
Microbiota–Liver Diseases Interactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
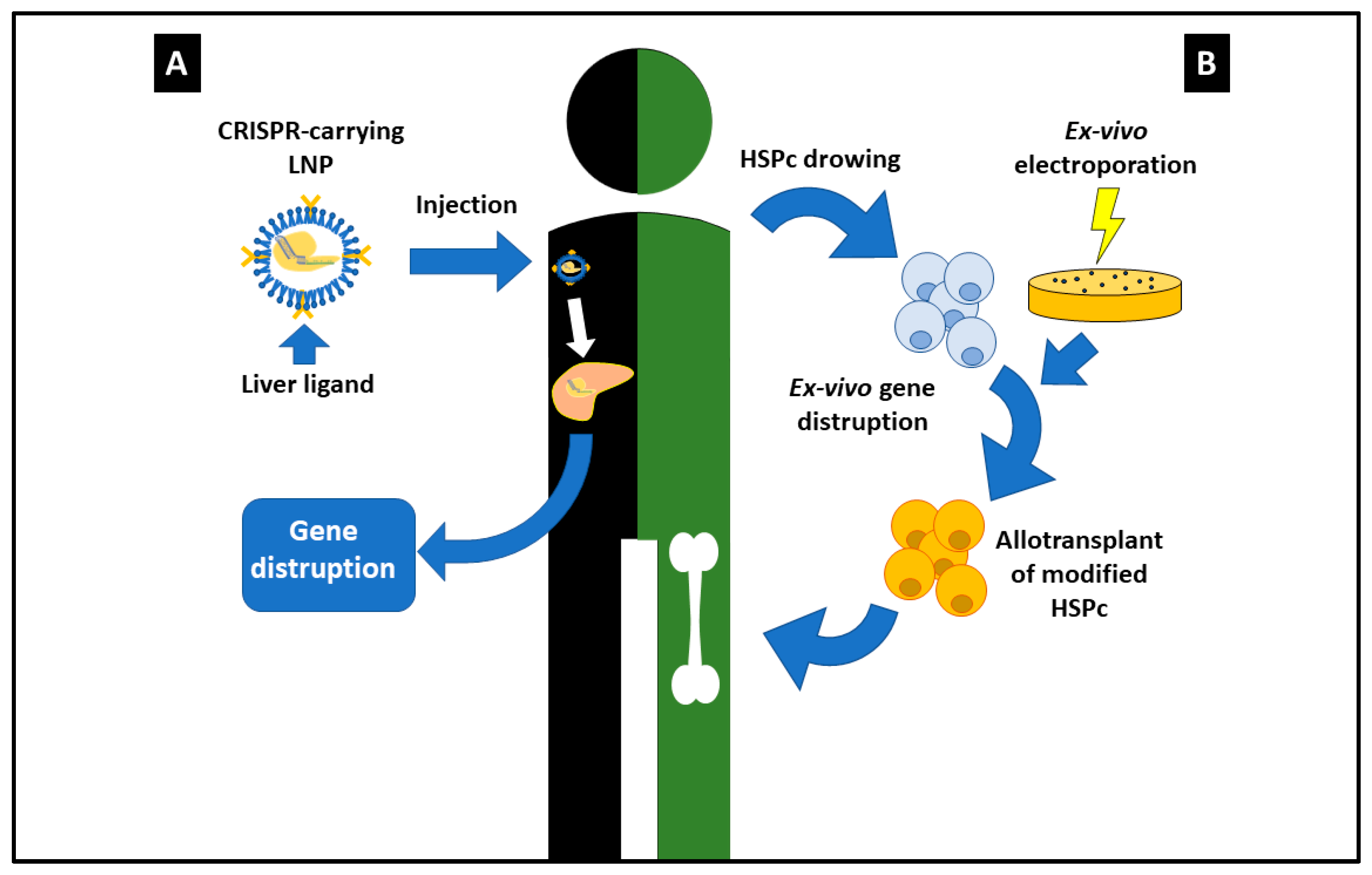
2. CRISPR/Cas9: The Most Outstanding Results
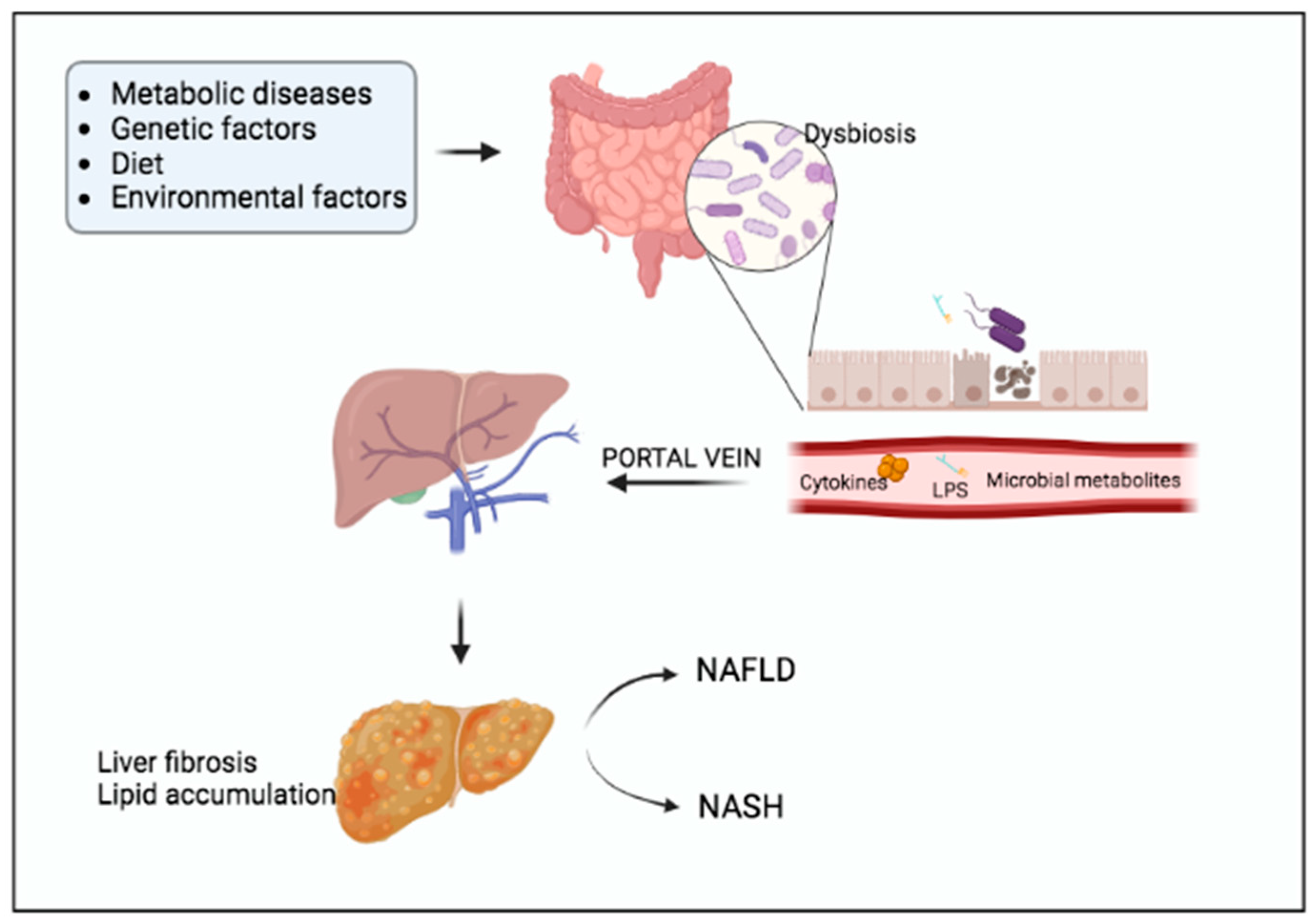
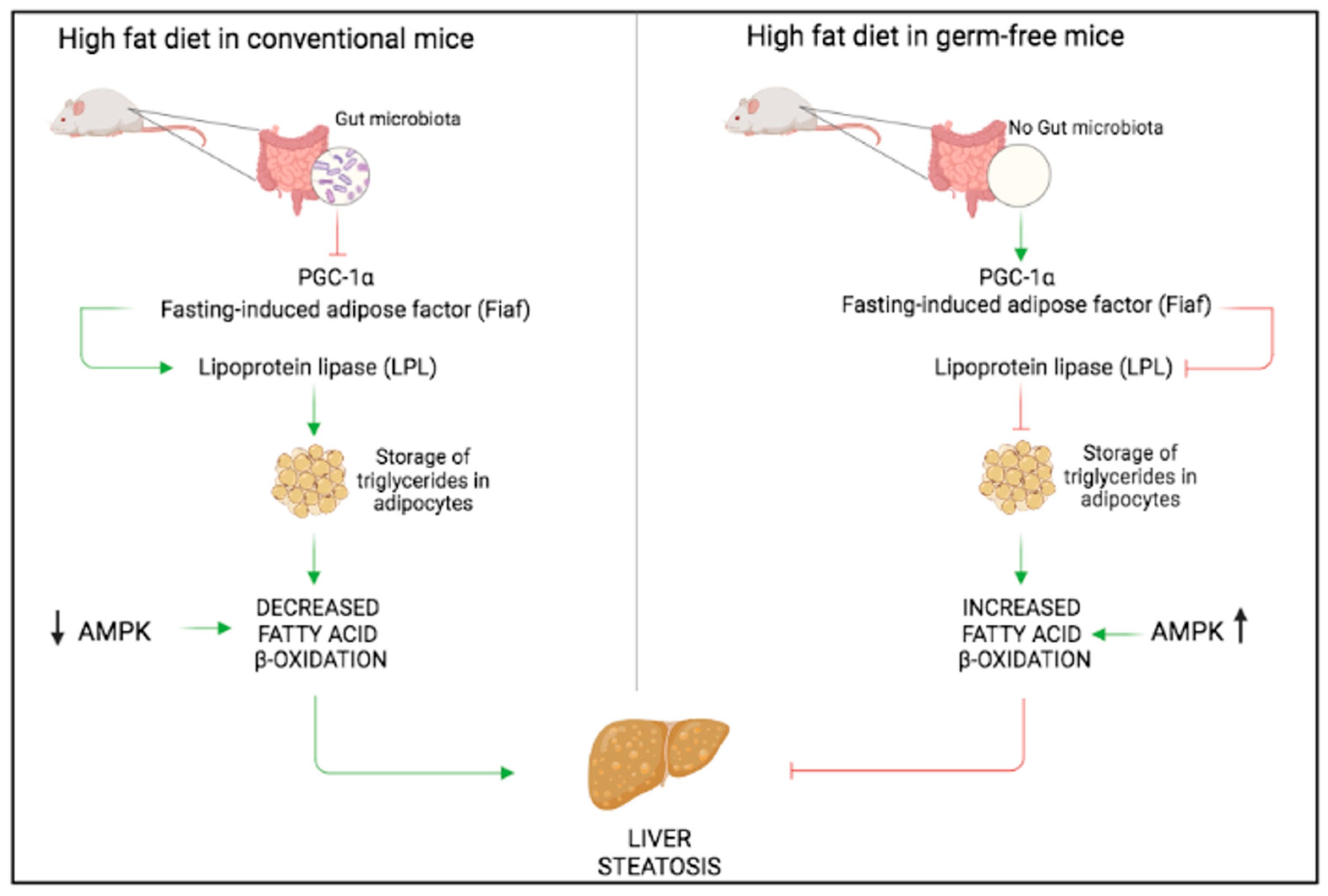
3. Non-Alcoholic Fatty Liver Disease (NAFLD)
4. Colorectal Cancer and Microbiota
5. Pathobionts
6. Segmented Filamentous Bacteria (SFB)
7. Multiple Mutations
8. An Attempt to Find a Rational to the Multiple Interactions between Gut Microbiota, Pathogens, and Host Interactions
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Zechner, E.L. Inflammatory disease caused by intestinal pathobionts. Curr. Opin. Microbiol. 2017, 35, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Schnabl, B. Intestinal microbiota and nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol. 2017, 33, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Salguero, M.V.; Al-Obaide, M.A.I.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Cassiman, D.; Blau, N. Clinical and biochemical footprints of inherited metabolic diseases. II. Metabolic liver diseases. Mol. Genet. Metab. 2019, 127, 117–121. [Google Scholar] [CrossRef]
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of gut microbiota with bile acid metabolism and its influ-ence on disease states. Appl. Microbiol. Biotechnol. 2017, 101, 47–64. [Google Scholar] [CrossRef]
- Long, S.L.; Gahan, C.G.; Joyce, S.A. Interactions between gut bacteria and bile in health and disease. Mol. Asp. Med. 2017, 56, 54–65. [Google Scholar] [CrossRef]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted Disruption of the Nuclear Receptor FXR/BAR Impairs Bile Acid and Lipid Homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef]
- Copple, B.L.; Li, T. Pharmacology of bile acid receptors: Evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol. Res. 2015, 104, 9–21. [Google Scholar] [CrossRef]
- Parséus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Ståhlman, M.; Greiner, T.U.; Perkins, R.; Bäckhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. Available online: https://pubmed.ncbi.nlm.nih.gov/24625896 (accessed on 18 November 2021). [CrossRef]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef]
- Fagiuoli, S.; Daina, E.; D’Antiga, L.; Colledan, M.; Remuzzi, G. Monogenic diseases that can be cured by liver transplantation. J. Hepatol. 2013, 59, 595–612. [Google Scholar] [CrossRef]
