits the metabolic side of SDH (SDH-A), SDHIs target the electron transfer from complex II (SDHB-D) to quinone, which is a minor contributor to MRC bioenergetics and mitochondrial ATP production.
- Heterogenous exposure to SDHIs resulting from the intake route, and degradation/elimination, would allow the compensation of inhibition of succinate oxidation in cells more exposed to SDHIs by a higher rate of oxidation in others.
10.2. To Identify
- Possible risks associated with these adaptive mechanisms: the response to partial SDH inhibition induces moderate and proportionate metabolic alterations, which may have insidious effects when present over the long term (chronic exposure). Altogether, if a marginal exposure to SDHIs is to be considered the main risk, it appears their role is as one of the many environmental factors adverse to the oxidative metabolism (sometimes called “mitochondrial fitness”), contributing to metabolic overload and aggravation of metabolic status. With oral exposure, a liver with risk of NAFLD appears to be the first target.
- Consequently, for SDHIs and other molecules interfering with mitochondrial function, metabolic disorders might well become an issue. Investigations may require specific adaptations of animal models.
- Clearly identified a lack of knowledge: (i) Interference of SDHI with metabolic status is hardly documented (Supplementary Materials S4). (ii) More extended studies of inhibition properties of SDHIs with regard to non-target species, and notably man, should be made public. A critical issue is the reversibility of the binding of SDHIs on SDH. (iii) The SDHI assimilation should be better evaluated; this includes the influence of diet as well as consideration of the formulations actually in use.
- Neglected metabolic schemes: while normal bioenergetics would be relatively indifferent to partial inhibition of SDH, tissue specific adaptations or events could greatly enhance sensitivity to SDH inhibition. In this respect, evaluation of active substances could hardly be considered satisfying if their interaction with life challenges, such as immune challenge, inflammation, and starvation are omitted.
10.3. To Determine Phenotypic Alterations That should Attract Attention
- Impaired gluconeogenesis and adaptation to starvation,
- Body weight gain (lipid accumulation) likely to be biphasic,
- Modification in brain structures (brainstem and basal ganglia),
- Retinopathy,
- Failure to comply with an energetic challenge: intense repeated neuronal stimulation, high oxygen demanding organs, oxygen limitation,
- Alteration in hypoxic signaling and response to ischemic reperfusion events, and,
- Increased index of oxidative stress and/or induction of antioxidant defenses.
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- Sierotzki, H.; Scalliet, G. A review of current knowledge of resistance aspects for the next-generation succinate dehydrogenase inhibitor fungicides. Phytopathology 2013, 103, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Coles, C.J.; Singer, T.P.; White, G.A.; Thorn, G.D. Studies on the binding of carboxin analogs to succinate dehydrogenase. J. Biol. Chem. 1978, 253, 5573–5578. [Google Scholar] [CrossRef] [PubMed]
- Bénit, P.; Kahn, A.; Chretien, D.; Bortoli, S.; Huc, L.; Schiff, M.; Gimenez-Roqueplo, A.-P.; Favier, J.; Gressens, P.; Rak, M.; et al. Evolutionarily conserved susceptibility of the mitochondrial respiratory chain to SDHI pesticides and its consequence on the impact of SDHIs on human cultured cells. PLoS ONE 2019, 14, e0224132. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.epa.gov/oppsrrd1/reregistration/REDs/rotenone_red.pdf (accessed on 20 December 2022).
