
Ketone Bodies and Cardiovascular Disease: An Alternate Fuel Source to the Rescue



Abstract
1. Introduction
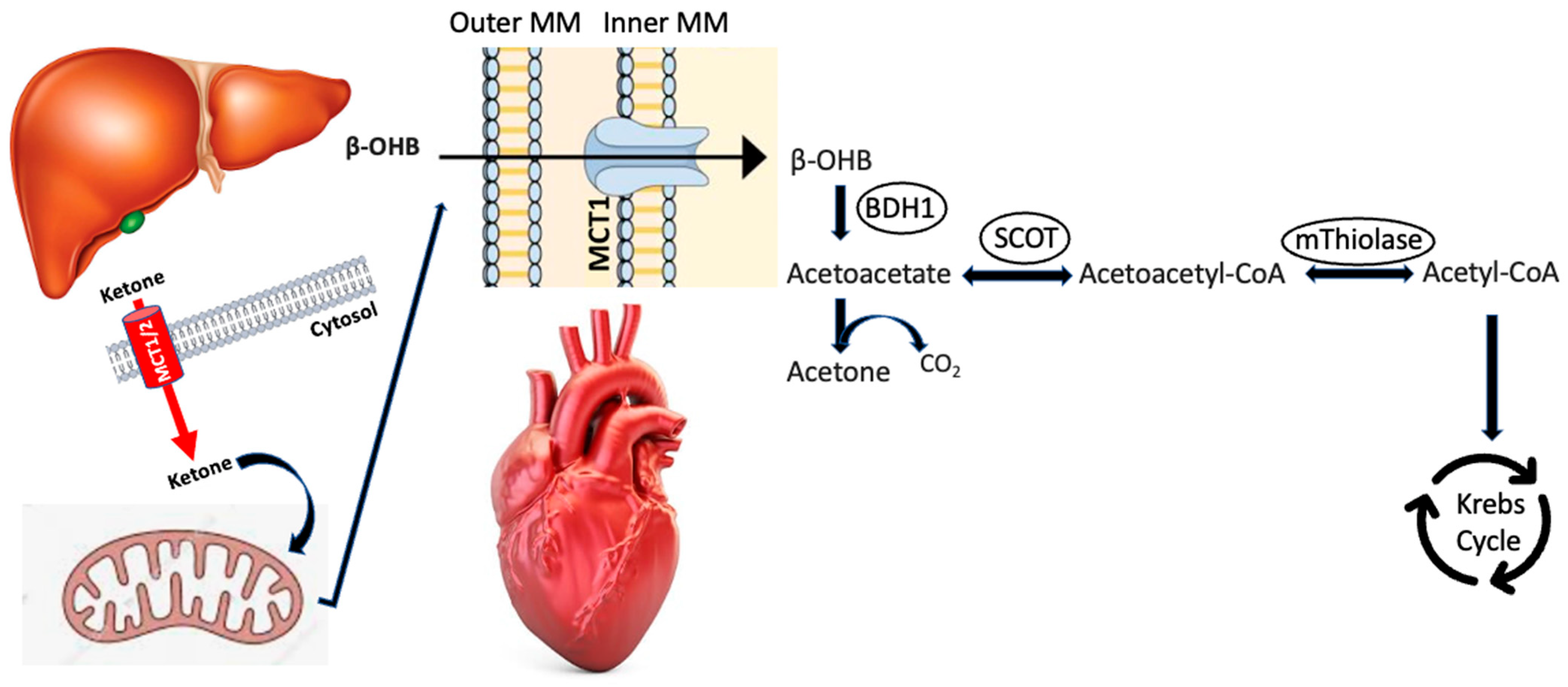
2. Ketogenesis/Ketolysis
3. Ketone Bodies in Cardiovascular Disease
3.1. Ketogenic Diet
3.2. Intermittent Fasting
3.3. Ketone Bodies in Heart Failure
3.4. Ketone Bodies as Biomarkers
3.5. Atherosclerotic Heart Disease/Coronary Artery Calcification
3.6. Myocardial Infarction
3.7. Cardiac Endothelial Cells
3.8. Cardiac Hypertrophy
4. Hypertension
Vascular Calcification
5. Atrial Fibrillation
6. Diabetic Heart
Insulin Resistance
7. Sodium-Glucose Cotransporter 2 (SGLT-2) Inhibitors
8. Ketone Bodies and the Heart/A Double-Edged Sword
Lean Mass Hyper-Responders
9. Mitochondria and Oxidative Stress
10. Cardiomyocyte Electrophysiology
11. Ketone Therapy
Role of Body Weight
12. Exercise
13. Controversial Issues
14. Conclusions
15. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Schulze, P.C.; Wu, J.M.F. Ketone bodies for the starving heart. Nat. Metab. 2020, 2, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Gibb, A.A.; Hill, B.G. Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ. Res. 2018, 123, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, T.; Wu, J.M.F.; Schulze, P.C. Mitochondrial Homeostasis Mediates Lipotoxicity in the Failing Myocardium. Int. J. Mol. Sci. 2021, 22, 1498. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, R.; Cipriani, F.; Masi, D.; Basciani, S.; Watanabe, M.; Lubrano, C.; Gnessi, L.; Mariani, S. Ketone Bodies and SIRT1, Synergic Epigenetic Regulators for Metabolic Health: A Narrative Review. Nutrients 2022, 14, 3145. [Google Scholar] [CrossRef]
- Bendridi, N.; Selmi, A.; Balcerczyk, A.; Pirola, L. Ketone Bodies as Metabolites and Signalling Molecules at the Crossroad between Inflammation and Epigenetic Control of Cardiometabolic Disorders. Int. J. Mol. Sci. 2022, 23, 14564. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Papazafiropoulou, A.K.; Georgopoulos, M.M.; Katsilambros, N.L. Ketone bodies and the heart. Arch. Med. Sci. Atheroscler. Dis. 2021, 6, e209–e214. [Google Scholar] [CrossRef]
- Lommi, J.; Kupari, M.; Koskinen, P.; Näveri, H.; Leinonen, H.; Pulkki, K.; Härkönen, M. Blood ketone bodies in congestive heart failure. J. Am. Coll. Cardiol. 1996, 28, 665–672. [Google Scholar] [CrossRef]
- Maejima, Y. SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 2019, 6, 186. [Google Scholar] [CrossRef]
- Dhillon, K.K.; Gupta, S. Biochemistry, Ketogenesis. [Updated 2022 Feb 10]. StatPearls, 2022. [Google Scholar]
- Saucedo-Orozco, H.; Voorrips, S.N.; Yurista, S.R.; de Boer, R.A.; Westenbrink, B.D. SGLT2 Inhibitors and Ketone Metabolism in Heart Failure. J. Lipid Atheroscler. 2022, 11, 1–19. [Google Scholar] [CrossRef]
- Cotter, D.G.; d’Avignon, D.A.; Wentz, A.E.; Weber, M.L.; Crawford, P.A. Obligate role for ketone body oxidation in neonatal metabolic homeostasis. J. Biol. Chem. 2011, 286, 6902–6910. [Google Scholar] [CrossRef]
- Halestrap, A.P. The monocarboxylate transporter family—Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family—Role and regulation. IUBMB Life 2012, 64, 109–119. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Biswas, D.; Pulinilkunnil, T.; Lopaschuk, G.D. Myocardial Ketones Metabolism in Heart Failure. J. Card. Fail. 2020, 26, 998–1005. [Google Scholar] [CrossRef]