- Cozmescu, A.C.; Counsell, J.; Gissen, P. Gene therapies targeting the liver. J. Hepatol. 2020, 74, 235–236. [Google Scholar] [CrossRef]
- Häberle, J.; Boddaert, N.; Burlina, A.; Chakrapani, A.; Dixon, M.; Huemer, M.; Karall, D.; Martinelli, D.; Crespo, P.S.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet. J. Rare Dis. 2012, 7, 32. [Google Scholar] [CrossRef]
- Martini, P.G.; Guey, L.T. A New Era for Rare Genetic Diseases: Messenger RNA Therapy. Hum. Gene Ther. 2019, 30, 1180–1189. [Google Scholar] [CrossRef]
- Richter, C.; Chang, J.T.; Fineran, P.C. Function and regulation of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated (Cas) systems. Viruses 2012, 4, 2291–2311. [Google Scholar] [CrossRef]
- Van Der Oost, J.; Westra, E.R.; Jackson, R.N.; Wiedenheft, B. Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nat. Rev. Microbiol. 2014, 12, 479–492. [Google Scholar] [CrossRef]
- Van Campenhout, C.; Cabochette, P.; Veillard, A.-C.; Laczik, M.; Zelisko-Schmidt, A.; Sabatel, C.; Dhainaut, M.; Vanhollebeke, B.; Gueydan, C.; Kruys, V. Guidelines for optimized gene knockout using CRISPR/Cas9. Biotechniques 2019, 66, 295–302. [Google Scholar] [CrossRef]
- Tian, M.; Xia, P.; Gou, X.; Yan, L.; Yu, H.; Zhang, X. CRISPR screen identified that UGT1A9 was required for bisphenols-induced mitochondria dyshomeostasis. Environ. Res. 2021, 205, 112427. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wu, L.; Zhang, S.-M.; Lu, M.; Cheung, W.K.; Cai, W.; Gale, M.; Xu, Q.; Yan, Q. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016, 44, e149. [Google Scholar] [CrossRef] [PubMed]
- Dallio, M.; Masarone, M.; Romeo, M.; Tuccillo, C.; Morisco, F.; Persico, M.; Loguercio, C.; Federico, A. PNPLA3, TM6SF2, and MBOAT7 Influence on Nutraceutical Therapy Response for Non-alcoholic Fatty Liver Disease: A Randomized Controlled Trial. Front. Med. 2021, 8, 734847. [Google Scholar] [CrossRef]
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Erconi, V.; Carli, F.; Fortunato, F.; Ronchi, D.; Piciotti, R.; Sabatini, S.; Macchi, C.; et al. TM6SF2/PNPLA3/MBOAT7 Loss-of-Function Genetic Variants Impact on NAFLD Development and Progression Both in Patients and in In Vitro Models. Cell. Mol. Gastroenterol. Hepatol. 2021, 13, 759–788. [Google Scholar] [CrossRef]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.-S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2020, 29, 571–586. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; De La Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Chopyk, D.M.; Grakoui, A. Contribution of the Intestinal Microbiome and Gut Barrier to Hepatic Disorders. Gastroenterology 2020, 159, 849–863. [Google Scholar] [CrossRef]
