
Neurotransmitters in Prevention and Treatment of Alzheimer’s Disease

Abstract
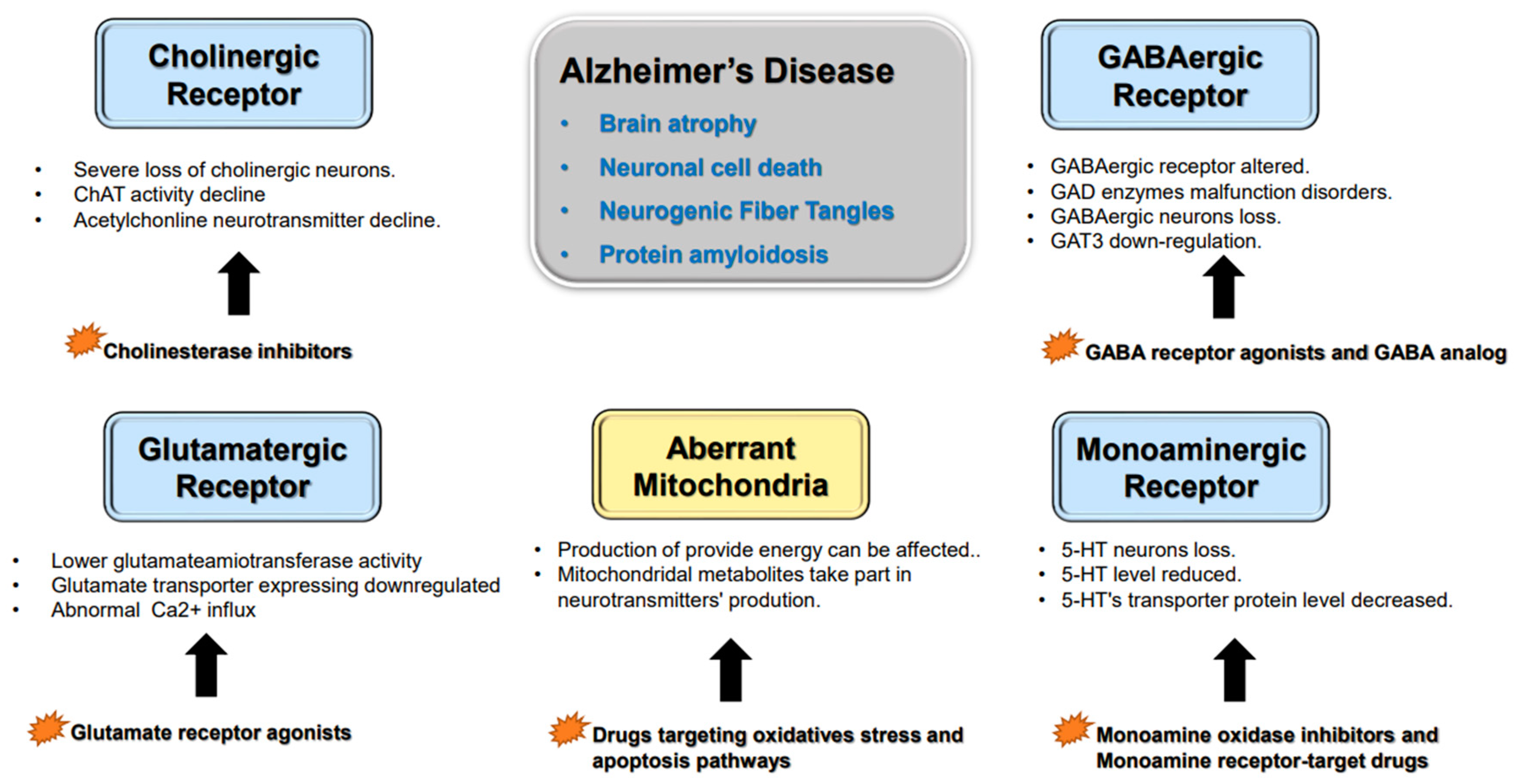
1. Alzheimer’s Disease’s Histopathological Variation
2. Neurotransmitter Abnormalities and Cognitive Dysfunction
2.1. Cholinergic Neurons
2.2. Glutamatergic Neurons
2.3. GABAergic Neurons
2.4. Monoaminergic Neurons
2.5. Other Factors Affecting Neurotransmitter Synthesis and Release
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Bae, H.-G.; Okun, E.; Thiruma, V. Arumugam, Dong-Gyu Jo, Physiology and pharmacology of amyloid precursor protein. Pharmacol. Ther. 2022, 235, 108122. [Google Scholar] [CrossRef] [PubMed]
- O’bryants, E.; Johnson, L.; Edwards, M.; Soares, H.; Devous, M.D.; Ross, S.; Rohlfing, G.; Hall, J.; for the Texas Alzheimer’s Research & Care Consortium. The link between C-reactive protein and Alzheimer’s disease among mexican Americans. J. Alzheimer’s Dis. Jad 2012, 34, 701–706. [Google Scholar]
- Zott, B.; Simon, M.M.; Hong, W.; Unger, F.; Chen-Engerer, H.J.; Frosch, M.P.; Sakmann, B.; Walsh, D.M.; Konnerth, A. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 2019, 365, 559–565. [Google Scholar] [CrossRef]
- Hoskin, J.L.; Sabbagh, M.N.; Al-Hasan, Y.; Decourt, B. Tau immunotherapies for Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 545–554. [Google Scholar] [CrossRef] [PubMed]
- McInnes, J.; Wierda, K.; Snellinx, A.; Bounti, L.; Wang, Y.C.; Stancu, I.C.; Apóstolo, N.; Gevaert, K.; Dewachter, I.; Spires-Jones, T.L.; et al. Synaptogyrin-3 Mediates Presynaptic Dysfunction Induced by Tau. Neuron 2018, 97, 823–835.e8. [Google Scholar] [CrossRef]
- Zubčić, K.; Hof, P.R.; Šimić, G.; Jazvinšćak Jembrek, M. The Role of Copper in Tau-Related Pathology in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 572308. [Google Scholar] [CrossRef]
- Yin, X.; Zhao, C.; Qiu, Y.; Zhou, Z.; Bao, J.; Qian, W. Dendritic/Post-synaptic Tau and Early Pathology of Alzheimer’s Disease. Front. Mol. Neurosci. 2021, 14, 671779. [Google Scholar] [CrossRef]
- Gomes, L.A.; Hipp, S.A.; Rijal Upadhaya, A.; Balakrishnan, K.; Ospitalieri, S.; Koper, M.J.; Largo-Barrientos, P.; Uytterhoeven, V.; Reichwald, J.; Rabe, S.; et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019, 138, 913–941. [Google Scholar] [CrossRef]
- Marinković, P.; Blumenstock, S.; Goltstein, P.; Korzhova, V.; Peters, F.; Knebl, A.; Herms, J. In vivo imaging reveals reduced activity of neuronal circuits in a mouse tauopathy model. Brain 2019, 142, 1051–1062. [Google Scholar] [CrossRef]
- Busche, M.A.; Wegmann, S.; Dujardin, S.; Commins, C.; Schiantarelli, J.; Klickstein, N.; Kamath, T.V.; Carlson, G.A.; Nelken, I.; Hyman, B.T. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 2019, 22, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, M.; Han, Y.; Zhang, H. GABAergic Inhibitory Interneuron Deficits in Alzheimer’s Disease: Implications for Treatment. Front. Neurosci. 2020, 14, 660. [Google Scholar] [CrossRef] [PubMed]
- Ozben, T.; Ozben, S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin. Biochem. 2019, 72, 87–89. [Google Scholar] [CrossRef]