- Chu, Y.; Zhang, C.; Xie, M. Beta-Hydroxybutyrate, Friend or Foe for Stressed Hearts. Front. Aging 2021, 2, 681513. [Google Scholar] [CrossRef]
- Chen, W.; Sharma, G.; Jiang, W.; Maptue, N.R.; Malloy, C.R.; Sherry, A.D.; Khemtong, C. Metabolism of hyperpolarized (13) C-acetoacetate to β-hydroxybutyrate detects real-time mitochondrial redox state and dysfunction in heart tissue. NMR Biomed. 2019, 32, e4091. [Google Scholar] [CrossRef]
- Abdurrachim, D.; Woo, C.C.; Teo, X.Q.; Chan, W.X.; Radda, G.K.; Lee, P.T.H. A new hyperpolarized (13)C ketone body probe reveals an increase in acetoacetate utilization in the diabetic rat heart. Sci. Rep. 2019, 9, 5532. [Google Scholar] [CrossRef]
- Squires, J.E.; Sun, J.; Caffrey, J.L.; Yoshishige, D.; Mallet, R.T. Acetoacetate augments beta-adrenergic inotropism of stunned myocardium by an antioxidant mechanism. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1340–H1347. [Google Scholar] [CrossRef]
- Yokokawa, T.; Yoshihisa, A.; Kanno, Y.; Abe, S.; Misaka, T.; Yamada, S.; Kaneshiro, T.; Sato, T.; Oikawa, M.; Kobayashi, A.; et al. Circulating acetoacetate is associated with poor prognosis in heart failure patients. Int. J. Cardiol. Heart Vasc. 2019, 25, 100432. [Google Scholar] [CrossRef]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes/Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Bassenge, E.; Wendt, V.E.; Schollmeyer, P.; Bluemchen, G.; Gudbjarnason, S.; Bing, R.J. Effect of Ketone Bodies on Cardiac Metabolism. Am. J. Physiol. 1965, 208, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Neumann, D.; Westenbrink, B.D.; Schianchi, F.; Wong, L.-Y.; Sun, A.; Strzelecka, A.; Glatz, J.F.C.; Luiken, J.J.F.P.; Nabben, M. Ketone Body Exposure of Cardiomyocytes Impairs Insulin Sensitivity and Contractile Function through Vacuolar-Type H(+)-ATPase Disassembly-Rescue by Specific Amino Acid Supplementation. Int. J. Mol. Sci. 2022, 23, 12909. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, F.; Peukert, K.; Surace, L.; Michla, M.; Nikolka, F.; Fox, M.; Weiss, P.; Feuerborn, C.; Maier, P.; Schulz, S.; et al. Impaired ketogenesis ties metabolism to T cell dysfunction in COVID-19. Nature 2022, 609, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef]
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef]
- Johnson, R.H.; Walton, J.; Krebs, H.A.; Williamson, D.H. Post-exercise ketosis. Lancet 1970, 1, 195. [Google Scholar] [CrossRef]
- Morris, A.A. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121. [Google Scholar] [CrossRef]
- Yurista, S.R.; Chen, S.; Welsh, A.; Tang, W.H.W.; Nguyen, C.T. Targeting Myocardial Substrate Metabolism in the Failing Heart: Ready for Prime Time? Curr. Heart Fail. Rep. 2022, 19, 180–190. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone bodies: From enemy to friend and guardian angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Weis, E.; Puchalska, P.; Nelson, A.B.; Taylor, J.; Moll, I.; Hasan, S.S.; Dewenter, M.; Hagenmüller, M.; Fleming, T.; Poschet, G.; et al. Ketone body oxidation increases cardiac endothelial cell proliferation. EMBO Mol. Med. 2022, 14, e14753. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, X.; Li, T.; Zhao, J.; Yang, Y.; Yao, Y.; Wang, L.; Yang, B.; Ren, G.; Tan, Y.; et al. Alternate-Day Ketogenic Diet Feeding Protects against Heart Failure through Preservation of Ketogenesis in the Liver. Oxidative Med. Cell. Longev. 2022, 2022, 4253651. [Google Scholar] [CrossRef]
