
Metabolism as a New Avenue for Hepatocellular Carcinoma Therapy

Abstract
{kind=link}
{kind=link}
{kind=link}
1. Hepatocellular Carcinoma
2. Cancer and Metabolism
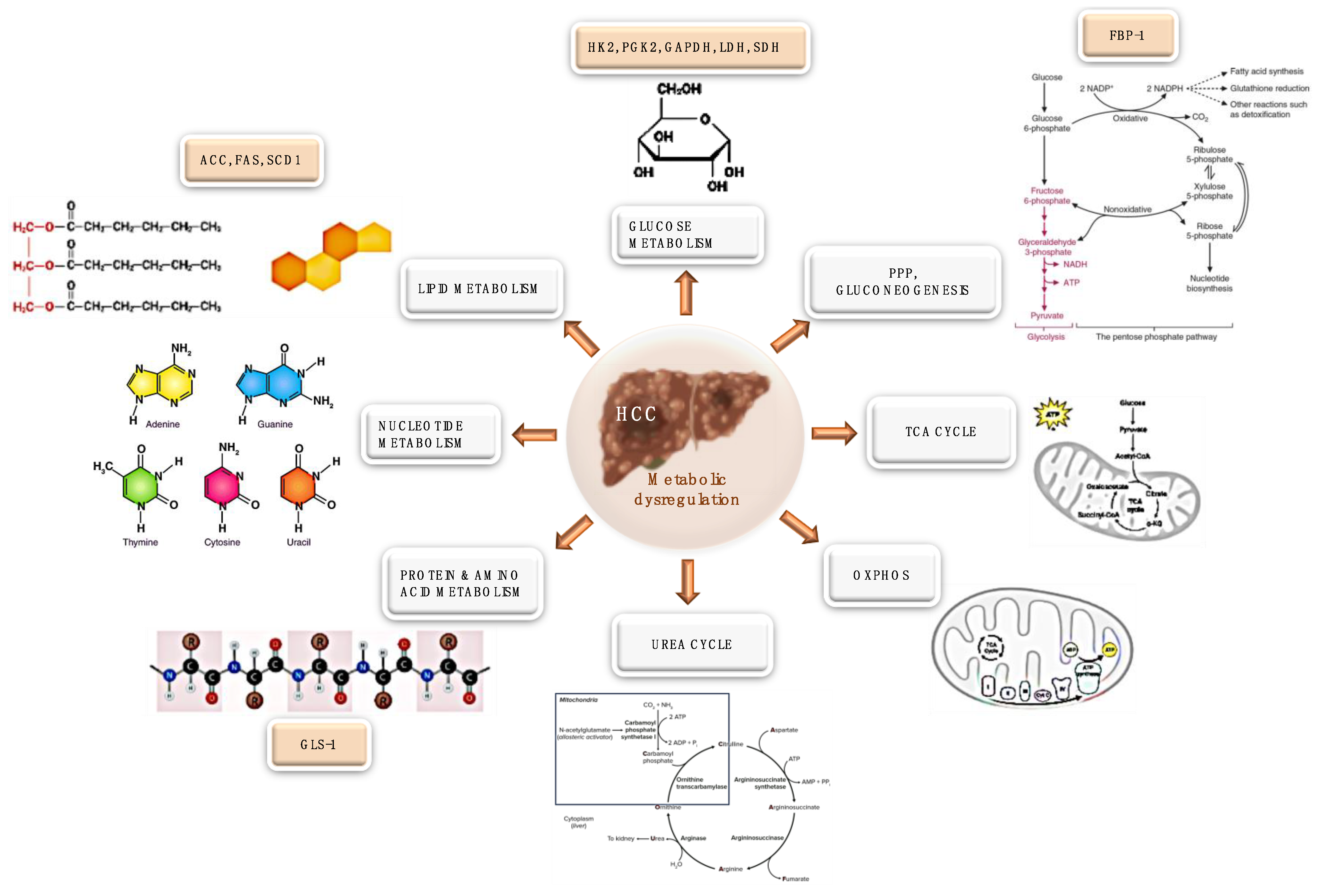
3. Hepatocellular Carcinoma and Metabolism
3.1. Glucose Metabolism Rewiring
3.2. Pentose Phosphate Pathway
3.3. TCA Cycle Rewiring
3.4. OXPHOS Rewiring
3.5. Lipid Metabolism Rewiring
3.6. Nucleotide Metabolism Rewiring
3.7. Protein and Amino Acid Metabolism Rewiring
3.8. Urea Cycle Rewiring
3.9. HCC Tumor Microenvironment Rewiring
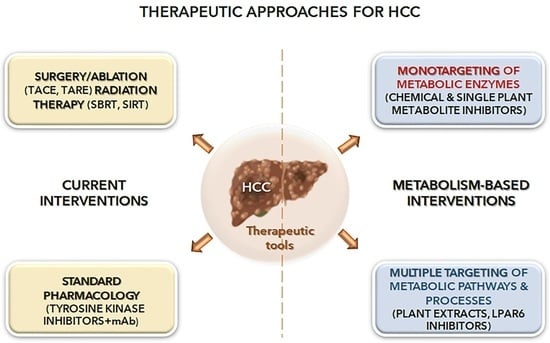
4. Metabolism-Based Pharmacology Approaches in HCC
4.1. Inhibition of the Glycolytic Pathway and Pentose Phosphate Pathway
4.2. Lipid Metabolism Rewiring Inhibition
4.3. Other Potential Metabolic Targets for HCC
5. Multitarget Metabolic Systems
5.1. Plant-Based Multi-Pathway Approach
5.2. Targets Exploiting Synergistic Effects on Metabolic Pathways
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef] [PubMed]
- Rumgay, H.; Ferlay, J.; de Martel, C.; Georges, D.; Ibrahim, A.S.; Zheng, R.; Wei, W.; Lemmens, V.; Soerjomataram, I. Global, regional and national burden of primary liver cancer by subtype. Eur. J. Cancer 2022, 161, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Agosti, P.; Sabba, C.; Mazzocca, A. Emerging metabolic risk factors in hepatocellular carcinoma and their influence on the liver microenvironment. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 607–617. [Google Scholar] [CrossRef]
- Kudo, M. Systemic Therapy for Hepatocellular Carcinoma: Latest Advances. Cancers 2018, 10, 412. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Szent-Gyorgyi, A.; Hegyeli, A.; Mc, L.J. Cancer therapy: A possible new approach. Science 1963, 140, 1391–1392. [Google Scholar] [CrossRef]
- Szent-Gyorgyi, A. The living state and cancer. Physiol. Chem. Phys. 1980, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Szent-Gyorgyi, A. Bioelectronics and cancer. J. Bioenerg. 1973, 4, 533–562. [Google Scholar] [CrossRef]
- Fendt, S.M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov. 2020, 10, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Hajaj, E.; Sciacovelli, M.; Frezza, C.; Erez, A. The context-specific roles of urea cycle enzymes in tumorigenesis. Mol. Cell 2021, 81, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Endicott, M.; Jones, M.; Hull, J. Amino acid metabolism as a therapeutic target in cancer: A review. Amino Acids 2021, 53, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707.e6. [Google Scholar] [CrossRef]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 698023. [Google Scholar] [CrossRef]
- Soukupova, J.; Malfettone, A.; Hyrossova, P.; Hernandez-Alvarez, M.I.; Penuelas-Haro, I.; Bertran, E.; Junza, A.; Capellades, J.; Giannelli, G.; Yanes, O.; et al. Role of the Transforming Growth Factor-beta in regulating hepatocellular carcinoma oxidative metabolism. Sci. Rep. 2017, 7, 12486. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Lim, J.J.; Jeoun, U.W.; Min, S.; Lee, E.B.; Kwon, S.M.; Lee, C.; Yoon, G. Lactate-mediated mitoribosomal defects impair mitochondrial oxidative phosphorylation and promote hepatoma cell invasiveness. J. Biol. Chem. 2017, 292, 20208–20217. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.L.; Li, F.; Yeo, J.Z.; Yong, K.J.; Bassal, M.A.; Ng, G.H.; Lee, M.Y.; Leong, C.Y.; Tan, H.K.; Wu, C.S.; et al. New High-Throughput Screening Identifies Compounds That Reduce Viability Specifically in Liver Cancer Cells That Express High Levels of SALL4 by Inhibiting Oxidative Phosphorylation. Gastroenterology 2019, 157, 1615–1629. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.S.; Seo, J.; Lee, S.H.; Kang, J.H.; Song, J.; Kim, S.Y. Targeting Mitochondrial Oxidative Phosphorylation Abrogated Irinotecan Resistance in NSCLC. Sci. Rep. 2018, 8, 15707. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.P.; Kapur, A.; Barroilhet, L.; Patankar, M.S. Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies Against Ovarian Cancer. Cancers 2018, 10, 337. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Xia, H.; Huang, Z.; Xu, Y.; Yam, J.W.P.; Cui, Y. Reprogramming of central carbon metabolism in hepatocellular carcinoma. Biomed. Pharmacother. 2022, 153, 113485. [Google Scholar] [CrossRef]