- Prakash, A.; Kalra, J.; Mani, V.; Ramasamy, K.; Majeed, A.B. Pharmacological approaches for Alzheimer’s disease: Neurotransmitter as drug targets. Expert Rev. Neurother. 2015, 15, 53–71. [Google Scholar] [CrossRef]
- Williams, C.L.; Smith, S.M. Calcium dependence of spontaneous neurotransmitter release. J. Neurosci. Res. 2018, 96, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Heine, M.; Heck, J.; Ciuraszkiewicz, A.; Bikbaev, A. Dynamic compartmentalization of calcium channel signalling in neurons. Neuropharmacology 2020, 169, 107556. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Halder, N.; Lal, G. Cholinergic System and Its Therapeutic Importance in Inflammation and Autoimmunity. Front. Immunol. 2021, 12, 660342. [Google Scholar] [CrossRef]
- Cho, G.W.; Kim, M.H.; Chai, Y.G.; Gilmor, M.L.; Levey, A.I.; Hersh, L.B. Phosphorylation of the rat vesicular acetylcholine transporter. J. Biol. Chem. 2000, 275, 19942–19948. [Google Scholar] [CrossRef]
- Gu, X.; Wang, X. An overview of recent analysis and detection of acetylcholine. Anal. Biochem. 2021, 632, 114381. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.F.; Grosch, J.; Fregni, F.; Paulus, W.; Nitsche, M.A. Focusing effect of acetylcholine on neuroplasticity in the human motor cortex. J. Neurosci. 2007, 27, 14442–14447. [Google Scholar] [CrossRef] [PubMed]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Haam, J.; Yakel, J.L. Cholinergic modulation of the hippocampal region and memory function. J. Neurochem. 2017, 142 (Suppl. S2), 111–121. [Google Scholar] [CrossRef] [PubMed]
- Revi, M. Alzheimer’s Disease Therapeutic Approaches. Adv. Exp. Med. Biol. 2020, 1195, 105–116. [Google Scholar] [PubMed]
- Dudai, A.; Doron, M.; Segev, I.; London, M. Synaptic Input and ACh Modulation Regulate Dendritic Ca2+ Spike Duration in Pyramidal Neurons, Directly Affecting Their Somatic OutputAmir Dudai, Michael Doron, Idan Segev, Michael London. J. Neurosci. 2022, 42, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef]
- Fang, X.; Zhang, J.; Zhao, J.; Wang, L. Effect of Resveratrol Combined with Donepezil Hydrochloride on Inflammatory Factor Level and Cognitive Function Level of Patients with Alzheimer’s Disease. J. Healthc. Eng. 2022, 2022, 9148650. [Google Scholar] [CrossRef]
- Levine, S.Z.; Goldberg, Y.; Yoshida, K.; Samara, M.; Cipriani, A.; Iwatsubo, T.; Leucht, S.; Furawaka, T.A. Quantifying the heterogeneity of cognitive functioning in Alzheimer’s disease to extend the placebo-treatment dichotomy: Latent class analysis of individual-participant data from five pivotal randomized clinical trials of donepezil. Eur. Psychiatry 2021, 64, e16. [Google Scholar] [CrossRef]