- Luong, T.V.; Abild, C.B.; Bangshaab, M.; Gormsen, L.C.; Søndergaard, E. Ketogenic Diet and Cardiac Substrate Metabolism. Nutrients 2022, 14, 1322. [Google Scholar] [CrossRef]
- Garza-González, S.; Nieblas, B.; Solbes-Gochicoa, M.M.; Altamirano, J.; García, N. Intermittent Fasting as Possible Treatment for Heart Failure. Curr. Vasc. Pharmacol. 2022, 20, 260–271. [Google Scholar]
- Kawakami, R.; Sunaga, H.; Iso, T.; Kaneko, R.; Koitabashi, N.; Obokata, M.; Harada, T.; Matsui, H.; Yokoyama, T.; Kurabayashi, M. Ketone body and FGF21 coordinately regulate fasting-induced oxidative stress response in the heart. Sci. Rep. 2022, 12, 7338. [Google Scholar] [CrossRef]
- Okoshi, K.; Cezar, M.D.M.; Polin, M.A.M.; Paladino, J.R., Jr.; Martinez, P.F.; Oliveira, S.A., Jr.; Lima, A.R.R.; Damatto, R.L.; Paiva, S.A.R.; Zornoff, L.A.M.; et al. Influence of intermittent fasting on myocardial infarction-induced cardiac remodeling. BMC Cardiovasc. Disord. 2019, 19, 126. [Google Scholar] [CrossRef]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef]
- Manla, Y.; Kuan, W.; Clark, A.L.; Cleland, J.G.F.; Pellicori, P. Ketone Bodies in Acute Heart Failure: Fuel for Thought. J. Card. Fail. 2023, 29, 42–44. [Google Scholar] [CrossRef]
- Yurista, S.R.; Nguyen, C.T.; Rosenzweig, A.; de Boer, R.A.; Westenbrink, B.D. Ketone bodies for the failing heart: Fuels that can fix the engine? Trends Endocrinol. Metab. 2021, 32, 814–826. [Google Scholar] [CrossRef]
- Flores-Guerrero, J.L.; Westenbrink, B.D.; Connelly, M.A.; Otvos, J.D.; Groothof, D.; Shalaurova, I.; Garcia, E.; Navis, G.; de Boer, R.A.; Bakker, S.J.L.; et al. Association of beta-hydroxybutyrate with development of heart failure: Sex differences in a Dutch population cohort. Eur. J. Clin. Investig. 2021, 51, e13468. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, Y.; Nagoshi, T.; Inoue, Y.; Tanaka, Y.; Takahashi, H.; Oi, Y.; Kimura, H.; Minai, K.; Yoshimura, M. Close linkage between blood total ketone body levels and B-type natriuretic peptide levels in patients with cardiovascular disorders. Sci Rep. 2021, 11, 6498. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Voorrips, S.; Boorsma, E.; Beusekamp, J.; De-Boer, R.; Connelly, M.; Dullaart, R.; Van-Der-Meer, P.; Van-Veldhuisen, D.; Voors, A.; Damman, K.; et al. Longitudinal Changes in Circulating Ketone Body Levels in Patients With Acute Heart Failure: A Post Hoc Analysis of the EMPA-Response-AHF Trial. J. Card. Fail. 2023, 29, 33–41. [Google Scholar] [CrossRef]
- Marcondes-Braga, F.G.; Gioli-Pereira, L.; Bernardez-Pereira, S.; Batista, G.L.; Mangini, S.; Issa, V.S.; Fernandes, F.; Bocchi, E.A.; Ayub-Ferreira, S.M.; Mansur, A.J.; et al. Exhaled breath acetone for predicting cardiac and overall mortality in chronic heart failure patients. ESC Heart Fail. 2020, 7, 1744–1752. [Google Scholar] [CrossRef]