- Lo, C.H.; Farina, F.; Morris, H.P.; Weinhouse, S. Glycolytic regulation in rat liver and hepatomas. Adv. Enzym. Regul. 1968, 6, 453–464. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Vaupel, P.; Hockel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal. 2007, 9, 1221–1235. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef]
- Wada, H.; Nagano, H.; Yamamoto, H.; Yang, Y.; Kondo, M.; Ota, H.; Nakamura, M.; Yoshioka, S.; Kato, H.; Damdinsuren, B.; et al. Expression pattern of angiogenic factors and prognosis after hepatic resection in hepatocellular carcinoma: Importance of angiopoietin-2 and hypoxia-induced factor-1 alpha. Liver Int. 2006, 26, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, G.; Zhu, H.; Dong, X.; Zhao, D.; Jiang, X.; Li, J.; Qiao, H.; Ni, S.; Sun, X. 2-Methoxyestradiol synergizes with sorafenib to suppress hepatocellular carcinoma by simultaneously dysregulating hypoxia-inducible factor-1 and -2. Cancer Lett. 2014, 355, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Korotchkina, L.G.; Patel, M.S. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J. Biol. Chem. 2001, 276, 37223–37229. [Google Scholar] [CrossRef]
- Bjornson, E.; Mukhopadhyay, B.; Asplund, A.; Pristovsek, N.; Cinar, R.; Romeo, S.; Uhlen, M.; Kunos, G.; Nielsen, J.; Mardinoglu, A. Stratification of Hepatocellular Carcinoma Patients Based on Acetate Utilization. Cell Rep. 2015, 13, 2014–2026. [Google Scholar] [CrossRef] [PubMed]
- Su, T.S.; Tsai, T.F.; Chi, C.W.; Han, S.H.; Chou, C.K. Elevation of facilitated glucose-transporter messenger RNA in human hepatocellular carcinoma. Hepatology 1990, 11, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Jeong, D.C.; Pak, K.; Han, M.E.; Kim, J.Y.; Liangwen, L.; Kim, H.J.; Kim, T.W.; Kim, T.H.; Hyun, D.W.; et al. SLC2A2 (GLUT2) as a novel prognostic factor for hepatocellular carcinoma. Oncotarget 2017, 8, 68381–68392. [Google Scholar] [CrossRef] [PubMed]
- Amann, T.; Hellerbrand, C. GLUT1 as a therapeutic target in hepatocellular carcinoma. Expert Opin. Ther. Targets 2009, 13, 1411–1427. [Google Scholar] [CrossRef]
- Gong, L.; Cui, Z.; Chen, P.; Han, H.; Peng, J.; Leng, X. Reduced survival of patients with hepatocellular carcinoma expressing hexokinase II. Med. Oncol. 2012, 29, 909–914. [Google Scholar] [CrossRef]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 446. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, S.; Wang, H.; Wu, J.; Chen, D.; Peng, B.; Zhou, Q. High expression of hexokinase domain containing 1 is associated with poor prognosis and aggressive phenotype in hepatocarcinoma. Biochem. Biophys. Res. Commun. 2016, 474, 673–679. [Google Scholar] [CrossRef]
- Liu, S.; Sun, Y.; Jiang, M.; Li, Y.; Tian, Y.; Xue, W.; Ding, N.; Sun, Y.; Cheng, C.; Li, J.; et al. Glyceraldehyde-3-phosphate dehydrogenase promotes liver tumorigenesis by modulating phosphoglycerate dehydrogenase. Hepatology 2017, 66, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Geschwind, J.F. Glyceraldehyde-3-phosphate dehydrogenase: A promising target for molecular therapy in hepatocellular carcinoma. Oncotarget 2012, 3, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Au, S.L.; Tse, A.P.; Xu, I.M.; Lai, R.K.; Chiu, D.K.; Wei, L.L.; Fan, D.N.; Tsang, F.H.; Lo, R.C.; et al. Switching of pyruvate kinase isoform L to M2 promotes metabolic reprogramming in hepatocarcinogenesis. PLoS ONE 2014, 9, e115036. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Bode, B.P.; Souba, W.W. Glutamine transport and human hepatocellular transformation. JPEN J. Parenter. Enter. Nutr. 1999, 23, S33–S37. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Mates, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Faloppi, L.; Scartozzi, M.; Bianconi, M.; Svegliati Baroni, G.; Toniutto, P.; Giampieri, R.; Del Prete, M.; De Minicis, S.; Bitetto, D.; Loretelli, C.; et al. The role of LDH serum levels in predicting global outcome in HCC patients treated with sorafenib: Implications for clinical management. BMC Cancer 2014, 14, 110. [Google Scholar] [CrossRef]