- Jia, J.; Wei, C.; Chen, W.; Jia, L.; Zhou, A.; Wang, F.; Tang, Y.; Xu, L. Safety and Efficacy of Donepezil 10 mg/day in Patients with Mild to Moderate Alzheimer’s Disease. J. Alzheimers Dis. 2020, 74, 199–211. [Google Scholar] [CrossRef]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, R.; Ohnishi, T.; Kobayashi, H.; Yamaoka, T.; Yajima, T.; Tanimura, A.; Kato, T.; Yoshizawa, K. Long-term effect of galantamine on cognitive function in patients with Alzheimer’s disease versus a simulated disease trajectory: An observational study in the clinical setting. Neuropsychiatr. Dis. Treat. 2017, 13, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Richarz, U.; Gaudig, M.; Rettig, K.; Schauble, B. Galantamine treatment in outpatients with mild Alzheimer’s disease. Acta Neurol. Scand. 2014, 129, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.R.; Grossberg, G.T.; Sadowsky, C.H.; Meng, X.; Somogyi, M. A 24-week, randomized, controlled trial of rivastigmine patch 13.3 mg/24 h versus 4.6 mg/24 h in severe Alzheimer’s dementia. CNS Neurosci Ther. 2013, 19, 745–752. [Google Scholar] [CrossRef]
- Farlow, M.R.; Sadowsky, C.H.; Velting, D.M.; Meng, X.; Islam, M.Z. Evaluating Response to High-Dose 13.3 mg/24 h Rivastigmine Patch in Patients with Severe Alzheimer’s Disease. CNS Neurosci. Ther. 2015, 21, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, R.; Ferreira, E.; Knowles, S.; Fux, C.; Rodin, A.; Winslow, W.; Oddo, S. Lifelong choline supplementation ameliorates Alzheimer’s disease pathology and associated cognitive deficits by attenuating microglia activation. Aging Cell 2019, 18, e13037. [Google Scholar] [CrossRef] [PubMed]
- Bruszt, N.; Bali, Z.K.; Tadepalli, S.A.; Nagy, L.V.; Hernádi, I. Potentiation of cognitive enhancer effects of Alzheimer’s disease medication memantine by alpha7 nicotinic acetylcholine receptor agonist PHA-543613 in the Morris water maze task. Psychopharmacology 2021, 238, 3273–3281. [Google Scholar] [CrossRef]
- Hung, S.Y.; Fu, W.M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 2017, 24, 47. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.L.; Perkins, A.J.; Gao, S.; Skaar, T.C.; Li, L.; Hendrie, H.C.; Fowler, N.; Callahan, C.M.; Boustani, M.A. Adherence and Tolerability of Alzheimer’s Disease Medications: A Pragmatic Randomized Trial. J. Am. Geriatr. Soc. 2017, 65, 1497–1504. [Google Scholar] [CrossRef]
- Walsh, S.; King, E.; Brayne, C. France removes state funding for dementia drugs. BMJ 2019, 367, l6930. [Google Scholar] [CrossRef]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The Dual Role of Glutamatergic Neurotransmission in Alzheimer’s Disease: From Pathophysiology to Pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Lira, M.; Mira, R.G.; Carvajal, F.J.; Zamorano, P.; Inestrosa, N.C.; Cerpa, W. Glutamatergic Receptor Trafficking and Delivery: Role of the Exocyst Complex. Cells 2020, 9, 2402. [Google Scholar] [CrossRef]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.D. Functional identification of activity-regulated, high-affinity glutamine transport in hippocampal neurons inhibited by riluzole. J. Neurochem. 2017, 142, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Moretto, E.; Murru, L.; Martano, G.; Sassone, J.; Passafaro, M. Glutamatergic synapses in neurodevelopmental disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2018, 84 Pt B, 328–342. [Google Scholar] [CrossRef]
