Implementation of Novel Affinity Ligand for Lentiviral Vector Purification

 ,
, 
 and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Ligand Discover and Initial Scouting
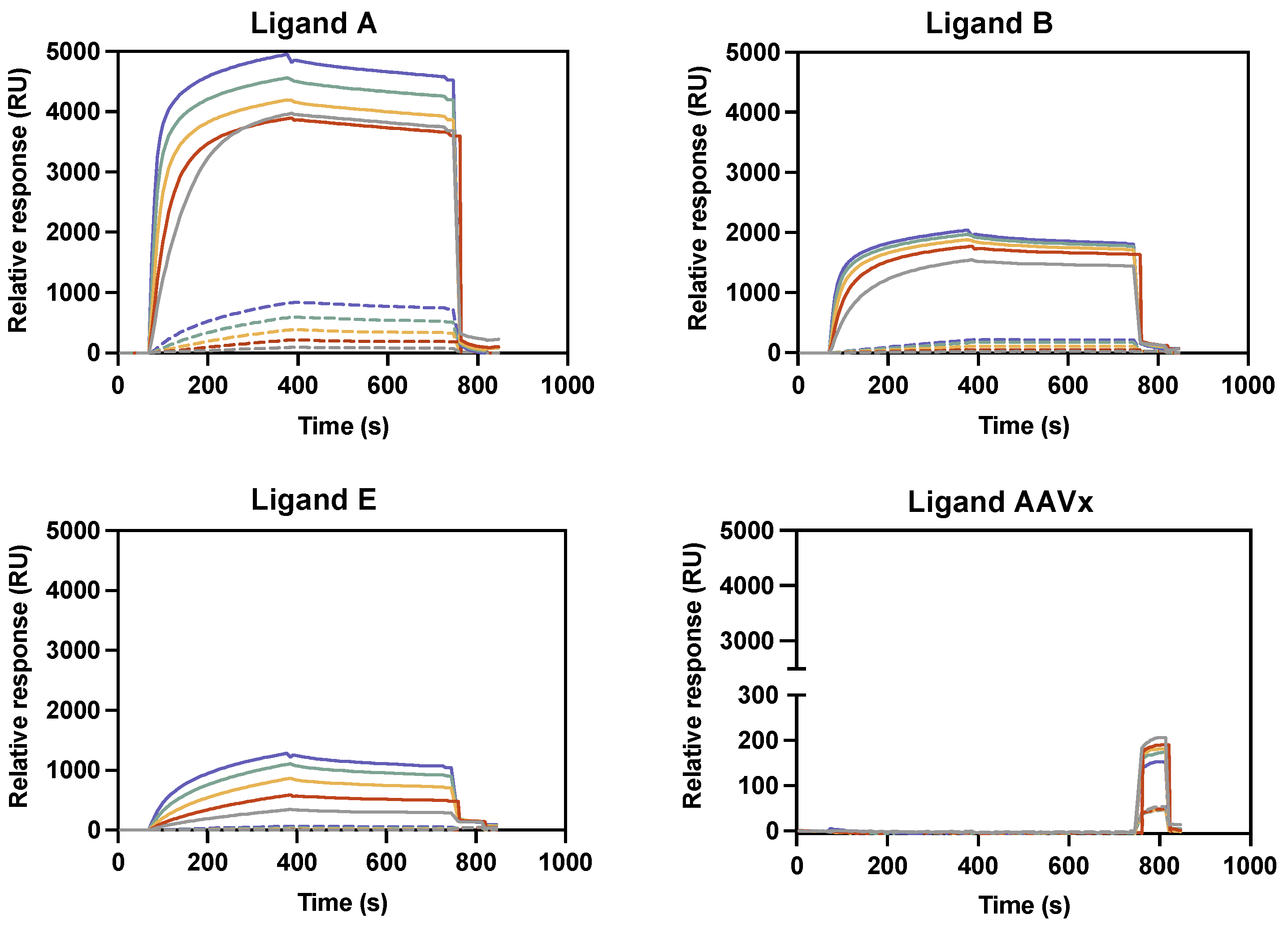