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Brouillet, E.; Guyot, M.-C.; Mittoux, V.; Altairac, S.; Condé, F.; Palfi, S.; Hantraye, P. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J. Neurochem. 1998, 70, 794–805. [Google Scholar] [CrossRef]
- Piruat, J.I.; Pintado, C.O.; Ortega-Sáenz, P.; Roche, M.; López-Barneo, J. The mitochondrial SDHD gene is required for early embryogenesis, and its partial deficiency results in persistent carotid body glomus cell activation with full responsiveness to hypoxia. Mol. Cell Biol. 2004, 24, 10933–10940. [Google Scholar] [CrossRef]
- Pagnamenta, A.T.; Hargreaves, I.P.; Duncan, A.J.; Taanman, J.-W.; Heales, S.J.; Land, J.M.; Bitner-Glindzicz, M.; Leonard, J.V.; Rahman, S. Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol. Genet. Metab. 2006, 89, 214–221. [Google Scholar] [CrossRef]
- Van Coster, R.; Seneca, S.; Smet, J.; Van Hecke, R.; Gerlo, E.; Devreese, B.; Van Beeumen, J.; Leroy, J.; De Meirleir, L.; Lissens, W. Homozygous Gly555Glu mutation in the nuclear-encoded 70 kDa flavoprotein gene causes instability of the respiratory chain complex II. Am. J. Med. Genet. A 2003, 120A, 13–18. [Google Scholar] [CrossRef]
- Bracko, O.; Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Tavazzi, B.; Artmann, J.; Wong, E.C.; Buxton, R.B.; Weller, M.; Luft, A.R.; et al. 3-Nitropropionic acid-induced ischemia tolerance in the rat brain is mediated by reduced metabolic activity and cerebral blood flow. J. Cereb. Blood Flow Metab. 2014, 34, 1522–1530. [Google Scholar] [CrossRef]
- Riepe, M.W.; Niemi, W.N.; Megow, D.; Ludolph, A.C.; Carpenter, D.O. Mitochondrial oxidation in rat hippocampus can be preconditioned by selective chemical inhibition of succinic dehydrogenase. Exp. Neurol. 1996, 138, 15–21. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515. [Google Scholar] [CrossRef] [PubMed]
- Kenney, W.C.; Mowery, P.C.; Seng, R.L.; Singer, T.P. Localization of the substrate and oxalacetate binding site of succinate dehydrogenase. J. Biol. Chem. 1976, 251, 2369–2373. [Google Scholar] [CrossRef] [PubMed]
- Hanstein, W.G.; Davis, K.A.; Ghalambor, M.A.; Hatefi, Y. Succinate dehydrogenase. II. Enzymatic properties. Biochemistry 1971, 10, 2517–2524. [Google Scholar] [CrossRef] [PubMed]
- Baginsky, M.L.; Hatefi, Y. Reconstitution of succinate-coenzyme Q reductase (complex II) and succinate oxidase activities by a highly purified, reactivated succinate dehydrogenase. J. Biol. Chem. 1969, 244, 5313–5319. [Google Scholar] [CrossRef]
- Medja, F.; Allouche, S.; Frachon, P.; Jardel, C.; Malgat, M.; Mousson de Camaret, B.; Slama, A.; Lunardi, J.; Mazat, J.P.; Lombès, A. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 2009, 9, 331–339. [Google Scholar] [CrossRef]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef]
- Haraux, F.; Lombès, A. Kinetic analysis of ATP hydrolysis by complex V in four murine tissues: Towards an assay suitable for clinical diagnosis. PLoS ONE 2019, 14, e0221886. [Google Scholar] [CrossRef]
- Watt, I.N.; Montgomery, M.G.; Runswick, M.J.; Leslie, A.G.W.; Walker, J.E. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc. Natl. Acad. Sci. USA 2010, 107, 16823–16827. [Google Scholar] [CrossRef]
- Anastacio, M.M.; Kanter, E.M.; Keith, A.D.; Schuessler, R.B.; Nichols, C.G.; Lawton, J.S. Inhibition of Succinate Dehydrogenase by Diazoxide Is Independent of the ATP-Sensitive Potassium Channel Subunit Sulfonylurea Type 1 Receptor. J. Am. Coll. Surg. 2013, 216, 1144–1149. [Google Scholar] [CrossRef]
- Quastel, J.H.; Wooldridge, W.R. Some properties of the dehydrogenating enzymes of bacteria. Biochem. J. 1928, 22, 689–702. [Google Scholar] [CrossRef]
