
“Pesto” Mutation: Phenotypic and Genotypic Characteristics of Eight GCK/MODY Ligurian Patients

 , , and
, , and
Abstract
1. Introduction
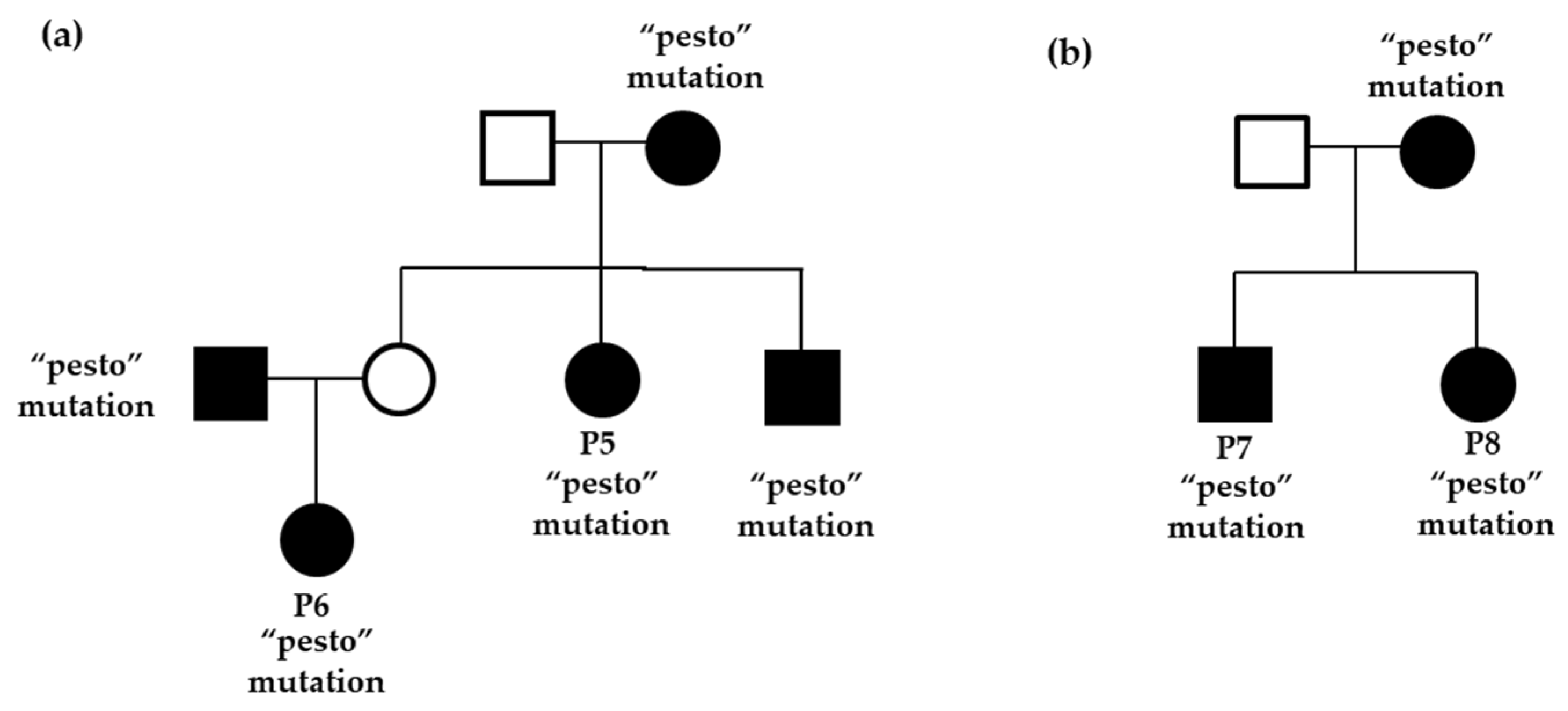
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lezzi, M.; Aloi, C.; Salina, A.; Fragola, M.; Bassi, M.; Strati, M.F.; d’Annunzio, G.; Minuto, N.; Maghnie, M. Diabetes Mellitus Diagnosed in Childhood and Adolescence with Negative Autoimmunity: Results of Genetic Investigation. Front. Endocrinol. 2022, 13, 894878. [Google Scholar] [CrossRef]
- Hattersley, A.T.; Greeley, S.A.W.; Polak, M.; Rubio-Cabezas, O.; Njølstad, P.R.; Mlynarski, W.; Castano, L.; Carlsson, A.; Raile, K.; Chi, D.V.; et al. ISPAD Clinical Practice Consensus Guidelines 2018: The Diagnosis and Management of Monogenic Diabetes in Children and Adolescents. Pediatr. Diabetes 2018, 19, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Nkonge, K.M.; Nkonge, D.K.; Nkonge, T.N. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin. Diabetes Endocrinol. 2020, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, M.; Pastore, C.; Giordano, P. Treatment Options for MODY Patients: A Systematic Review of Literature. Diabetes Ther. 2020, 11, 1667–1685. [Google Scholar] [CrossRef] [PubMed]
- Sperling, M.A.; Garg, A.; Cowie, C.C.; Casagrande, S.S.; Menke, A.; Cissel, M.A.; Eberhardt, M.S.; Meigs, J.B.; Gregg, E.W.; Knowler, W.C.; et al. Monogenic Forms of Diabetes. In Diabetes in America, 3rd ed.; National Institute of Diabetes and Digestive and Kidney Diseases (US): Bethesda, MD, USA, 2018; Chapter 7. [Google Scholar]
- Kawakita, R.; Hosokawa, Y.; Fujimaru, R.; Tamagawa, N.; Urakami, T.; Takasawa, K.; Moriya, K.; Mizuno, H.; Maruo, Y.; Takuwa, M.; et al. Molecular and clinical characterization of glucokinase maturity-onset diabetes of the young (GCK-MODY) in Japanese patients. Diabet. Med. 2014, 31, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Sanyoura, M.; Philipson, L.H.; Naylor, R. Monogenic Diabetes in Children and Adolescents: Recognition and Treatment Options. Curr. Diabetes Rep. 2018, 18, 58. [Google Scholar] [CrossRef]
- Delvecchio, M.; Mozzillo, E.; Salzano, G.; Iafusco, D.; Frontino, G.; Patera, P.I.; Rabbone, I.; Cherubini, V.; Grasso, V.; Tinto, N.; et al. Monogenic Diabetes Accounts for 6.3% of Cases Referred to 15 Italian Pediatric Diabetes Centers During 2007 to 2012. J. Clin. Endocrinol. Metab. 2017, 102, 1826–1834. [Google Scholar] [CrossRef]