- Kupari, M.; Lommi, J.; Ventilä, M.; Karjalainen, U. Breath acetone in congestive heart failure. Am. J. Cardiol. 1995, 76, 1076–1078. [Google Scholar] [CrossRef]
- Gouzi, F.; Ayache, D.; Hédon, C.; Molinari, N.; Vicet, A. Breath acetone concentration: Too heterogeneous to constitute a diagnosis or prognosis biomarker in heart failure? A systematic review and meta-analysis. J. Breath Res. 2021, 16, 016001. [Google Scholar] [CrossRef]
- Lohano, P.D.; Ibrahim, M.; Raza, S.J.; Gowa, M.; Baloch, S.H. Comparing Finger-stick Βeta-hydroxybutyrate with Dipstick Urine Tests in the Detection of Ketone Bodies in the Diagnosis of Children with Diabetic Ketoacidosis. J. Coll. Physicians Surg. Pak. 2022, 32, 483–486. [Google Scholar]
- Brooke, J.; Stiell, M.; Ojo, O. Evaluation of the Accuracy of Capillary Hydroxybutyrate Measurement Compared with Other Measurements in the Diagnosis of Diabetic Ketoacidosis: A Systematic Review. Int. J. Environ. Res. Public Health 2016, 13, 837. [Google Scholar] [CrossRef]
- Yokokawa, T.; Sato, T.; Suzuki, S.; Oikawa, M.; Yoshihisa, A.; Kobayashi, A.; Yamaki, T.; Kunii, H.; Nakazato, K.; Suzuki, H.; et al. Change of Exhaled Acetone Concentration Levels in Patients with Acute Decompensated Heart Failure. Int. Heart J. 2018, 59, 808–812. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Z.; Zhang, Y.; Chen, J.; Li, Y.; Wu, F.; Wang, W.; Cui, Z.J.; Chen, G. Ketone Body 3-Hydroxybutyrate Ameliorates Atherosclerosis via Receptor Gpr109a-Mediated Calcium Influx. Adv. Sci. Weinh. 2021, 8, 2003410. [Google Scholar] [CrossRef]
- Cho, I.Y.; Chang, Y.; Sung, E.; Kim, Y.; Kang, J.-H.; Shin, H.; Wild, S.H.; Byrne, C.D.; Ryu, S. Fasting ketonuria is inversely associated with coronary artery calcification in non-diabetic individuals. Atherosclerosis 2022, 348, 1–7. [Google Scholar] [CrossRef]
- de Koning, M.L.Y.; Westenbrink, B.D.; Assa, S.; Garcia, E.; Connelly, M.; van Veldhuisen, D.; Dullaart, R.; Lipsic, E.; van der Harst, P. Association of Circulating Ketone Bodies with Functional Outcomes after ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2021, 78, 1421–1432. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef]
- Costa, T.J.; Linder, B.A.; Hester, S.; Fontes, M.; Pernomian, L.; Wenceslau, C.F.; Robinson, A.T.; McCarthy, C.G. The janus face of ketone bodies in hypertension. J. Hypertens. 2022, 40, 2111–2119. [Google Scholar] [CrossRef]
- Pietschner, R.; Kolwelter, J.; Bosch, A.; Striepe, K.; Jung, S.; Kannenkeril, D.; Ott, C.; Schiffer, M.; Achenbach, S.; Schmieder, R.E. Effect of empagliflozin on ketone bodies in patients with stable chronic heart failure. Cardiovasc. Diabetol. 2021, 20, 219. [Google Scholar] [CrossRef]
- Lan, Z.; Chen, A.; Li, L.; Ye, Y.; Liang, Q.; Dong, Q.; Wang, S.; Fu, M.; Li, Y.; Liu, X.; et al. Downregulation of HDAC9 by the ketone metabolite β-hydroxybutyrate suppresses vascular calcification. J. Pathol. 2022, 258, 213–226. [Google Scholar] [CrossRef]