- The Lancet Gastroenterology & Hepatology. Headway and hurdles in non-alcoholic fatty liver disease. Lancet Gastroenterol. Hepatol. 2020, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Wong, V.W.-S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Prim. 2015, 1, 15080. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Chu, H.; Duan, Y.; Schnabl, B. Gut microbiota in liver disease: Too much is harmful, nothing at all is not helpful either. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G563–G573. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Seki, E.; Brenner, D.A.; Friedman, S.; Cohen, J.I.; Nagy, L.; Szabo, G.; Zakhari, S. Innate immunity in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G516–G525. [Google Scholar] [CrossRef]
- Okubo, H.; Sakoda, H.; Kushiyama, A.; Fujishiro, M.; Nakatsu, Y.; Fukushima, T.; Matsunaga, Y.; Kamata, H.; Asahara, T.; Yoshida, Y.; et al. Lactobacillus casei strain Shirota protects against nonalcoholic steatohepatitis development in a rodent model. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G911–G918. [Google Scholar] [CrossRef]
- Craven, L.; Rahman, A.; Nair Parvathy, S.; Beaton, M.; Silverman, J.; Qumosani, K.; Hramiak, I.; Hegele, R.; Joy, T.; Meddings, J.; et al. Allogenic Fecal Microbiota Transplantation in Patients With Nonalcoholic Fatty Liver Disease Improves Abnormal Small Intestinal Permeability: A Randomized Control Trial. Am. J. Gastroenterol. 2020, 115, 1055–1065. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef]
- Zhou, D.; Pan, Q.; Shen, F.; Cao, H.-X.; Ding, W.-J.; Chen, Y.-W.; Fan, J.-G. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci. Rep. 2017, 7, 1529. [Google Scholar] [CrossRef]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Guan, X.-X.; Tang, Y.-J.; Sun, J.-F.; Wang, X.-K.; Wang, W.-D.; Fan, J.-M. Clinical effects and gut microbiota changes of using probiotics, prebiotics or synbiotics in inflammatory bowel disease: A systematic review and meta-analysis. Eur. J. Nutr. 2021, 60, 2855–2875. [Google Scholar] [CrossRef]