- Faloppi, L.; Bianconi, M.; Memeo, R.; Casadei Gardini, A.; Giampieri, R.; Bittoni, A.; Andrikou, K.; Del Prete, M.; Cascinu, S.; Scartozzi, M. Lactate Dehydrogenase in Hepatocellular Carcinoma: Something Old, Something New. BioMed Res. Int. 2016, 2016, 7196280. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, J.; Li, J.; Feng, J.; Chen, Z.; Wang, X. Plasma metabolomic analysis of human hepatocellular carcinoma: Diagnostic and therapeutic study. Oncotarget 2016, 7, 47332–47342. [Google Scholar] [CrossRef]
- Gao, H.J.; Zhao, M.C.; Zhang, Y.J.; Zhou, D.S.; Xu, L.; Li, G.B.; Chen, M.S.; Liu, J. Monocarboxylate transporter 4 predicts poor prognosis in hepatocellular carcinoma and is associated with cell proliferation and migration. J. Cancer Res. Clin. Oncol. 2015, 141, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, J.; Xing, J.; Li, W.; Li, H.; Ke, X.; Zhang, J.; Ren, T.; Shang, Y.; Yang, H.; et al. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J. Hepatol. 2014, 61, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Megger, D.A.; Hammad, S.; Sitek, B.; Roessler, S.; Ebert, M.P.; Meyer, C.; Dooley, S. Identification of the Consistently Altered Metabolic Targets in Human Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 303–323.e301. [Google Scholar] [CrossRef]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef]
- Xu, I.M.; Lai, R.K.; Lin, S.H.; Tse, A.P.; Chiu, D.K.; Koh, H.Y.; Law, C.T.; Wong, C.M.; Cai, Z.; Wong, C.C.; et al. Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. USA 2016, 113, E725–E734. [Google Scholar] [CrossRef]
- Yang, C.; Wang, S.; Ruan, H.; Li, B.; Cheng, Z.; He, J.; Zuo, Q.; Yu, C.; Wang, H.; Lv, Y.; et al. Downregulation of PDK4 Increases Lipogenesis and Associates with Poor Prognosis in Hepatocellular Carcinoma. J. Cancer 2019, 10, 918–926. [Google Scholar] [CrossRef]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef]
- Tseng, P.L.; Wu, W.H.; Hu, T.H.; Chen, C.W.; Cheng, H.C.; Li, C.F.; Tsai, W.H.; Tsai, H.J.; Hsieh, M.C.; Chuang, J.H.; et al. Decreased succinate dehydrogenase B in human hepatocellular carcinoma accelerates tumor malignancy by inducing the Warburg effect. Sci. Rep. 2018, 8, 3081. [Google Scholar] [CrossRef]
- Gerresheim, G.K.; Roeb, E.; Michel, A.M.; Niepmann, M. Hepatitis C Virus Downregulates Core Subunits of Oxidative Phosphorylation, Reminiscent of the Warburg Effect in Cancer Cells. Cells 2019, 8, 1410. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zheng, X.; Wang, J.; Yang, T.; Dai, W.; Song, S.; Fang, L.; Wang, Y.; Gu, J. O-GlcNAcylation on Rab3A attenuates its effects on mitochondrial oxidative phosphorylation and metastasis in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 970. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Chen, Y.; Wu, Z.; Xu, Q.; Chen, M.; Shao, M.; Cao, X.; Zhou, Y.; Xie, M.; Shi, Y.; et al. Mitochondrial miR-181a-5p promotes glucose metabolism reprogramming in liver cancer by regulating the electron transport chain. Carcinogenesis 2020, 41, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Gao, P. Diabetes Mellitus and Risk of Hepatocellular Carcinoma. BioMed Res. Int. 2017, 2017, 5202684. [Google Scholar] [CrossRef]
- Gan, L.; Liu, Z.; Sun, C. Obesity linking to hepatocellular carcinoma: A global view. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 97–102. [Google Scholar] [CrossRef]
- Batchuluun, B.; Pinkosky, S.L.; Steinberg, G.R. Lipogenesis inhibitors: Therapeutic opportunities and challenges. Nat. Rev. Drug Discov. 2022, 21, 283–305. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Mounier, C.; Bouraoui, L.; Rassart, E. Lipogenesis in cancer progression (review). Int. J. Oncol. 2014, 45, 485–492. [Google Scholar] [CrossRef]
- Cao, D.; Song, X.; Che, L.; Li, X.; Pilo, M.G.; Vidili, G.; Porcu, A.; Solinas, A.; Cigliano, A.; Pes, G.M.; et al. Both de novo synthetized and exogenous fatty acids support the growth of hepatocellular carcinoma cells. Liver Int. 2017, 37, 80–89. [Google Scholar] [CrossRef]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 2015, 5, 14752. [Google Scholar] [CrossRef] [PubMed]