- Potter, V.R.; Dubois, K.P. Studies on the mechanism of hydrogen transport in animal tissues: VI. inhibitor studies with succinic dehydrogenase. J. Gen. Physiol. 1943, 26, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Alston, T.A.; Mela, L.; Bright, H.J. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc. Natl. Acad. Sci. USA 1977, 74, 3767–3771. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal Structure of Mitochondrial Respiratory Membrane Protein Complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef]
- Molinié, T.; Cougouilles, E.; David, C.; Cahoreau, E.; Portais, J.-C.; Mourier, A. MDH2 produced OAA is a metabolic switch rewiring the fuelling of respiratory chain and TCA cycle. Biochim. Biophys. Acta Bioenerg. 2022, 1863, 148532. [Google Scholar] [CrossRef]
- Belikova, Y.O.; Kotlyar, A.B.; Vinogradov, A.D. Oxidation of malate by the mitochondrial succinate-ubiquinone reductase. Biochim. Biophys. Acta 1988, 936, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.F.; Lin, F.; Yuan, J.; Cui, J.C. Toxic effects and potential mechanisms of Fluxapyroxad to zebrafish (Danio rerio) embryos. Sci. Total Environ. 2021, 769, 144519. [Google Scholar] [CrossRef]
- Burnichon, N.; Brière, J.-J.; Libé, R.; Vescovo, L.; Rivière, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Bénit, P.; Tzagoloff, A.; et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef]
- van Nederveen, F.H.; Gaal, J.; Favier, J.; Korpershoek, E.; Oldenburg, R.A.; de Bruyn, E.M.; Sleddens, H.F.; Derkx, P.; Rivière, J.; Dannenberg, H. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: A retrospective and prospective analysis. Lancet Oncol. 2009, 10, 764–771. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kalinin, D.V.; Pavlov, V.S.; Lukyanova, E.N.; Golovyuk, A.L.; Fedorova, M.S.; Pudova, E.A.; Savvateeva, M.V.; Stepanov, O.A.; Poloznikov, A.A.; et al. Immunohistochemistry and Mutation Analysis of SDHx Genes in Carotid Paragangliomas. Int. J. Mol. Sci. 2020, 21, 6950. [Google Scholar] [CrossRef]
- Prag, H.A.; Aksentijevic, D.; Dannhorn, A.; Giles, A.V.; Mulvey, J.F.; Sauchanka, O.; Du, L.; Bates, G.; Reinhold, J.; Kula-Alwar, D.; et al. Ischemia-Selective Cardioprotection by Malonate for Ischemia/Reperfusion Injury. Circ. Res. 2022, 131, 528–541. [Google Scholar] [CrossRef]
- Bouillaud, F.; Ransy, C.; Moreau, M.; Benhaim, J.; Lombès, A.; Haouzi, P. Methylene blue induced O2 consumption is not dependent on mitochondrial oxidative phosphorylation: Implications for salvage pathways during acute mitochondrial poisoning. Respir. Physiol. Neurobiol. 2022, 304, 103939. [Google Scholar] [CrossRef] [PubMed]
- Ransy, C.; Vaz, C.; Lombès, A.; Bouillaud, F. Use of H2O2 to Cause Oxidative Stress, the Catalase Issue. Int. J. Mol. Sci. 2020, 21, 9149. [Google Scholar] [CrossRef] [PubMed]
- Hirai, D.M.; Colburn, T.D.; Craig, J.C.; Hotta, K.; Kano, Y.; Musch, T.I.; Poole, D.C. Skeletal muscle interstitial O2 pressures: Bridging the gap between the capillary and myocyte. Microcirculation 2019, 26. [Google Scholar] [CrossRef]
- Bouillaud, F.; Hammad, N.; Schwartz, L. Warburg Effect, Glutamine, Succinate, Alanine, When Oxygen Matters. Biology 2021, 10, 1000. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [PubMed]
- Hochachka, P.W.; Lutz, P.L. Mechanism, origin, and evolution of anoxia tolerance in animals. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2001, 130, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Hochachka, P.W.; Mustafa, T. Invertebrate facultative anaerobiosis. Science 1972, 178, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Hochachka, P.W. Defense strategies against hypoxia and hypothermia. Science 1986, 231, 234–241. [Google Scholar] [CrossRef]