- Nair, J.D.; Wilkinson, K.A.; Henley, J.M.; Mellor, J.R. Kainate receptors and synaptic plasticity. Neuropharmacology 2021, 1, 108540. [Google Scholar] [CrossRef]
- Ribeiro, F.M.; Vieira, L.B.; Pires, R.G.; Olmo, R.P.; Ferguson, S.S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol. Res. 2017, 115, 179–191. [Google Scholar] [CrossRef]
- Conway, M.E. Alzheimer’s disease: Targeting the glutamatergic system. Biogerontology 2020, 21, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Dickson, W.; Atiya, S.; Fogarty, J.; Montero-Odasso, M.; Pasternak, S.H.; Brymer, C.; Borrie, M.J.; Bartha, R. Reduced Hippocampal Glutamate and Posterior Cingulate N-Acetyl Aspartate in Mild Cognitive Impairment and Alzheimer’s Disease Is Associated with Episodic Memory Performance and White Matter Integrity in the Cingulum: A Pilot Study. J. Alzheimer’s Dis. 2020, 73, 1385–1405. [Google Scholar]
- Olajide, O.J.; Gbadamosi, I.T.; Yawson, E.O.; Arogundade, T.; Lewu, F.S.; Ogunrinola, K.Y.; Adigun, O.O.; Bamisi, O.; Lambe, E.; Arietarhire, L.O.; et al. Hippocampal Degeneration and Behavioral Impairment During Alzheimer-Like Pathogenesis Involves Glutamate Excitotoxicity. J. Mol. Neurosci. 2021, 71, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Fuchsberger, T.; Yuste, R.; Martinez-Bellver, S.; Blanco-Gandia, M.C.; Torres-Cuevas, I.; Blasco-Serra, A.; Arango, R.; Miñarro, J.; Rodríguez-Arias, M.; Teruel-Marti, V.; et al. Oral Monosodium Glutamate Administration Causes Early Onset of Alzheimer’s Disease-Like Pathophysiology in APP/PS1 Mice. J. Alzheimer’s Dis. 2019, 72, 957–975. [Google Scholar] [CrossRef] [PubMed]
- Fotuhi, S.N.; Khalaj-Kondori, M.; Feizi, M.A.; Talebi, M. Memory-related process in physiological status and alzheimer’s disease. Mol. Biol. Rep. 2020, 47, 4651–4657. [Google Scholar] [CrossRef]
- Kadoyama, K.; Matsuura, K.; Takano, M.; Otani, M.; Tomiyama, T.; Mori, H.; Matsuyama, S. Proteomic analysis involved with synaptic plasticity improvement by GABAA receptor blockade in hippocampus of a mouse model of Alzheimer’s disease. Neurosci. Res. 2021, 165, 61–68. [Google Scholar] [CrossRef]
- Dennis, J. Selkoe, Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar]
- Kishi, T.; Matsunaga, S.; Oya, K.; Nomura, I.; Ikuta, T.; Iwata, N. Memantine for Alzheimer’s Disease: An Updated Systematic Review and Meta-analysis. J. Alzheimers Dis. 2017, 60, 401–425. [Google Scholar] [CrossRef]
- Tariot, P.N.; Farlow, M.R.; Grossberg, G.T.; Graham, S.M.; McDonald, S.; Gergel, I.; Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: A randomized controlled trial. JAMA 2004, 291, 317–324. [Google Scholar] [CrossRef]
- Li, P.; Xu, J.; Gu, H.; Peng, H.; Yin, Y.; Zhuang, J. Memantine ameliorates cognitive deficit in AD mice via enhancement of entorhinal–CA1 projection. BMC Neurosci. 2021, 22, 41. [Google Scholar] [CrossRef]
- Nogo, D.; Nazal, H.; Song, Y.; Teopiz, K.M.; Ho, R.; McIntyre, R.S.; Lui, L.M.W.; Rosenblat, J.D. A review of potential neuropathological changes associated with ketamine. Expert Opin. Drug Saf. 2022, 21, 813–831. [Google Scholar] [CrossRef] [PubMed]