- Šimčíková, D.; Kocková, L.; Vackářová, K.; Těšínský, M.; Heneberg, P. Evidence-based tailoring of bioinformatics approaches to optimize methods that predict the effects of nonsynonymous amino acid substitutions in glucokinase. Sci. Rep. 2017, 7, 9499. [Google Scholar] [CrossRef]
- Tinto, N.; Zagari, A.; Capuano, M.; De Simone, A.; Capobianco, V.; Daniele, G.; Giugliano, M.; Spadaro, R.; Franzese, A.; Sacchetti, L. Glucokinase gene mutations: Structural and genotype-phenotype analyses in MODY children from South Italy. PLoS ONE 2008, 4, e1870. [Google Scholar] [CrossRef]
- Gašperíková, D.; Tribble, N.D.; Staník, J.; Hučková, M.; Mišovicová, N.; van de Bunt, M.; Valentínová, L.; Barrow, B.A.; Barák, L.U.; Dobránsky, R.; et al. Identification of a novel b-cell glucokinase (GCK) promoter mutation (−71G>C) that modulates GCK gene expression through loss of allele-specific Sp1 binding causing mild fasting hyperglycemia in humans. Diabetes 2009, 58, 1929–1935. [Google Scholar] [CrossRef]
- Ghadir, E.A.; Saab, R.; Molnes, J.; Hess, O.; Abu-Ras, R.; Darawshi, H.; Njølstad, P.R.; Tenenbaum-Rakover, Y. Maturity onset diabetes of the young type 2 (GCK_MODY): Insight from an extended family. Diabetes Res. Clin. Pract. 2021, 175, 108791. [Google Scholar] [CrossRef]
- Delvecchio, M.; Ludovico, O.; Bellacchio, E.; Stallone, R.; Palladino, T.; Mastroianno, S.; Zelante, L.; Sacco, M.; Trischitta, V.; Carella, M. MODY type 2 P59S GCK mutant: Foundereffect in South of Italy. Clin. Genet. 2013, 83, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Dusatkova, P.; Pruhova, S.; Borowiec, M.; Vesela, K.; Antosik, K.; Lebl, J.; Mlynarski, W.; Cinek, O. Ancestral mutations may cause a significant proportion of GCK-MODY. Pediatr. Diabetes 2012, 6, 489–498. [Google Scholar] [CrossRef]
- Pruhova, S.; Dusatkova, P.; Sumnik, Z.; Kolouskova, S.; Pedersen, O.; Hansen, T.; Cinek, O.; Lebl, J. Glucokinase diabetes in 103 families from a country-based study in the Czech Republic: Geographically restricted distribution of two prevalent GCK mutations. Pediatr. Diabetes 2010, 8, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Saker, P.J.; Hattersley, A.T.; Betal, B.; Hammersley, M.S.; McLellan, J.A.; Lo, Y.M.; Olds, R.J.; Gillmer, M.D.; Holman, R.R.; Turner, R.C. High prevalence of a missense mutation of the glucokinase gene in gestational diabetic patients due to a founder-effect in a local population. Diabetologia 1996, 39, 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Estalella, I.; Rica, I.; Perez de Nanclares, G.; Bilbao, J.R.; Vazquez, J.A.; San Pedro, J.I.; Busturia, M.A.; Castaño, L. Mutations in GCK and HNF-1alpha explain the majority of cases with clinical diagnosis of MODY in Spain. Clin. Endocrinol. 2007, 67, 538–546. [Google Scholar] [CrossRef]