- Brown, S.M.; Larsen, N.K.; Thankam, F.G.; Agrawal, D.K. Fetal cardiomyocyte phenotype, ketone body metabolism, and mitochondrial dysfunction in the pathology of atrial fibrillation. Mol. Cell. Biochem. 2021, 476, 1165–1178. [Google Scholar] [CrossRef]
- Mayr, M.; Yusuf, S.; Weir, G.; Chung, Y.-L.; Mayr, U.; Yin, X.; Ladroue, C.; Madhu, B.; Roberts, N.; De Souza, A.; et al. Combined metabolomic and proteomic analysis of human atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 585–594. [Google Scholar] [CrossRef]
- Xu, S.; Tao, H.; Cao, W.; Cao, L.; Lin, Y.; Zhao, S.-M.; Xu, W.; Cao, J.; Zhao, J.-Y. Ketogenic diets inhibit mitochondrial biogenesis and induce cardiac fibrosis. Signal Transduct. Target. Ther. 2021, 6, 54. [Google Scholar] [CrossRef]
- Ma, C.; Zheng, X.; Yang, Y.; Bu, P. The effect of black tea supplementation on blood pressure: A systematic review and dose-response meta-analysis of randomized controlled trials. Food Funct. 2021, 12, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Ihara, K.; Sasano, T. Role of Inflammation in the Pathogenesis of Atrial Fibrillation. Front Physiol. 2022, 13, 862164. [Google Scholar] [CrossRef] [PubMed]
- Brahma, M.K.; Ha, C.; Pepin, M.E.; Mia, S.; Sun, Z.; Chatham, J.C.; Habegger, K.M.; Abel, E.D.; Paterson, A.J.; Young, M.E.; et al. Increased Glucose Availability Attenuates Myocardial Ketone Body Utilization. J. Am. Heart Assoc. 2020, 9, e013039. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the Empa-Reg Outcome Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care. 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Harada, E.; Nakagawa, H.; Morikawa, Y.; Shono, M.; Kugimiya, F.; Yoshimura, M.; Yasue, H. The diabetic heart utilizes ketone bodies as an energy source. Metabolism 2017, 77, 65–72. [Google Scholar] [CrossRef]
- Mishra, P.K. Why the diabetic heart is energy inefficient: A ketogenesis and ketolysis perspective. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H751–H755. [Google Scholar] [CrossRef]
- Schugar, R.C.; Moll, A.R.; André d’Avignon, D.; Weinheimer, C.J.; Kovacs, A.; Crawford, P.A. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol. Metab. 2014, 3, 754–769. [Google Scholar] [CrossRef]
- Wang, X.; Ni, J.; Guo, R.; Li, L.; Su, J.; He, F.; Fan, G. SGLT2 inhibitors break the vicious circle between heart failure and insulin resistance: Targeting energy metabolism. Heart Fail. Rev. 2022, 27, 961–980. [Google Scholar] [CrossRef]
- Verma, S. Potential Mechanisms of Sodium-Glucose Co-Transporter 2 Inhibitor-Related Cardiovascular Benefits. Am. J. Cardiol. 2019, 124 (Suppl. S1), S36–S44. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Lopaschuk, G.D. Branched-Chain Amino Acid Metabolism in the Failing Heart. Cardiovasc. Drugs Ther. 2022, 2022, 12. [Google Scholar] [CrossRef]
- Bloomgarden, Z. Diabetes and branched-chain amino acids: What is the link? J. Diabetes 2018, 10, 350–352. [Google Scholar] [CrossRef]
- White, P.J.; Newgard, C.B. Branched-chain amino acids in disease. Science 2019, 363, 582–583. [Google Scholar] [CrossRef]
- Yoon, M.S. The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism. Nutrients 2016, 8, 405. [Google Scholar] [CrossRef]