- Fleissner, C.K.; Huebel, N.; El-Bary, M.M.A.; Loh, G.; Klaus, S.; Blaut, M. Absence of intestinal microbiota does not protect mice from diet-induced obesity. Br. J. Nutr. 2010, 104, 919–929. [Google Scholar] [CrossRef]
- Marra, M.V.; Simmons, S.F.; Shotwell, M.S.; Hudson, A.; Hollingsworth, E.K.; Long, E.; Kuertz, B.; Silver, H.J. Elevated Serum Osmolality and Total Water Deficit Indicate Impaired Hydration Status in Residents of Long-Term Care Facilities Regardless of Low or High Body Mass Index. J. Acad. Nutr. Diet. 2016, 116, 828–836.e2. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Novikova, D.S.; Garabadzhiu, A.V.; Melino, G.; Barlev, N.A.; Tribulovich, V.G. AMP-activated protein kinase: Structure, function, and role in pathological processes. Biochem. Moscow 2015, 80, 127–144. [Google Scholar] [CrossRef]
- Rabot, S.; Membrez, M.; Bruneau, A.; Gérard, P.; Harach, T.; Moser, M.; Raymond, F.; Mansourian, R.; Chou, C.J. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010, 24, 4948–4959. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- O’Brien, J.M. Environmental and Heritable Factors in the Causation of Cancer. Surv. Ophthalmol. 2000, 45, 167–168. [Google Scholar] [CrossRef]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef]
- Chung, H.; Pamp, S.J.; Hill, J.A.; Surana, N.K.; Edelman, S.M.; Troy, E.B.; Reading, N.C.; Villablanca, E.J.; Wang, S.; Mora, J.R.; et al. Gut Immune Maturation Depends on Colonization with a Host-Specific Microbiota. Cell 2012, 149, 1578–1593. [Google Scholar] [CrossRef]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of Fecal Samples From Patients With Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology 2017, 153, 1621–1633.e6. [Google Scholar] [CrossRef] [PubMed]
- Repass, J.; Iorns, E.; Denis, A.; Williams, S.R.; Perfito, N.; Errington, T.M. Replication Study: Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Elife 2018, 7, e25801. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, L.; Allgayer, H. Viruses in colorectal cancer. Mol. Oncol. 2021, 16, 1423–1450. [Google Scholar] [CrossRef] [PubMed]
- Biesaga, B.; Janecka-Widła, A.; Kołodziej-Rzepa, M.; Słonina, D.; Darasz, Z.; Gasińska, A. The prevalence of HPV infection in rectal cancer—Report from South-Central Poland (Cracow region). Pathol. Res. Pract. 2019, 215, 152513. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Duhaime, M.B.; Ruffin, M.T.; Koumpouras, C.C.; Schloss, P.D. Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. Mbio 2018, 9, e02248-18. [Google Scholar] [CrossRef]