- Dogra, S.; Stansley, B.J.; Xiang, Z.; Qian, W.; Gogliotti, R.G.; Nicoletti, F.; Lindsley, C.W.; Niswender, C.M.; Joffe, M.E.; Conn, P.J. Activating mGlu3 Metabotropic Glutamate Receptors Rescues Schizophrenia-like Cognitive Deficits Through Metaplastic Adaptations Within the Hippocampus. Biol. Psychiatry 2021, 90, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Chaki, S. mGlu2/3 receptor antagonists. Adv. Pharmacol. 2019, 86, 97–120. [Google Scholar]
- Engin, E.; Benham, R.S.; Rudolph, U. An Emerging Circuit Pharmacology of GABAA Receptors. Trends Pharmacol. Sci. 2018, 39, 710–732. [Google Scholar] [CrossRef] [PubMed]
- Rowley, N.M.; Madsen, K.K.; Schousboe, A.; White, H.S. Glutamate and GABA synthesis, release, transport and metabolism as targets for seizure control. Neurochem. Int. 2012, 61, 546–558. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Fischer, F.P.; Kasture, A.S.; Hummel, T.; Sucic, S. Molecular and Clinical Repercussions of GABA Transporter 1 Variants Gone Amiss: Links to Epilepsy and Developmental Spectrum Disorders. Front. Mol. Biosci. 2022, 9, 834498. [Google Scholar] [CrossRef] [PubMed]
- Ghit, A.; Assal, D.; Al-Shami, A.S.; Hussein, D.E.E. GABAA receptors: Structure, function, pharmacology, and related disorders. J. Genet. Eng. Biotechnol. 2021, 19, 123. [Google Scholar] [CrossRef]
- Gassmann, M.; Bettler, B. Regulation of neuronal GABAB receptor functions by subunit composition. Nat. Rev. Neurosci. 2012, 13, 380–394. [Google Scholar] [CrossRef]
- Salcedo, C.; Wagner, A.; Andersen, J.V.; Vinten, K.T.; Waagepetersen, H.S.; Schousboe, A.; Freude, K.K.; Aldana, B.I. Downregulation of GABA Transporter 3 (GAT3) is Associated with Deficient Oxidative GABA Metabolism in Human Induced Pluripotent Stem Cell-Derived Astrocytes in Alzheimer’s Disease. Neurochem. Res. 2021, 46, 2676–2686. [Google Scholar] [CrossRef]
- Salazar, A.M.; Leisgang, A.M.; Ortiz, A.A.; Murtishaw, A.S.; Kinney, J.W. Alterations of GABA B receptors in the APP/PS1 mouse model of Alzheimer’s disease. Neurobiol. Aging 2021, 97, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Li, H.L.; Tian, N.; Liu, F.; Wang, L.; Yin, Y.; Yue, L.; Ma, L.; Wan, Y.; Wang, J.Z. Interneuron Accumulation of Phosphorylated tau Impairs Adult Hippocampal Neurogenesis by Suppressing GABAergic Transmission. Cell Stem Cell 2020, 26, 331–345.e6. [Google Scholar] [CrossRef] [PubMed]
- Buddhala, C.; Hsu, C.C.; Wu, J.Y. A novel mechanism for GABA synthesis and packaging into synaptic vesicles. Neurochem. Int. 2009, 55, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Dade, M.; Berzero, G.; Izquierdo, C.; Giry, M.; Benazra, M.; Delattre, J.Y.; Psimaras, D.; Alentorn, A. Neurological Syndromes Associated with Anti-GAD Antibodies. Int. J. Mol. Sci. 2020, 21, 3701. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.O.; Paulsen, O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 2007, 30, 343–349. [Google Scholar] [CrossRef]