- Prattichizzo, F.; De Nigris, V.; Micheloni, S.; La Sala, L.; Ceriello, A. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is low-grade inflammation the neglected component? Diabetes Obes. Metab. 2018, 20, 2515–2522. [Google Scholar] [CrossRef]
- Wallenius, K.; Kroon, T.; Hagstedt, T.; Löfgren, L.; Sörhede-Winzell, M.; Boucher, J.; Lindén, D.; Oakes, N.D. The SGLT2 inhibitor dapagliflozin promotes systemic FFA mobilization, enhances hepatic β-oxidation, and induces ketosis. J. Lipid Res. 2022, 63, 100176. [Google Scholar] [CrossRef]
- Ferrannini, E.; Baldi, S.; Scozzaro, T.; Tsimihodimos, V.; Tesfaye, F.; Shaw, W.; Rosenthal, N.; Figtree, G.A.; Neal, B.; Mahaffey, K.W.; et al. Fasting Substrate Concentrations Predict Cardiovascular Outcomes in the CANagliflozin cardioVascular Assessment Study (CANVAS). Diabetes Care 2022, 45, 1893–1899. [Google Scholar] [CrossRef]
- Selvaraj, S.; Fu, Z.; Jones, P.; Kwee, L.C.; Windsor, S.L.; Ilkayeva, O.; Newgard, C.B.; Margulies, K.B.; Husain, M.; Inzucchi, S.E.; et al. Metabolomic Profiling of the Effects of Dapagliflozin in Heart Failure With Reduced Ejection Fraction: DEFINE-HF. Circulation 2022, 146, 808–818. [Google Scholar] [CrossRef]
- Monami, M.; Nreu, B.; Zannoni, S.; Lualdi, C.; Mannucci, E. Effects of SGLT-2 inhibitors on diabetic ketoacidosis: A meta-analysis of randomised controlled trials. Diabetes Res. Clin. Pract. 2017, 130, 53–60. [Google Scholar] [CrossRef]
- Liu, J.; Li, L.; Li, S.; Wang, Y.; Qin, X.; Deng, K.; Liu, Y.; Zou, K.; Sun, X. Sodium-glucose co-transporter-2 inhibitors and the risk of diabetic ketoacidosis in patients with type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2020, 22, 1619–1627. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Biomath, D.; Devins, T.; Johansen, O.E.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Kruljac, I.; Ćaćić, M.; Ćaćić, P.; Ostojić, V.; Štefanović, M.; Šikić, A.; Vrkljan, M. Diabetic ketosis during hyperglycemic crisis is associated with decreased all-cause mortality in patients with type 2 diabetes mellitus. Endocrine 2017, 55, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bharmal, S.H.; Kimita, W.; Petrov, M.S. Effect of acute ketosis on lipid profile in prediabetes: Findings from a cross-over randomized controlled trial. Cardiovasc. Diabetol. 2022, 21, 138. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, A.; Coderre, L. Ketone bodies alter dinitrophenol-induced glucose uptake through AMPK inhibition and oxidative stress generation in adult cardiomyocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1325–E1332. [Google Scholar] [CrossRef]
- Norwitz, N.G.; Mindrum, M.R.; Giral, P.; Kontush, A.; Soto-Mota, A.; Wood, T.R.; D’Agostino, D.P.; Manubolu, V.S.; Budoff, M.; Krauss, R.M. Elevated LDL-cholesterol levels among lean mass hyper-responders on low-carbohydrate ketogenic diets deserve urgent clinical attention and further research. J. Clin. Lipidol. 2022, 16, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Norwitz, N.G.; Feldman, D.; Soto-Mota, A.; Kalayjian, T.; Ludwig DSElevated, L.D.L. Cholesterol with a Carbohydrate-Restricted Diet: Evidence for a “Lean Mass Hyper-Responder” Phenotype. Curr. Dev. Nutr. 2022, 6, nzab144. [Google Scholar] [CrossRef]