- Gao, R.; Kong, C.; Li, H.; Huang, L.; Qu, X.; Qin, N.; Qin, H. Dysbiosis signature of mycobiota in colon polyp and colorectal cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2457–2468. [Google Scholar] [CrossRef]
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef]
- Durack, J.; Lynch, S.V. The gut microbiome: Relationships with disease and opportunities for therapy. J. Exp. Med. 2019, 216, 20–40. [Google Scholar] [CrossRef]
- Chow, J.; Tang, H.; Mazmanian, S.K. Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr. Opin. Immunol. 2011, 23, 473–480. [Google Scholar] [CrossRef]
- Fine, R.L.; Vieira, S.M.; Gilmore, M.S.; Kriegel, M.A. Mechanisms and consequences of gut commensal translocation in chronic diseases. Gut Microbes 2019, 11, 217–230. [Google Scholar] [CrossRef]
- Proctor, L.M.; Creasy, H.H.; Fettweis, J.M.; Lloyd-Price, J.; Mahurkar, A.; Zhou, W.; Buck, G.A.; Snyder, M.P.; Strauss, J.F.; Weinstock, G.M.; et al. The Integrative Human Microbiome Project. Nature 2019, 569, 641–648. [Google Scholar] [CrossRef]
- Vieira, S.M.; Hiltensperger, M.; Kumar, V.; Zegarra-Ruiz, D.; Dehner, C.; Khan, N.; Costa, F.R.C.; Tiniakou, E.; Greiling, T.; Ruff, W.; et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 2018, 359, 1156–1161. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Takiishi, T.; Fenero, C.I.M.; Câmara, N.O.S. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers 2017, 5, e1373208. [Google Scholar] [CrossRef]
- Stepankova, R.; Powrie, F.; Kofronova, O.; Kozakova, H.; Hudcovic, T.; Hrncir, T.; Uhlig, H.; Read, S.; Rehakova, Z.; Benada, O.; et al. Segmented filamentous bacteria in a defined bacterial cocktail induce intestinal inflammation in SCID mice reconstituted with CD45RBhigh CD4+ T cells. Inflamm. Bowel Dis. 2007, 13, 1202–1211. [Google Scholar] [CrossRef]
- Cheng, H.; Guan, X.; Chen, D.; Ma, W. The th17/treg cell balance: A gut microbiota-modulated story. Microorganisms 2019, 7, 583. [Google Scholar] [CrossRef]
- Mathelier, A.; Shi, W.; Wasserman, W.W. Identification of altered cis-regulatory elements in human disease. Trends Genet. 2015, 31, 67–76. [Google Scholar] [CrossRef]