- Kang, Y.J.; Lewis, H.E.S.; Young, M.W.; Govindaiah, G.; Greenfield, L.J., Jr.; Garcia-Rill, E.; Lee, S.H. Cell Type-specific Intrinsic Perithreshold Oscillations in Hippocampal GABAergic Interneurons. Neuroscience 2018, 376, 80–93. [Google Scholar] [CrossRef]
- Hollnagel, J.O.; Elzoheiry, S.; Gorgas, K.; Kins, S.; Beretta, C.A.; Kirsch, J.; Kuhse, J.; Kann, O.; Kiss, E. Early alterations in hippocampal perisomatic GABAergic synapses and network oscillations in a mouse model of Alzheimer’s disease amyloidosis. PLoS ONE 2019, 14, e0209228. [Google Scholar] [CrossRef]
- Stoiljkovic, M.; Gutierrez, K.O.; Kelley, C.; Horvath, T.L.; Hajós, M. TREM2 Deficiency Disrupts Network Oscillations Leading to Epileptic Activity and Aggravates Amyloid-β-Related Hippocampal Pathophysiology in Mice. J. Alzheimers Dis. 2022, 88, 837–847. [Google Scholar] [CrossRef]
- Jafari, Z.; Kolb, B.E.; Mohajerani, M.H. Neural oscillations and brain stimulation in Alzheimer’s disease. Prog. Neurobiol. 2020, 194, 101878. [Google Scholar] [CrossRef]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef]
- Wang, S.; Li, K.; Zhao, S.; Zhang, X.; Yang, Z.; Zhang, J.; Zhang, T. Early-stage dysfunction of hippocampal theta and gamma oscillations and its modulation of neural network in a transgenic 5xFAD mouse model. Neurobiol. Aging 2020, 94, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Wong, A.H.C. GABAergic inhibitory neurons as therapeutic targets for cognitive impairment in schizophrenia. Acta Pharmacol. Sin. 2018, 39, 733–753. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, P.; Garrett, D.D.; Polk, T.A. Dynamic Recovery: GABA Agonism Restores Neural Variability in Older, Poorer Performing Adults. J. Neurosci. 2021, 41, 9350–9360. [Google Scholar] [CrossRef]
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973. [Google Scholar] [CrossRef] [PubMed]
- Harp, T.; Barnes, N.M. Central 5-HT receptors and their function; present and future. Neuropharmacology 2020, 177, 108155. [Google Scholar]
- Voronova, I.P. 5-HT Receptors and Temperature Homeostasis. Biomolecules 2021, 11, 1914. [Google Scholar] [CrossRef]
- Liu, C.; Kaeser, P.S. Mechanisms and regulation of dopamine release. Curr. Opin. Neurobiol. 2019, 57, 46–53. [Google Scholar] [CrossRef]
- Klein, M.O.; Battagello, D.S.; Cardoso, A.R.; Hauser, D.N.; Bittencourt, J.C.; Correa, R.G. Dopamine: Functions, Signaling, and Association with Neurological Diseases. Cell Mol. Neurobiol. 2019, 39, 31–59. [Google Scholar] [CrossRef]
- Martel, J.C.; Gatti McArthur, S. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 1003. [Google Scholar] [CrossRef]
- Joshi, A.; Wang, D.-H.; Watterson, S.; McClean, P.L.; Behera, C.K.; Sharp, T.; Wong-Lin, K. Opportunities for multiscale computational modelling of serotonergic drug effects in Alzheimer’s disease. Neuropharmacology 2020, 174, 108118. [Google Scholar] [CrossRef]
- Bethea, C.L.; Reddy, A.P.; Christian, F.L. How Studies of the Serotonin System in Macaque Models of Menopause Relate to Alzheimer’s Disease1. J. Alzheimer’s Dis. 2017, 57, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Morgese, M.G.; Trabace, L. Monoaminergic System Modulation in Depression and Alzheimer’s Disease: A New Standpoint? Front. Pharmacol. 2019, 10, 483. [Google Scholar] [CrossRef] [PubMed]