- Sagen, J.V.; Bjorkhaug, L.; Molnes, J.; Raeder, H.; Grevle, L.; Søvik, O.; Molven, A.; Njølstad, P.R. Diagnostic screening of MODY2/GCK mutations in the Norwegian MODY Registry. Pediatr. Diabetes 2008, 9, 442–449. [Google Scholar] [CrossRef]
- Henderson, M.; Levy, E.; Delvin, E.; Losekoot, M.; Lambert, M. Prevalence and clinical phenotype of the p.Val226Met glucokinase gene mutation in French Canadians in Quebec, Canada. Mol. Genet. Metab. 2007, 90, 87–92. [Google Scholar] [CrossRef]
- Aloi, C.; Salina, A.; Minuto, N.; Tallone, R.; Lugani, F.; Mascagni, A.; Mazza, O.; Cassanello, M.; Maghnie, M.; d’Annunzio, G. Glucokinasemutations in pediatric patients with impaired fasting glucose. Acta Diabetol. 2017, 10, 913–923. [Google Scholar] [CrossRef]
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum. Mutat. 2009, 11, 1512–1526. [Google Scholar] [CrossRef]
- López-Garrido, M.P.; Herranz-Antolín, S.; Alija-Merillas, M.J.; Giralt, P.; Escribano, J. Co-inheritance of HNF1a and GCK mutations in a family with maturity-onset diabetes of the young (MODY): Implications for genetic testing. Clin. Endocrinol. 2013, 3, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Fabiano, M.C.; Verrienti, A.; Carbone, A.; Sponziello, M.; Bellitti, P.; Bruno, R. Genotype-Phenotype Correlation in a MODY 2 Family: An Under-Diagnosed Disease. J. Diabetes Mellit. 2016, 6, 263–268. [Google Scholar] [CrossRef]
- Lunt, H.; Heenan, H.; Chan, H. Exploring Phenotype-Genotype Correlations Using Interstitial Glucose Results in a Family With a Glucokinase Mutation. J. Diabetes Sci. Technol. 2018, 12, 1248–1249. [Google Scholar] [CrossRef] [PubMed]
- Greeley, S.A.W.; Polak, M.; Njølstad, P.R.; Barbetti, F.; Williams, R.; Castano, L.; Raile, K.; Chi, D.V.; Habeb, A.; Hattersley, A.T.; et al. ISPAD Clinical Practice Consensus Guidelines 2022: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr. Diabetes 2022, 8, 1188–1211. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Cabezas, O.; Hattersley, A.T.; Njølstad, P.R.; Mlynarski, W.; Ellard, S.; White, N.; Chi, D.V.; Craig, M.E. ISPAD Clinical Practice Consensus Guidelines 2014. The diagnosis and management of monogenic diabetes in children and adolescents; International Society for Pediatric and Adolescent Diabetes. Pediatr. Diabetes 2014, 20, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 5, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG Recommendations for Reporting of Incidental Findings in Clinical Exome and Genome Sequencing. Genet. Med. 2013, 7, 565–574. [Google Scholar] [CrossRef]