- Kerner, J.; Hoppel, C. Fatty acid import into mitochondria. Biochim. Biophys. Acta 2000, 1486, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Giudetti, A.M.; Bellanti, F.; Priore, P.; Rollo, T.; Tamborra, R.; Siculella, L.; Vendemiale, G.; Altomare, E.; Gnoni, G.V. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid beta-oxidation in rats fed a methionine-choline deficient diet. PLoS ONE 2011, 6, e24084. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Romano, A.D.; Giudetti, A.M.; Capitanio, N.; Petrella, A.; Vendemiale, G.; Altomare, E. Alterations of hepatic ATP homeostasis and respiratory chain during development of non-alcoholic steatohepatitis in a rodent model. Eur. J. Clin. Investig. 2008, 38, 245–252. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; Zhu, G.Q.; Wang, Y.; Zheng, J.N.; Ruan, L.Y.; Cheng, Z.; Hu, B.; Fu, S.W.; Zheng, M.H. Systematic review with network meta-analysis: Statins and risk of hepatocellular carcinoma. Oncotarget 2016, 7, 21753–21762. [Google Scholar] [CrossRef]
- Wada, A.; Fukui, K.; Sawai, Y.; Imanaka, K.; Kiso, S.; Tamura, S.; Shimomura, I.; Hayashi, N. Pamidronate induced anti-proliferative, apoptotic, and anti-migratory effects in hepatocellular carcinoma. J. Hepatol. 2006, 44, 142–150. [Google Scholar] [CrossRef]
- Singh, S.; Singh, P.P.; Singh, A.G.; Murad, M.H.; Sanchez, W. Statins are associated with a reduced risk of hepatocellular cancer: A systematic review and meta-analysis. Gastroenterology 2013, 144, 323–332. [Google Scholar] [CrossRef]
- Ridruejo, E.; Romero-Caimi, G.; Obregon, M.J.; Kleiman de Pisarev, D.; Alvarez, L. Potential Molecular Targets of Statins in the Prevention of Hepatocarcinogenesis. Ann. Hepatol. 2018, 17, 490–500. [Google Scholar] [CrossRef]
- Honda, Y.; Aikata, H.; Honda, F.; Nakano, N.; Nakamura, Y.; Hatooka, M.; Morio, K.; Kobayashi, T.; Fukuhara, T.; Nagaoki, Y.; et al. Clinical outcome and prognostic factors in hepatocellular carcinoma patients with bone metastases medicated with zoledronic acid. Hepatol. Res. 2017, 47, 1053–1060. [Google Scholar] [CrossRef]
- Yamada, S.; Takashina, Y.; Watanabe, M.; Nagamine, R.; Saito, Y.; Kamada, N.; Saito, H. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget 2018, 9, 9925–9939. [Google Scholar] [CrossRef]
- Lozano, E.; Sanchez-Vicente, L.; Monte, M.J.; Herraez, E.; Briz, O.; Banales, J.M.; Marin, J.J.; Macias, R.I. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol. Cancer Res. 2014, 12, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Stanca, E.; Serviddio, G.; Bellanti, F.; Vendemiale, G.; Siculella, L.; Giudetti, A.M. Down-regulation of LPCAT expression increases platelet-activating factor level in cirrhotic rat liver: Potential antiinflammatory effect of silybin. Biochim. Biophys. Acta 2013, 1832, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Tan, Y.X.; Yin, P.Y.; Ye, G.Z.; Gao, P.; Lu, X.; Wang, H.Y.; Xu, G.W. Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res. 2013, 73, 4992–5002. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Dong, H.; Robertson, K.; Liu, C. DNA methylation suppresses expression of the urea cycle enzyme carbamoyl phosphate synthetase 1 (CPS1) in human hepatocellular carcinoma. Am. J. Pathol. 2011, 178, 652–661. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Wheeler, D.A.; Roberts, L.R. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Yeh, H.W.; Lee, S.S.; Chang, C.Y.; Hu, C.M.; Jou, Y.S. Pyrimidine metabolic rate limiting enzymes in poorly-differentiated hepatocellular carcinoma are signature genes of cancer stemness and associated with poor prognosis. Oncotarget 2017, 8, 77734–77751. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Peng, X.; Chen, Z.; Farshidfar, F.; Xu, X.; Lorenzi, P.L.; Wang, Y.; Cheng, F.; Tan, L.; Mojumdar, K.; Du, D.; et al. Molecular Characterization and Clinical Relevance of Metabolic Expression Subtypes in Human Cancers. Cell Rep. 2018, 23, 255–269. [Google Scholar] [CrossRef]
- Carr, B.I.; Guerra, V. Serum albumin levels in relation to tumor parameters in hepatocellular carcinoma patients. Int. J. Biol. Mrk. 2017, 32, e391–e396. [Google Scholar] [CrossRef]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef]