- Korábečný, J.; Nepovimová, E.; Cikánková, T.; Špilovská, K.; Vašková, L.; Mezeiová, E.; Kuča, K.; Hroudová, J. Newly Developed Drugs for Alzheimer’s Disease in Relation to Energy Metabolism, Cholinergic and Monoaminergic Neurotransmission. Neuroscience 2018, 370, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.P.; Yin, X.; Sawant, N.; Reddy, P.H. Protective effects of antidepressant citalopram against abnormal APP processing and amyloid beta-induced mitochondrial dynamics, biogenesis, mitophagy and synaptic toxicities in Alzheimer’s disease. Hum. Mol. Genet. 2021, 30, 847–864. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Smith, J.A.; Chang, R.; Bourdet, D.L.; Tsuruda, P.R.; Obedencio, G.P.; Beattie, D.T. 5-HT (4) receptor agonist mediated enhancement of cognitive function in vivo and amyloid precursor protein processing in vitro: A pharmacodynamic and pharmacokinetic assessment. Neuropharmacology 2011, 61, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Martínez, J.; Marmisolle, I.; Tarallo, D.; Quijano, C. Mitochondrial Bioenergetics and Dynamics in Secretion Processes. Front. Endocrinol. 2020, 11, 319. [Google Scholar] [CrossRef]
- Raefsky, S.M.; Mattson, M.P. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free. Radic. Biol. Med. 2017, 102, 203–216. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Regulation and roles of mitophagy at synapses. Mech. Ageing Dev. 2020, 187, 111216. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]


{kind=link}
| Author | Type | Study Design and Patients | Dosage | Conclusion | |
|---|---|---|---|---|---|
| Number of Patients | Group | ||||
| Stephen Z. Levine et al., 2021 [26] | Donepezil | 2191 AD patients | 1339 female and 852 male | Control group: 760 and experimental group: 1431 | Logistic modeling showed that donepezil compared to placebo was significantly positively associated with membership in the improvers class, and negatively with high scorers. |
| Jianping Jia et al., 2019 [27] | Donepezil | 241 AD patients | Mild to moderate AD | Donepezil 5 mg/day for at least 4 weeks. All patients received donepezil 10 mg/day for 20 weeks | Single-arm, prospective, multicenter trial. Donepezil 10 mg/day treatment can be tolerated and is also effective in Chinese patients with mild-to-moderate AD, and thus can be used to treat these patients when their response to donepezil 5 mg/day treatment diminishes. |
| Litao Wang et al., 2021 [25] | Donepezil | 90 AD patients | Control group (CG) and experimental group (EG). Patients in CG received donepezil hydrochloride treatment, and on this basis, those in EG received additional RES treatment | Compared with the CG after treatment, the EG obtained significantly higher rates, MMSE scores, and FIM scores (p < 0.05) and evidently lower clinical indicators and ADAS-cog scores (p < 0.001), and between the CG and EG, no obvious difference in the total incidence rate of adverse reactions was observed after treatment (p > 0.05). Conclusion: combining RES with donepezil hydrochloride has significant clinical efficacy in treating AD, which can effectively improve patients’ inflammatory factor indicators, promote their cognitive function, and facilitate patient prognosis. | |