2.1.1. Binding Reactivity Using Biotin–Ligand Conjugates
2.1.2. VSVG-LVs Release Efficiency Using Biotin–Ligand Conjugates
2.2. Generation of VSVG-LVs Affinity Chromatography Prototype
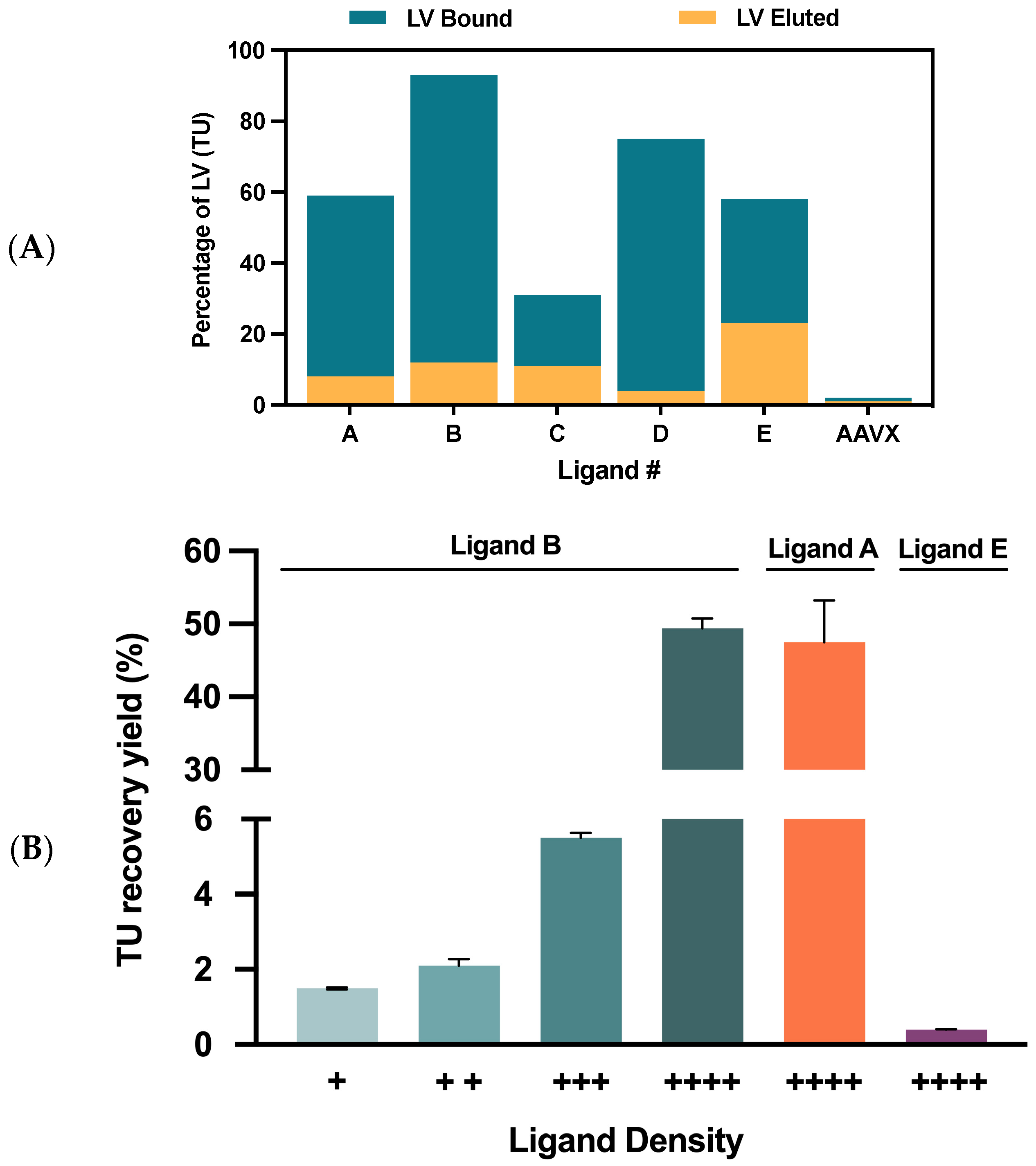
2.2.1. VSVG-LVs Depletion Using Streptavidin Agarose Beads