- Stepien, M.; Duarte-Salles, T.; Fedirko, V.; Floegel, A.; Barupal, D.K.; Rinaldi, S.; Achaintre, D.; Assi, N.; Tjonneland, A.; Overvad, K.; et al. Alteration of amino acid and biogenic amine metabolism in hepatobiliary cancers: Findings from a prospective cohort study. Int. J. Cancer 2016, 138, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Cheng, J.; Fan, C.; Shi, X.; Cao, Y.; Sun, B.; Ding, H.; Hu, C.; Dong, F.; Yan, X. Serum Metabolomics to Identify the Liver Disease-Specific Biomarkers for the Progression of Hepatitis to Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 18175. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zeng, J.; Geng, P.; Fang, C.; Wang, Y.; Sun, M.; Wang, C.; Wang, J.; Yin, P.; Hu, C.; et al. Global Metabolic Profiling Identifies a Pivotal Role of Proline and Hydroxyproline Metabolism in Supporting Hypoxic Response in Hepatocellular Carcinoma. Clin. Cancer Res. 2018, 24, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Chittezhath, M.; Shalova, I.N.; Lim, J.Y. Macrophage polarization and plasticity in health and disease. Immunol. Res. 2012, 53, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Sica, A.; Lewis, C.E. Plasticity of macrophage function during tumor progression: Regulation by distinct molecular mechanisms. J. Immunol. 2008, 180, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Forgues, M.; Ye, Q.H.; Jia, H.L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.X.; Tang, Z.Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Q.; Peng, S.; Liu, X. The combination of the glycolysis inhibitor 2-DG and sorafenib can be effective against sorafenib-tolerant persister cancer cells. OncoTargets Ther. 2019, 12, 5359–5373. [Google Scholar] [CrossRef]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Ishige, N. 2-Deoxyglucose and sorafenib synergistically suppress the proliferation and motility of hepatocellular carcinoma cells. Oncol. Lett. 2017, 13, 800–804. [Google Scholar] [CrossRef]
- Sun, X.; Sun, G.; Huang, Y.; Hao, Y.; Tang, X.; Zhang, N.; Zhao, L.; Zhong, R.; Peng, Y. 3-Bromopyruvate regulates the status of glycolysis and BCNU sensitivity in human hepatocellular carcinoma cells. Biochem. Pharmacol. 2020, 177, 113988. [Google Scholar] [CrossRef]
- Pereira da Silva, A.P.; El-Bacha, T.; Kyaw, N.; dos Santos, R.S.; da-Silva, W.S.; Almeida, F.C.; Da Poian, A.T.; Galina, A. Inhibition of energy-producing pathways of HepG2 cells by 3-bromopyruvate. Biochem. J. 2009, 417, 717–726. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.F.; Kunjithapatham, R.; Buijs, M.; Vossen, J.A.; Tchernyshyov, I.; Cole, R.N.; Syed, L.H.; Rao, P.P.; Ota, S.; et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is pyruvylated during 3-bromopyruvate mediated cancer cell death. Anticancer Res. 2009, 29, 4909–4918. [Google Scholar]
- Liberti, M.V.; Dai, Z.; Wardell, S.E.; Baccile, J.A.; Liu, X.; Gao, X.; Baldi, R.; Mehrmohamadi, M.; Johnson, M.O.; Madhukar, N.S.; et al. A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab. 2017, 26, 648–659.e8. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jin, J.; Yu, H.; Zhao, Z.; Ma, D.; Zhang, C.; Jiang, H. Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2. J. Exp. Clin. Cancer Res. 2017, 36, 44. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, K.; Wang, F.; Dai, W.; Li, S.; Feng, J.; Wu, L.; Liu, T.; Xu, S.; Xia, Y.; et al. Methyl jasmonate leads to necrosis and apoptosis in hepatocellular carcinoma cells via inhibition of glycolysis and represses tumor growth in mice. Oncotarget 2017, 8, 45965–45980. [Google Scholar] [CrossRef]
- Liu, T.; Li, S.; Wu, L.; Yu, Q.; Li, J.; Feng, J.; Zhang, J.; Chen, J.; Zhou, Y.; Ji, J.; et al. Experimental Study of Hepatocellular Carcinoma Treatment by Shikonin through Regulating PKM2. J Hepatocell. Carcinoma 2020, 7, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Dai, W.; Zhang, Q.; Feng, J.; Wu, L.; Liu, T.; Yu, Q.; Xu, S.; Wang, W.; et al. Genistein suppresses aerobic glycolysis and induces hepatocellular carcinoma cell death. Br. J. Cancer 2017, 117, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.X.; Li, M.H.; Tao, L.; Ruan, L.Y.; Hong, W.; Chen, C.; Zhao, W.L.; Xu, H.; Chen, J.F.; Wang, J.S. Anti-Cancer Effects of Emodin on HepG2 Cells as Revealed by (1)H NMR Based Metabolic Profiling. J. Proteome Res. 2018, 17, 1943–1952. [Google Scholar] [CrossRef]
- Wei, L.; Harriman, G.; Ghoshal, S.; Moaven, O.; Greenwood, J.; Bhat, S.; Westlin, W.F.; Harwood, H.J.; Kapeller, R.; Tanabe, K.K.; et al. Combination therapy with a liver selective acetyl-CoA carboxylase inhibitor ND-654 and sorafenib improves efficacy in the treatment of cirrhotic rats with hepatocellular carcinoma. Cancer Res. 2016, 76, 3781. [Google Scholar] [CrossRef]