- Mindrum, M.R. Let’s Be Clear about Expected Cardiovascular Risk: A Commentary on the Massive Rise in LDL Cholesterol Induced by Carbohydrate Restriction in the Proposed “Lean Mass Hyper-Responder” Phenotype. Curr. Dev. Nutr. 2022, 6, nzac042. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Klos, M.L.; Hou, W.; Nsengimana, B.; Weng, S.; Yan, C.; Xu, S.; Devaney, E.; Han, S. Differential Effects of Beta-Hydroxybutyrate Enantiomers on Induced Pluripotent Stem Derived Cardiac Myocyte Electrophysiology. Biomolecules 2022, 12, 1500. [Google Scholar] [CrossRef]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 2019, 4, e124079. [Google Scholar] [CrossRef]
- Falkenhain, K.; Daraei, A.; Forbes, S.C.; Little, J.P. Effects of Exogenous Ketone Supplementation on Blood Glucose: A Systematic Review and Meta-analysis. Adv. Nutr. 2022, 13, 1697–1714. [Google Scholar] [CrossRef]
- Trang, N.N.; Lee, T.W.; Kao, Y.H.; Chao, T.F.; Lee, T.I.; Chen, Y.J. Ketogenic diet modulates cardiac metabolic dysregulation in streptozocin-induced diabetic rats. J. Nutr. Biochem. 2023, 111, 109161. [Google Scholar] [CrossRef]
- Takahara, S.; Soni, S.; Phaterpekar, K.; Kim, T.T.; Maayah, Z.H.; Levasseur, J.L.; Silver, H.L.; Freed, D.H.; Ferdaoussi, M.; Dyck, J.R. Chronic exogenous ketone supplementation blunts the decline of cardiac function in the failing heart. ESC Heart Fail. 2021, 8, 5606–5612. [Google Scholar] [CrossRef]
- Takahara, S.; Soni, S.; Maayah, Z.H.; Ferdaoussi, M.; Dyck, J.R.B. Ketone therapy for heart failure: Current evidence for clinical use. Cardiovasc. Res. 2022, 118, 977–987. [Google Scholar] [CrossRef]
- Soni, S.; Martens, M.D.; Takahara, S.; Silver, H.L.; Maayah, Z.H.; Ussher, J.R.; Ferdaoussi, M.; Dyck, J.R. Exogenous ketone ester administration attenuates systemic inflammation and reduces organ damage in a lipopolysaccharide model of sepsis. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166507. [Google Scholar] [CrossRef]
- Nielsen, R.; Møller, N.; Gormsen, L.C.; Tolbod, L.P.; Hansson, N.H.; Sorensen, J.; Harms, H.J.; Frøkiær, J.; Eiskjaer, H.; Jespersen, N.R.; et al. Cardiovascular Effects of Treatment With the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019, 139, 2129–2141. [Google Scholar] [CrossRef]
- Byrne, N.J.; Soni, S.; Takahara, S.; Ferdaoussi, M.; Al Batran, R.; Darwesh, A.M.; Levasseur, J.L.; Beker, D.; Vos, D.Y.; Schmidt, M.A.; et al. Chronically Elevating Circulating Ketones Can Reduce Cardiac Inflammation and Blunt the Development of Heart Failure. Circ. Heart Fail. 2020, 13, e006573. [Google Scholar] [CrossRef]
- Paoli, A. Ketogenic diet for obesity: Friend or foe? Int. J. Environ. Res. Public Health 2014, 11, 2092–2107. [Google Scholar] [CrossRef]
- Luo, W.; Zhang, J.; Xu, D.; Zhou, Y.; Qu, Z.; Yang, Q.; Lv, Q. Low carbohydrate ketogenic diets reduce cardiovascular risk factor levels in obese or overweight patients with T2DM: A meta-analysis of randomized controlled trials. Front. Nutr. 2022, 9, 1092031. [Google Scholar] [CrossRef]
- Tragni, E.; Vigna, L.; Ruscica, M.; Macchi, C.; Casula, M.; Santelia, A.; Catapano, A.; Magni, P. Reduction of Cardio-Metabolic Risk and Body Weight through a Multiphasic Very-Low Calorie Ketogenic Diet Program in Women with Overweight/Obesity: A Study in a Real-World Setting. Nutrients 2021, 13, 1804. [Google Scholar] [CrossRef]