- Saito, Y.; Koya, J.; Araki, M.; Kogure, Y.; Shingaki, S.; Tabata, M.; McClure, M.B.; Yoshifuji, K.; Matsumoto, S.; Isaka, Y.; et al. Landscape and function of multiple mutations within individual oncogenes. Nature 2020, 582, 95–99. [Google Scholar] [CrossRef]
- Cheng, Z.; Vermeulen, M.; Rollins-Green, M.; DeVeale, B.; Babak, T. Cis-regulatory mutations with driver hallmarks in major cancers. iScience 2021, 24, 102144. [Google Scholar] [CrossRef]
- Pirofski, L.-A.; Casadevall, A. Q&A: What is a pathogen? A question that begs the point. BMC Biol. 2012, 10, 6. [Google Scholar] [CrossRef]
- Nguyen, Y.; Sperandio, V. Enterohemorrhagic E. coli (EHEC) pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 90. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.; Anderson, R. HIV-Associated Bacterial Pneumonia. Clin. Chest Med. 2013, 34, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef]
- Antunes, L.C.M.; McDonald, J.A.K.; Schroeter, K.; Carlucci, C.; Ferreira, R.B.R.; Wang, M.; Yurist-Doutsch, S.; Hira, G.; Jacobson, K.; Davies, J.; et al. Antivirulence Activity of the Human Gut Metabolome. Mbio 2014, 5, e01183-14. [Google Scholar] [CrossRef]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Mi-crobiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef]
- Paredes-Sabja, D.; Shen, A.; Sorg, J. Clostridium difficile spore biology: Sporulation, germination, and spore structural proteins. Trends Microbiol. 2014, 22, 406–416. [Google Scholar] [CrossRef]
- Fournier, P.-E.; Minnick, M.F.; Lepidi, H.; Salvo, E.; Raoult, D. Experimental Model of Human Body Louse Infection Using Green Fluorescent Protein-Expressing Bartonella quintana. Infect. Immun. 2001, 69, 1876–1879. [Google Scholar] [CrossRef]
- Poindexter, H.A.; Washington, T.D. Microbial opportunism in clinical medicine. J. Natl. Med. Assoc. 1974, 66, 284–291. [Google Scholar]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Schwenger, K.J.; Clermont-Dejean, N.; Allard, J.P. The role of the gut microbiome in chronic liver disease: The clinical evidence revised. JHEP Rep. 2019, 1, 214–226. [Google Scholar] [CrossRef]
- Wang, R.; Tang, R.; Li, B.; Ma, X.; Schnabl, B.; Tilg, H. Gut microbiome, liver immunology, and liver diseases. Cell. Mol. Immunol. 2021, 18, 4–17. [Google Scholar] [CrossRef]
- Li, R.; Mao, Z.; Ye, X.; Zuo, T. Human Gut Microbiome and Liver Diseases: From Correlation to Causation. Microorganisms 2021, 9, 1017. [Google Scholar] [CrossRef]