- Zhou, W.; Simpson, P.J.; McFadden, J.M.; Townsend, C.A.; Medghalchi, S.M.; Vadlamudi, A.; Pinn, M.L.; Ronnett, G.V.; Kuhajda, F.P. Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells. Cancer Res. 2003, 63, 7330–7337. [Google Scholar]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef]
- Ma, M.K.F.; Lau, E.Y.T.; Leung, D.H.W.; Lo, J.; Ho, N.P.Y.; Cheng, L.K.W.; Ma, S.; Lin, C.H.; Copland, J.A.; Ding, J.; et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J. Hepatol. 2017, 67, 979–990. [Google Scholar] [CrossRef]
- Xiong, T.; Li, Z.; Huang, X.; Lu, K.; Xie, W.; Zhou, Z.; Tu, J. TO901317 inhibits the development of hepatocellular carcinoma by LXRalpha/Glut1 decreasing glycometabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G598–G607. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Morales, A.; Llacuna, L.; Lluis, J.M.; Terrones, O.; Basanez, G.; Antonsson, B.; Prieto, J.; Garcia-Ruiz, C.; Colell, A.; et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. 2008, 68, 5246–5256. [Google Scholar] [CrossRef]
- Zheng, Y.H.; Zhou, Q.Q.; Zhao, C.; Li, J.J.; Yu, Z.P.; Zhu, Q.D. ATP citrate lyase inhibitor triggers endoplasmic reticulum stress to induce hepatocellular carcinoma cell apoptosis via p-eIF2 alpha/ATF4/CHOP axis. J. Cell. Mol. Med. 2021, 25, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, D.; Pedrelli, M.; Hurt-Camejo, E.; Parini, P. Lipids around the Clock: Focus on Circadian Rhythms and Lipid Metabolism. Biology 2015, 4, 104–132. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, D.; Bruscalupi, G. Circadian Rhythms and Hormonal Homeostasis: Pathophysiological Implications. Biology 2017, 6, 10. [Google Scholar] [CrossRef]
- Gnocchi, D.; Custodero, C.; Sabba, C.; Mazzocca, A. Circadian rhythms: A possible new player in non-alcoholic fatty liver disease pathophysiology. J. Mol. Med. 2019, 97, 741–759. [Google Scholar] [CrossRef]
- Jin, X.; Pan, Y.; Wang, L.; Zhang, L.; Ravichandran, R.; Potts, P.R.; Jiang, J.; Wu, H.; Huang, H. MAGE-TRIM28 complex promotes the Warburg effect and hepatocellular carcinoma progression by targeting FBP1 for degradation. Oncogenesis 2017, 6, e312. [Google Scholar] [CrossRef]
- Yang, J.; Jin, X.; Yan, Y.; Shao, Y.; Pan, Y.; Roberts, L.R.; Zhang, J.; Huang, H.; Jiang, J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 2017, 7, 43864. [Google Scholar] [CrossRef]
- Schmidt, D.M.Z.; McCafferty, D.G. trans-2-phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. [Google Scholar] [CrossRef]
- Pan, D.N.; Mao, C.X.; Wang, Y.X. Suppression of Gluconeogenic Gene Expression by LSD1-Mediated Histone Demethylation. PLoS ONE 2013, 8, e66294. [Google Scholar] [CrossRef]
- Ma, R.H.; Zhang, W.G.; Tang, K.; Zhang, H.F.; Zhang, Y.; Li, D.P.; Li, Y.; Xu, P.W.; Luo, S.Q.; Cai, W.Q.; et al. Switch of glycolysis to gluconeogenesis by dexamethasone for treatment of hepatocarcinoma. Nat. Commun. 2013, 4, 2508. [Google Scholar] [CrossRef] [PubMed]
- Shukla, K.; Ferraris, D.V.; Thomas, A.G.; Stathis, M.; Duvall, B.; Delahanty, G.; Alt, J.; Rais, R.; Rojas, C.; Gao, P.; et al. Design, Synthesis, and Pharmacological Evaluation of Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl Sulfide 3 (BPTES) Analogs as Glutaminase Inhibitors. J. Med. Chem. 2012, 55, 10551–10563. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, D.; Xu, N.; Fang, J.Z.; Yu, Y.; Hou, W.; Ruan, H.Q.; Zhu, P.P.; Ma, R.C.; Lu, S.Y.; et al. Novel 1,3,4-Selenadiazole-Containing Kidney-Type Glutaminase Inhibitors Showed Improved Cellular Uptake and Antitumor Activity. J. Med. Chem. 2019, 62, 589–603. [Google Scholar] [CrossRef]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Wang, S.Y.; Zaal, E.A.; Wang, C.; Wu, H.Q.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.Z.; Lieftink, C.; et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. eLife 2020, 9, e56749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.C.; Wang, Q.C.; Lin, Z.B.; Yang, P.J.; Dou, K.F.; Zhang, R.H. Berberine Inhibits Growth of Liver Cancer Cells by Suppressing Glutamine Uptake. OncoTargets Ther. 2019, 12, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2022. [Google Scholar] [CrossRef]