- Kang, J.; Ratamess, N.A.; Faigenbaum, A.D.; Bush, J.A. Ergogenic Properties of Ketogenic Diets in Normal-Weight Individuals: A Systematic Review. J. Am. Coll. Nutr. 2020, 39, 665–675. [Google Scholar] [CrossRef]
- Selvaraj, S.; Kelly, D.P.; Margulies, K.B. Implications of Altered Ketone Metabolism and Therapeutic Ketosis in Heart Failure. Circulation 2020, 141, 1800–1812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Guo, X.; Chen, L.; Chen, T.; Yu, J.; Wu, C.; Zheng, J. Ketogenic Diets and Cardio-Metabolic Diseases. Front. Endocrinol. Lausanne 2021, 12, 753039. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Ciesielski, O.; Balcerczyk, A. Fat not so bad? The role of ketone bodies and ketogenic diet in the treatment of endothelial dysfunction and hypertension. Biochem. Pharmacol. 2022, 206, 115346. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, M.; Li, X.; Wang, Y.; Du, R. Exercise Ameliorates Atherosclerosis via Up-Regulating Serum β-Hydroxybutyrate Levels. Int. J. Mol. Sci. 2022, 23, 3788. [Google Scholar] [CrossRef]
- Coppola, G.; Natale, F.; Torino, A.; Capasso, R.; D’Aniello, A.; Pironti, E.; Santoro, E.; Calabrò, R.; Verrotti, A. The impact of the ketogenic diet on arterial morphology and endothelial function in children and young adults with epilepsy: A case-control study. Seizure 2014, 23, 260–265. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, X.; Jia, P.; You, Y.; Cheng, Y.; Deng, H.; Luo, S.; Huang, B. Ketogenic diet aggravates hypertension via NF-κB-mediated endothelial dysfunction in spontaneously hypertensive rats. Life Sci. 2020, 258, 118124. [Google Scholar] [CrossRef]
- Jain, S.K.; McVie, R.; Bocchini, J.A., Jr. Hyperketonemia (ketosis), oxidative stress and type 1 diabetes. Pathophysiology 2006, 13, 163–170. [Google Scholar] [CrossRef]
- Nasser, S.; Vialichka, V.; Biesiekierska, M.; Balcerczyk, A.; Pirola, L. Effects of ketogenic diet and ketone bodies on the cardiovascular system: Concentration matters. World J. Diabetes 2020, 11, 584–595. [Google Scholar] [CrossRef]


{kind=link}
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manolis, A.S.; Manolis, T.A.; Manolis, A.A. Ketone Bodies and Cardiovascular Disease: An Alternate Fuel Source to the Rescue. Int. J. Mol. Sci. 2023, 24, 3534. https://doi.org/10.3390/ijms24043534
Manolis AS, Manolis TA, Manolis AA. Ketone Bodies and Cardiovascular Disease: An Alternate Fuel Source to the Rescue. International Journal of Molecular Sciences. 2023; 24(4):3534. https://doi.org/10.3390/ijms24043534
Chicago/Turabian StyleManolis, Antonis S., Theodora A. Manolis, and Antonis A. Manolis. 2023. "Ketone Bodies and Cardiovascular Disease: An Alternate Fuel Source to the Rescue" International Journal of Molecular Sciences 24, no. 4: 3534. https://doi.org/10.3390/ijms24043534
APA StyleManolis, A. S., Manolis, T. A., & Manolis, A. A. (2023). Ketone Bodies and Cardiovascular Disease: An Alternate Fuel Source to the Rescue. International Journal of Molecular Sciences, 24(4), 3534. https://doi.org/10.3390/ijms24043534