- Jiang, L.; Schnabl, B. Gut microbiota in liver disease: What do we know and what do we not know? Physiology 2020, 35, 261–274. [Google Scholar] [CrossRef]
- Jiminez, J.A.; Uwiera, T.C.; Abbott, D.W.; Uwiera, R.R.E.; Inglis, G.D. Butyrate Supplementation at High Concentrations Alters Enteric Bacterial Communities and Reduces Intestinal Inflammation in Mice Infected with Citrobacter rodentium. mSphere 2017, 2, e00243-17. [Google Scholar] [CrossRef]
- Zwartjes, M.; Gerdes, V.; Nieuwdorp, M. The Role of Gut Microbiota and Its Produced Metabolites in Obesity, Dyslipidemia, Adipocyte Dysfunction, and Its Interventions. Metabolites 2021, 11, 531. [Google Scholar] [CrossRef]
- Smits, L.P.; Bouter, K.E.; de Vos, W.M.; Borody, T.J.; Nieuwdorp, M. Therapeutic Potential of Fecal Microbiota Transplantation. Gastroenterology 2013, 145, 946–953. [Google Scholar] [CrossRef]
- de Groot, P.F.; Frissen, M.N.; de Clercq, N.C.; Nieuwdorp, M. Fecal microbiota transplantation in metabolic syndrome: History, present and future. Gut Microbes 2017, 8, 253–267. [Google Scholar] [CrossRef]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef]
- Vos, W.M. Fame and future of faecal transplantations—Developing next-generation therapies with synthetic microbiomes. Microb. Biotechnol. 2013, 6, 316–325. [Google Scholar] [CrossRef]
- Li, S.S.; Zhu, A.; Benes, V.; Costea, P.I.; Hercog, R.; Hildebrand, F.; Huerta-Cepas, J.; Nieuwdorp, M.; Salojärvi, J.; Voigt, A.Y.; et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 2016, 352, 586–589. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capparelli, R.; Cuomo, P.; Gentile, A.; Iannelli, D. Microbiota–Liver Diseases Interactions. Int. J. Mol. Sci. 2023, 24, 3883. https://doi.org/10.3390/ijms24043883
Capparelli R, Cuomo P, Gentile A, Iannelli D. Microbiota–Liver Diseases Interactions. International Journal of Molecular Sciences. 2023; 24(4):3883. https://doi.org/10.3390/ijms24043883
Chicago/Turabian StyleCapparelli, Rosanna, Paola Cuomo, Antonio Gentile, and Domenico Iannelli. 2023. "Microbiota–Liver Diseases Interactions" International Journal of Molecular Sciences 24, no. 4: 3883. https://doi.org/10.3390/ijms24043883
APA StyleCapparelli, R., Cuomo, P., Gentile, A., & Iannelli, D. (2023). Microbiota–Liver Diseases Interactions. International Journal of Molecular Sciences, 24(4), 3883. https://doi.org/10.3390/ijms24043883