2.2.2. Matrix and Final Ligand Evaluation
2.3. Chromatography Optimization for the Lead Candidate
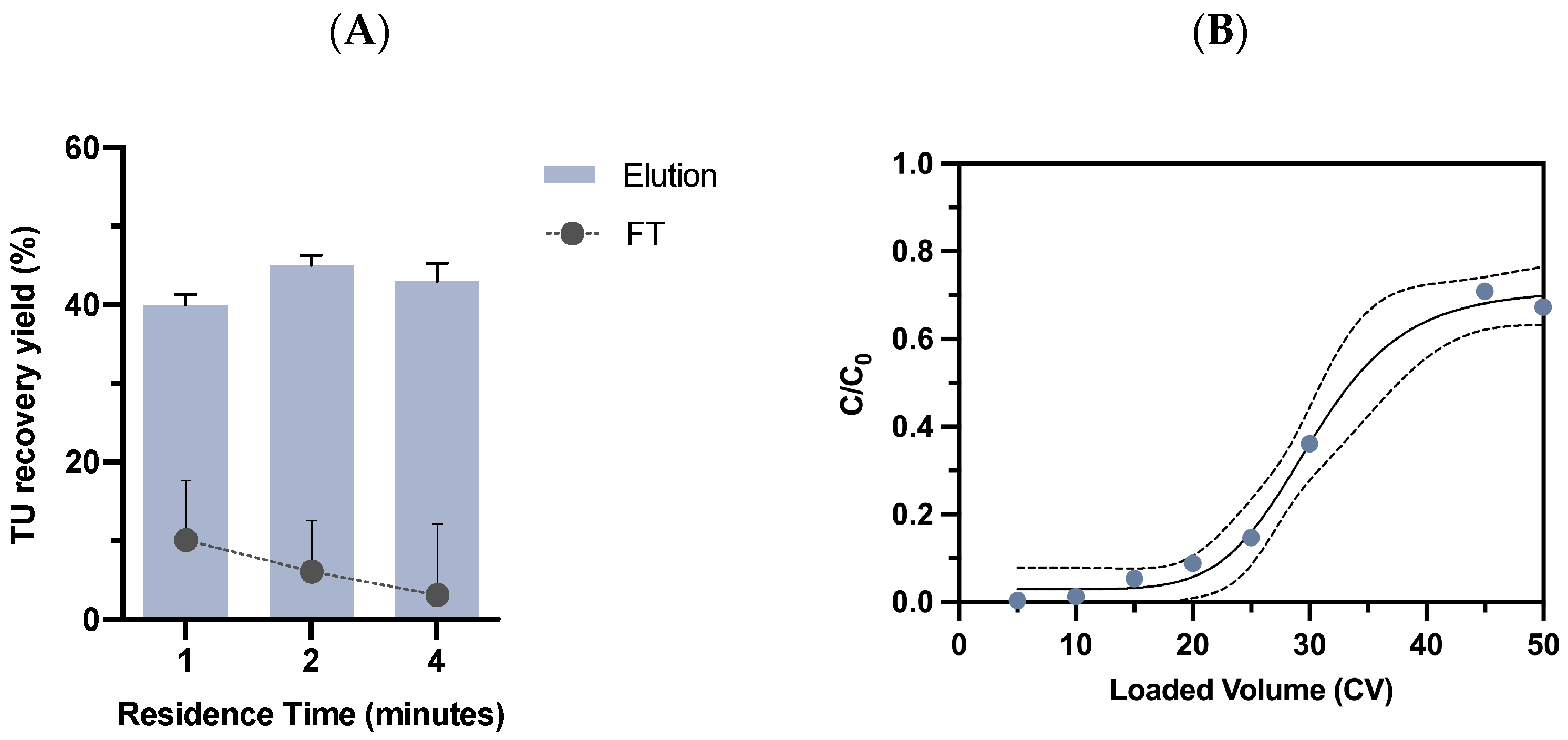
2.3.1. Residence Time Evaluation
2.3.2. Dynamic Binding Capacity Determination