- ACMG Board of Directors. ACMG Policy Statement: Updated Recommendations Regarding Analysis and Reporting of Secondary Findings in Clinical Genome-Scale Sequencing. Genet. Med. 2015, 1, 68–69. [Google Scholar] [CrossRef]


{kind=link}
| P | Sex (M/F) | Birth Weight (Grams) | E.G (Weeks) | Age at Hyperglycaemia Diagnosis (Years) | 1st Fasting Glucose Level (mg/dL) | BMI (kg/m2) | OGTT 120 min (mg/dL) | HbA1c (%) | GDM | Affected Family | Pharmacological Treatment (Within 1 Year of Diagnosis) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Member | HbA1c (%) | |||||||||||
| 1 | M | 2700 | 38 | 15 | 131 | 20 | 169 | 6.7 | no | father | 6.5 | insulin |
| 2 | M | 3250 | 40 | 4 | 120 | 15 | 160 | 6.4 | no | mother | nd | no |
| 3 | M | 2900 | 41 + 2 | 3 | 115 | 14.1 | nd | 6.2 | no | father | 6.7 | no |
| 4 | F | 1665 | 31 + 6 | 6 months | 151 | 14.6 | nd | 6.2 | yes | mother | 5.4 | no |
| 5 * | F | 3100 | 40 | 20 | 130 | 18 | nd | 7.2 | yes | mother | nd | insulin |
| 6 | F | 2680 | 40 | 3 | 96 | 14.4 | 171 | 6,5 | no | father | 6.5 | no |
| 7 | M | 2500 | 37 | 7 | 117 | 13.1 | 164 | 6.8 | yes | mother | 6.8 | no |
| 8 | F | 2500 | 39 | 11 | 118 | 20 | 250 | 6.2 | yes | nd | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salina, A.; Bassi, M.; Aloi, C.; Strati, M.F.; Bocciardi, R.; d’Annunzio, G.; Maghnie, M.; Minuto, N. “Pesto” Mutation: Phenotypic and Genotypic Characteristics of Eight GCK/MODY Ligurian Patients. Int. J. Mol. Sci. 2023, 24, 4034. https://doi.org/10.3390/ijms24044034
Salina A, Bassi M, Aloi C, Strati MF, Bocciardi R, d’Annunzio G, Maghnie M, Minuto N. “Pesto” Mutation: Phenotypic and Genotypic Characteristics of Eight GCK/MODY Ligurian Patients. International Journal of Molecular Sciences. 2023; 24(4):4034. https://doi.org/10.3390/ijms24044034
Chicago/Turabian StyleSalina, Alessandro, Marta Bassi, Concetta Aloi, Marina Francesca Strati, Renata Bocciardi, Giuseppe d’Annunzio, Mohamad Maghnie, and Nicola Minuto. 2023. "“Pesto” Mutation: Phenotypic and Genotypic Characteristics of Eight GCK/MODY Ligurian Patients" International Journal of Molecular Sciences 24, no. 4: 4034. https://doi.org/10.3390/ijms24044034
APA StyleSalina, A., Bassi, M., Aloi, C., Strati, M. F., Bocciardi, R., d’Annunzio, G., Maghnie, M., & Minuto, N. (2023). “Pesto” Mutation: Phenotypic and Genotypic Characteristics of Eight GCK/MODY Ligurian Patients. International Journal of Molecular Sciences, 24(4), 4034. https://doi.org/10.3390/ijms24044034