| M. Gaudig et al., 2014 [30] | Galantamine | 75 patients | 55% women; mean ADAS-cog: 22.3; mean age: 70.2 years | Total daily dose of 24 mg galantamine at final visit | ADAS-cog/11, Bayer-ADL scale (self- and caregivers’ ratings), 10-item NPI and CGI-change, safety and tolerability measures. Galantamine was generally safe and well tolerated during the 3-year observation period. Cognition, behavior, and activities of daily living improved during the 12 months of treatment. At the 3-year follow-up, worsening of all outcomes was measured; however, cognition remained improved compared with an untreated population. |
| U Richarz et al., 2014 [30] | Galantamine | 661 AD patients; 554 were assessed for efficacy | Patients with mild-to-moderate AD received flexible dosing of galantamine (16–24 mg/day) during this study | Galantamine was regarded as generally safe. Importantly, this study revealed that galantamine improved cognitive function above the predicted level in 70% of the patients. | |
| Martin R. Farlow et al., 2013–2015 [31,32,33,34,35,36,37,38,39,40,41,42] | Rivastigmine | 1014 patients | 716 were randomized to 13.3 mg/24 h (N = 356) or 4.6 mg/24 h (N = 360) patch group | Severe Impairment Battery (SIB) and AD Cooperative Study Activities of Daily Living scale, Severe Impairment Version and ADCS. The 13.3 mg/24 h patch demonstrated superior efficacy to the 4.6 mg/24 h patch on SIB and ADCS-ADL-SIV, a without marked increase in AEs, suggesting the higher dose patch has a favorable benefit-to-risk profile in severe AD. | |
| Rivastigmine | 1014 patients | 716 were randomized to 13.3 mg/24 h (N = 356) or 4.6 mg/24 h (N = 360) patch group | A significant therapeutic effect of the high- dose rivastigmine patch on ADCS-CGIC response was observed. The 13.3 mg/24 h patch was identified as a predictor of “improvement” or “improvement or no change”. Patients with minimal worsening/improvement/no change after treatment initiation may be more likely to respond to the treatment following long-term therapy. | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Zou, Y.; Wang, L. Neurotransmitters in Prevention and Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 3841. https://doi.org/10.3390/ijms24043841
Yang Z, Zou Y, Wang L. Neurotransmitters in Prevention and Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(4):3841. https://doi.org/10.3390/ijms24043841
Chicago/Turabian StyleYang, Zhenqi, Yong Zou, and Lifeng Wang. 2023. "Neurotransmitters in Prevention and Treatment of Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 4: 3841. https://doi.org/10.3390/ijms24043841
APA StyleYang, Z., Zou, Y., & Wang, L. (2023). Neurotransmitters in Prevention and Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences, 24(4), 3841. https://doi.org/10.3390/ijms24043841