- Goodman, M.; Liu, Z.; Zhu, P.; Li, J. AMPK Activators as a Drug for Diabetes, Cancer and Cardiovascular Disease. Pharm. Regul. Aff. 2014, 3, 118. [Google Scholar] [CrossRef]
- Tseng, H.I.; Zeng, Y.S.; Lin, Y.C.J.; Huang, J.W.; Lin, C.L.; Lee, M.H.; Yang, F.W.; Fang, T.P.; Mar, A.C.; Su, J.C. A novel AMPK activator shows therapeutic potential in hepatocellular carcinoma by suppressing HIF1 alpha-mediated aerobic glycolysis. Mol. Oncol. 2022, 16, 2274–2294. [Google Scholar] [CrossRef]
- Vara-Ciruelos, D.; Russell, F.M.; Hardie, D.G. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? (dagger). Open Biol. 2019, 9, 190099. [Google Scholar] [CrossRef]
- Lepropre, S.; Kautbally, S.; Octave, M.; Ginion, A.; Onselaer, M.B.; Steinberg, G.R.; Kemp, B.E.; Hego, A.; Wera, O.; Brouns, S.; et al. AMPK-ACC signaling modulates platelet phospholipids and potentiates thrombus formation. Blood 2018, 132, 1180–1192. [Google Scholar] [CrossRef]
- Ciavarella, A.; Gnocchi, D.; Custodero, C.; Lenato, G.M.; Fiore, G.; Sabba, C.; Mazzocca, A. Translational insight into prothrombotic state and hypercoagulation in nonalcoholic fatty liver disease. Thromb. Res. 2021, 198, 139–150. [Google Scholar] [CrossRef]
- Kwan, Y.P.; Saito, T.; Ibrahim, D.; Al-Hassan, F.M.; Ein Oon, C.; Chen, Y.; Jothy, S.L.; Kanwar, J.R.; Sasidharan, S. Evaluation of the cytotoxicity, cell-cycle arrest, and apoptotic induction by Euphorbia hirta in MCF-7 breast cancer cells. Pharm. Biol. 2016, 54, 1223–1236. [Google Scholar] [CrossRef]
- Karna, P.; Chagani, S.; Gundala, S.R.; Rida, P.C.; Asif, G.; Sharma, V.; Gupta, M.V.; Aneja, R. Benefits of whole ginger extract in prostate cancer. Br. J. Nutr. 2012, 107, 473–484. [Google Scholar] [CrossRef]
- Jo, K.J.; Cha, M.R.; Lee, M.R.; Yoon, M.Y.; Park, H.R. Methanolic extracts of Uncaria rhynchophylla induce cytotoxicity and apoptosis in HT-29 human colon carcinoma cells. Plant Foods Hum. Nutr. 2008, 63, 77–82. [Google Scholar] [CrossRef]
- Gill, M.S.A.; Saleem, H.; Ahemad, N. Plant Extracts and their Secondary Metabolites as Modulators of Kinases. Curr. Top. Med. Chem. 2020, 20, 1093–1104. [Google Scholar] [CrossRef]
- Encalada, M.A.; Hoyos, K.M.; Rehecho, S.; Berasategi, I.; de Ciriano, M.G.; Ansorena, D.; Astiasaran, I.; Navarro-Blasco, I.; Cavero, R.Y.; Calvo, M.I. Anti-proliferative effect of Melissa officinalis on human colon cancer cell line. Plant Foods Hum. Nutr. 2011, 66, 328–334. [Google Scholar] [CrossRef]
- Pirola, L.; Frojdo, S. Resveratrol: One molecule, many targets. IUBMB Life 2008, 60, 323–332. [Google Scholar] [CrossRef]
- Massimi, M.; Tomassini, A.; Sciubba, F.; Sobolev, A.P.; Devirgiliis, L.C.; Miccheli, A. Effects of resveratrol on HepG2 cells as revealed by H-1-NMR based metabolic profiling. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 1–8. [Google Scholar] [CrossRef]
- Teng, W.D.; Yin, W.J.; Zhao, L.; Ma, C.W.; Huang, J.Q.; Ren, F.Z. Resveratrol metabolites ameliorate insulin resistance in HepG2 hepatocytes by modulating IRS-1/AMPK. RSC Adv. 2018, 8, 36034–36042. [Google Scholar] [CrossRef]
- Gnocchi, D.; Del Coco, L.; Girelli, C.R.; Castellaneta, F.; Cesari, G.; Sabba, C.; Fanizzi, F.P.; Mazzocca, A. 1H-NMR metabolomics reveals a multitarget action of Crithmum maritimum ethyl acetate extract in inhibiting hepatocellular carcinoma cell growth. Sci. Rep. 2021, 11, 1259. [Google Scholar] [CrossRef]
- Gnocchi, D.; Sabba, C.; Mazzocca, A. The Edible Plant Crithmum maritimum Shows Nutraceutical Properties by Targeting Energy Metabolism in Hepatic Cancer. Plant Foods Hum. Nutr. 2022, 77, 481–483. [Google Scholar] [CrossRef]
- Gnocchi, D.; Sabba, C.; Mazzocca, A. Crithmum maritimum Improves Sorafenib Sensitivity by Decreasing Lactic Acid Fermentation and Inducing a Pro-Hepatocyte Marker Profile in Hepatocellular Carcinoma. Plant Foods Hum. Nutr. 2022. [Google Scholar] [CrossRef]
- Gnocchi, D.; Castellaneta, F.; Cesari, G.; Fiore, G.; Sabba, C.; Mazzocca, A. Treatment of liver cancer cells with ethyl acetate extract of Crithmum maritimum permits reducing sorafenib dose and toxicity maintaining its efficacy. J. Pharm. Pharm. 2021, 73, 1369–1376. [Google Scholar] [CrossRef]