- Goldberg, N.D.; Passonneau, J.V.; Lowry, O.H. Effects of changes in brain metabolism on the levels of citric acid cycle intermediates. J. Biol. Chem. 1966, 241, 3997–4003. [Google Scholar] [CrossRef]
- McNamara, J.W.; Porrello, E.R. From Fragrances to Heart Regeneration: Malonate Repairs Broken Hearts. Circulation 2021, 143, 1987–1990. [Google Scholar] [CrossRef]
- Bisbach, C.M.; Hass, D.T.; Robbings, B.M.; Rountree, A.M.; Sadilek, M.; Sweet, I.R.; Hurley, J.B. Succinate Can Shuttle Reducing Power from the Hypoxic Retina to the O2-Rich Pigment Epithelium. Cell Rep. 2020, 31, 107606. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Johnson-Cadwell, L.; Vesce, S.; Jekabsons, M.; Yadava, N. Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J. Neurosci. Res. 2007, 85, 3206–3212. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, M.M.; Komaromy-Hiller, G.; Shushan, B.; Urry, F.M.; Roberts, W.L. Analysis of Dicarboxylic Acids by Tandem Mass Spectrometry. High-Throughput Quantitative Measurement of Methylmalonic Acid in Serum, Plasma, and Urine. Clinic. Chem. 2001, 47, 1993–2002. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Moore, H.B.; Moore, E.E.; Reisz, J.A.; Wither, M.J.; Ghasasbyan, A.; Chandler, J.; Silliman, C.C.; Hansen, K.C.; Banerjee, A. Plasma succinate is a predictor of mortality in critically injured patients. J. Trauma Acute Care Surg. 2017, 83, 491–495. [Google Scholar] [CrossRef]
- Osuna-Prieto, F.J.; Martinez-Tellez, B.; Ortiz-Alvarez, L.; Di, X.; Jurado-Fasoli, L.; Xu, H.; Ceperuelo-Mallafré, V.; Núñez-Roa, C.; Kohler, I.; Segura-Carretero, A.; et al. Elevated plasma succinate levels are linked to higher cardiovascular disease risk factors in young adults. Cardiovasc. Diabetol. 2021, 20, 151. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Harmon, C.; Jedrychowski, M.P.; Xiao, H.; Garrity, R.; Tran, N.V.; Bradshaw, G.A.; Fu, A.; Szpyt, J.; Reddy, A.; et al. UCP1 governs liver extracellular succinate and inflammatory pathogenesis. Nat. Metab. 2021, 3, 604–617. [Google Scholar] [CrossRef]
- Ceperuelo-Mallafré, V.; Llauradó, G.; Keiran, N.; Benaiges, E.; Astiarraga, B.; Martínez, L.; Pellitero, S.; González-Clemente, J.M.; Rodríguez, A.; Fernández-Real, J.M.; et al. Preoperative Circulating Succinate Levels as a Biomarker for Diabetes Remission After Bariatric Surgery. Diabetes Care 2019, 42, 1956–1965. [Google Scholar] [CrossRef]
- Helmy, N.; Prip-Buus, C.; Vons, C.; Lenoir, V.; Abou-Hamdan, A.; Guedouari-Bounihi, H.; Lombès, A.; Bouillaud, F. Oxidation of hydrogen sulfide by human liver mitochondria. Nitric Oxide 2014, 41, 105–112. [Google Scholar] [CrossRef]
- Goubern, M.; Andriamihaja, M.; Nübel, T.; Blachier, F.; Bouillaud, F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007, 21, 1699–1706. [Google Scholar] [CrossRef]
- Lagoutte, E.; Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Blachier, F.; Bouillaud, F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 2010, 1797. [Google Scholar] [CrossRef]
- Nespolo, R.F.; Mejias, C.; Bozinovic, F. Why bears hibernate? Redefining the scaling energetics of hibernation. Proc. R. Soc. B. 2022, 289, 20220456. [Google Scholar] [CrossRef] [PubMed]
- Nespolo, R.F.; Mejias, C.; Bozinovic, F. Isometric scaling of hibernation: When trees do not let the forest be seen. Proc. R. Soc. B. 2022, 289, 20221719. [Google Scholar] [CrossRef] [PubMed]