- Lippolis, R.; Gnocchi, D.; Santacroce, L.; Siciliano, R.A.; Mazzeo, M.F.; Scacco, S.; Sabba, C.; Mazzocca, A. A distinctive protein signature induced by lysophosphatidic acid receptor 6 (LPAR6) expression in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2020, 526, 1150–1156. [Google Scholar] [CrossRef]
- Gnocchi, D.; Kurzyk, A.; Mintrone, A.; Lentini, G.; Sabba, C.; Mazzocca, A. Inhibition of LPAR6 overcomes sorafenib resistance by switching glycolysis into oxidative phosphorylation in hepatocellular carcinoma. Biochimie 2022, 202, 180–189. [Google Scholar] [CrossRef]
- Gnocchi, D.; Kapoor, S.; Nitti, P.; Cavalluzzi, M.M.; Lentini, G.; Denora, N.; Sabba, C.; Mazzocca, A. Novel lysophosphatidic acid receptor 6 antagonists inhibit hepatocellular carcinoma growth through affecting mitochondrial function. J. Mol. Med. 2020, 98, 179–191. [Google Scholar] [CrossRef]
- Gnocchi, D.; Cavalluzzi, M.M.; Mangiatordi, G.F.; Rizzi, R.; Tortorella, C.; Spennacchio, M.; Lentini, G.; Altomare, A.; Sabba, C.; Mazzocca, A. Xanthenylacetic Acid Derivatives Effectively Target Lysophosphatidic Acid Receptor 6 to Inhibit Hepatocellular Carcinoma Cell Growth. ChemMedChem 2021, 16, 2121–2129. [Google Scholar] [CrossRef]
- Mazzocca, A.; Dituri, F.; De Santis, F.; Filannino, A.; Lopane, C.; Betz, R.C.; Li, Y.Y.; Mukaida, N.; Winter, P.; Tortorella, C.; et al. Lysophosphatidic acid receptor LPAR6 supports the tumorigenicity of hepatocellular carcinoma. Cancer Res. 2015, 75, 532–543. [Google Scholar] [CrossRef]
- Mazzocca, A.; Dituri, F.; Lupo, L.; Quaranta, M.; Antonaci, S.; Giannelli, G. Tumor-secreted lysophostatidic acid accelerates hepatocellular carcinoma progression by promoting differentiation of peritumoral fibroblasts in myofibroblasts. Hepatology 2011, 54, 920–930. [Google Scholar] [CrossRef]
- Gnocchi, D.; Sabba, C.; Mazzocca, A. Lactic acid fermentation: A maladaptive mechanism and an evolutionary throwback boosting cancer drug resistance. Biochimie 2023. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, M.A.; de la Cruz-Ojeda, P.; Lopez-Grueso, M.J.; Navarro-Villaran, E.; Requejo-Aguilar, R.; Castejon-Vega, B.; Negrete, M.; Gallego, P.; Vega-Ochoa, A.; Victor, V.M.; et al. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox Biol. 2020, 36, 101510. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, M.A.; de la Cruz-Ojeda, P.; Gallego, P.; Navarro-Villaran, E.; Stankova, P.; Del Campo, J.A.; Kucera, O.; Elkalaf, M.; Maseko, T.E.; Cervinkova, Z.; et al. Dose-dependent regulation of mitochondrial function and cell death pathway by sorafenib in liver cancer cells. Biochem. Pharm. 2020, 176, 113902. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, M.A.; Chapresto-Garzon, R.; Cadenas, M.; Navarro-Villaran, E.; Negrete, M.; Gomez-Bravo, M.A.; Victor, V.M.; Padillo, F.J.; Muntane, J. Differential effectiveness of tyrosine kinase inhibitors in 2D/3D culture according to cell differentiation, p53 status and mitochondrial respiration in liver cancer cells. Cell Death Dis. 2020, 11, 339. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Pioronska, W.; Battello, N.; Zimmer, A.D.; Dewidar, B.; Han, M.; Pereira, S.; Blagojevic, B.; Castven, D.; Charlestin, V.; et al. Severe metabolic alterations in liver cancer lead to ERK pathway activation and drug resistance. EBioMedicine 2020, 54, 102699. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnocchi, D.; Sabbà, C.; Massimi, M.; Mazzocca, A. Metabolism as a New Avenue for Hepatocellular Carcinoma Therapy. Int. J. Mol. Sci. 2023, 24, 3710. https://doi.org/10.3390/ijms24043710
Gnocchi D, Sabbà C, Massimi M, Mazzocca A. Metabolism as a New Avenue for Hepatocellular Carcinoma Therapy. International Journal of Molecular Sciences. 2023; 24(4):3710. https://doi.org/10.3390/ijms24043710
Chicago/Turabian StyleGnocchi, Davide, Carlo Sabbà, Mara Massimi, and Antonio Mazzocca. 2023. "Metabolism as a New Avenue for Hepatocellular Carcinoma Therapy" International Journal of Molecular Sciences 24, no. 4: 3710. https://doi.org/10.3390/ijms24043710
APA StyleGnocchi, D., Sabbà, C., Massimi, M., & Mazzocca, A. (2023). Metabolism as a New Avenue for Hepatocellular Carcinoma Therapy. International Journal of Molecular Sciences, 24(4), 3710. https://doi.org/10.3390/ijms24043710