- Choukér, A.; Ngo-Anh, T.J.; Biesbroek, R.; Heldmaier, G.; Heppener, M.; Bereiter-Hahn, J. European space agency’s hibernation (torpor) strategy for deep space missions: Linking biology to engineering. Neurosci. Biobehav. Rev. 2021, 131, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Reddon, H.; Patel, Y.; Turcotte, M.; Pigeyre, M.; Meyre, D. Revisiting the evolutionary origins of obesity: Lazy versus peppy-thrifty genotype hypothesis. Obes. Rev. 2018, 19, 1525–1543. [Google Scholar] [CrossRef]
- Neel, J.V. Diabetes mellitus: A ‘thrifty’ genotype rendered detrimental by ‘progress’? Am. J. Hum. Genet. 1962, 14, 353–362. [Google Scholar]
- Johnson, R.J.; Sánchez-Lozada, L.G.; Nakagawa, T.; Rodriguez-Iturbe, B.; Tolan, D.; Gaucher, E.A.; Andrews, P.; Lanaspa, M.A. Do thrifty genes exist? Revisiting uricase. Obesity (Silver Spring) 2022, 30, 1917–1926. [Google Scholar] [CrossRef]
- Weyer, C.; Vozarova, B.; Ravussin, E.; Tataranni, P.A. Changes in energy metabolism in response to 48 h of overfeeding and fasting in Caucasians and Pima Indians. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 593–600. [Google Scholar] [CrossRef]
- Williams, D.R. Epidemiological and geographic factors in diabetes. Eye 1993, 7 (Pt 2), 202–204. [Google Scholar] [CrossRef]
- Ravussin, E. Energy metabolism in obesity. Studies in the Pima Indians. Diabetes Care 1993, 16, 232–238. [Google Scholar] [CrossRef]
- Schulz, L.O.; Bennett, P.H.; Ravussin, E.; Kidd, J.R.; Kidd, K.K.; Esparza, J.; Valencia, M.E. Effects of traditional and western environments on prevalence of type 2 diabetes in Pima Indians in Mexico and the U.S. Diabetes Care 2006, 29, 1866–1871. [Google Scholar] [CrossRef]
- Kalman, R.; Ziv, E.; Shafrir, E.; Bar-On, H.; Perez, R. Psammomys obesus and the albino rat--two different models of nutritional insulin resistance, representing two different types of human populations. Lab. Anim. 2001, 35, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Yanicostas, C.; Soussi-Yanicostas, N. SDHI Fungicide Toxicity and Associated Adverse Outcome Pathways: What Can Zebrafish Tell Us? Int. J. Mol. Sci. 2021, 22, 12362. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lei, L.; Liu, M.; Song, Y.; Lu, S.; Li, D.; Shi, H.; Raley-Susman, K.M.; He, D. Single and mixture toxicity of strobilurin and SDHI fungicides to Xenopus tropicalis embryos. Ecotoxicol. Environ. Saf. 2018, 153, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.R.; Smith-Vaniz, W.F. Rotenone: An Essential but Demonized Tool for Assessing Marine Fish Diversity. BioScience 2008, 58, 165–170. [Google Scholar] [CrossRef]
- Lennon, R.E.; Vézina, C.; Antimycin, A. A Piscicidal Antibiotic. In Advances in Applied Microbiology; Elsevier: Amsterdam, The Netherlands, 1973; Volume 16, pp. 55–96. [Google Scholar] [CrossRef]
- Madeira, D.; Andrade, J.; Leal, M.C.; Ferreira, V.; Rocha, R.J.M.; Rosa, R.; Calado, R. Synergistic Effects of Ocean Warming and Cyanide Poisoning in an Ornamental Tropical Reef Fish. Front. Mar. Sci. 2020, 7, 246. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouillaud, F. Inhibition of Succinate Dehydrogenase by Pesticides (SDHIs) and Energy Metabolism. Int. J. Mol. Sci. 2023, 24, 4045. https://doi.org/10.3390/ijms24044045
Bouillaud F. Inhibition of Succinate Dehydrogenase by Pesticides (SDHIs) and Energy Metabolism. International Journal of Molecular Sciences. 2023; 24(4):4045. https://doi.org/10.3390/ijms24044045
Chicago/Turabian StyleBouillaud, Frederic. 2023. "Inhibition of Succinate Dehydrogenase by Pesticides (SDHIs) and Energy Metabolism" International Journal of Molecular Sciences 24, no. 4: 4045. https://doi.org/10.3390/ijms24044045
APA StyleBouillaud, F. (2023). Inhibition of Succinate Dehydrogenase by Pesticides (SDHIs) and Energy Metabolism. International Journal of Molecular Sciences, 24(4), 4045. https://doi.org/10.3390/ijms24044045