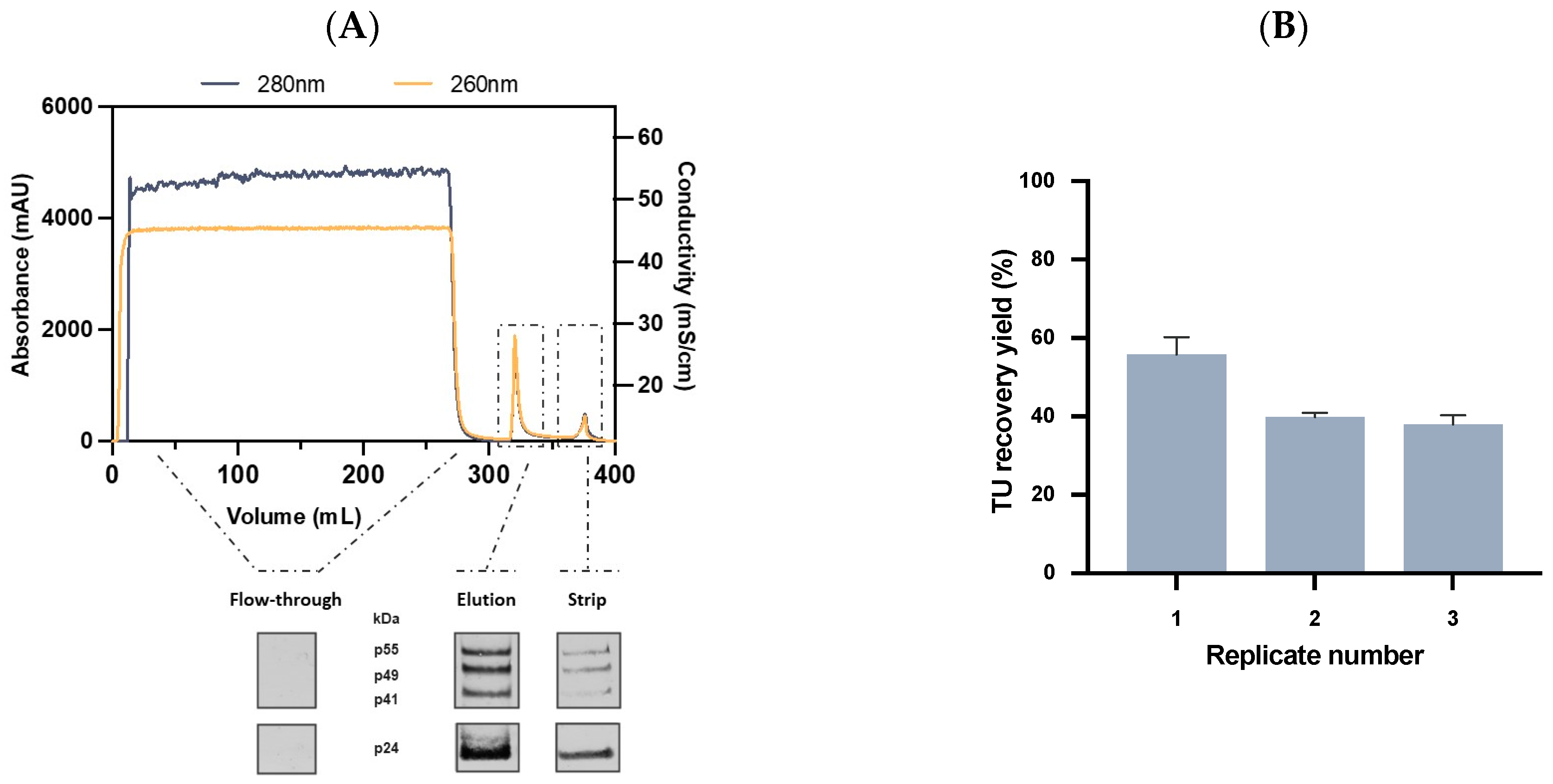
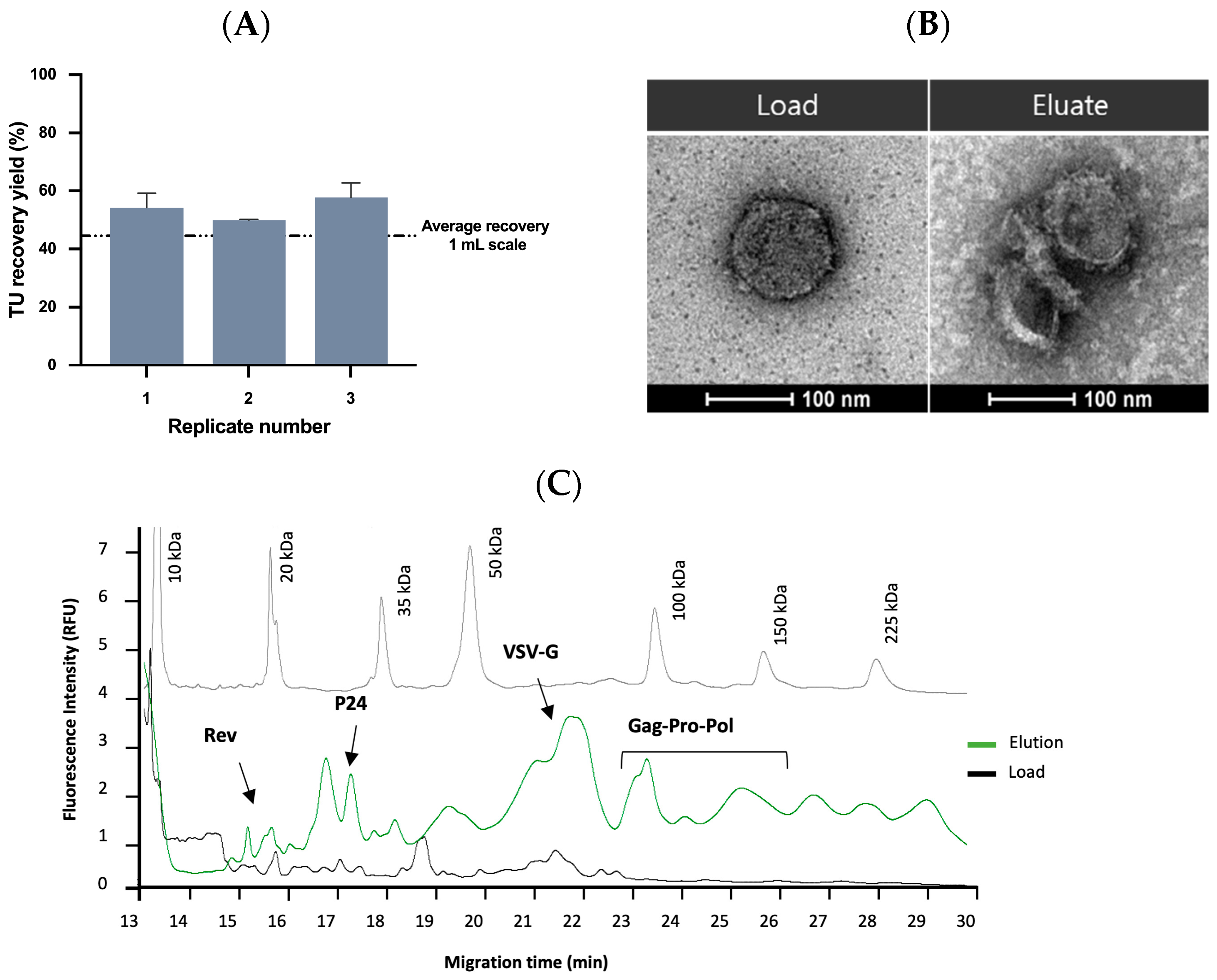
2.3.3. Affinity Chromatography Performance and Scalability Evaluation
2.4. Quality Control and Vector Characterization
3. Materials and Methods
3.1. Lentiviral Vector Production
3.1.1. Cell Line Maintenance and Transfection
3.1.2. Nuclease Treatment and Virus Clarification
3.2. Discovery, Selection, and Production of VHH Fragments
3.3. Surface Plasmon Resonance (SPR)
3.3.1. Binding Profiles
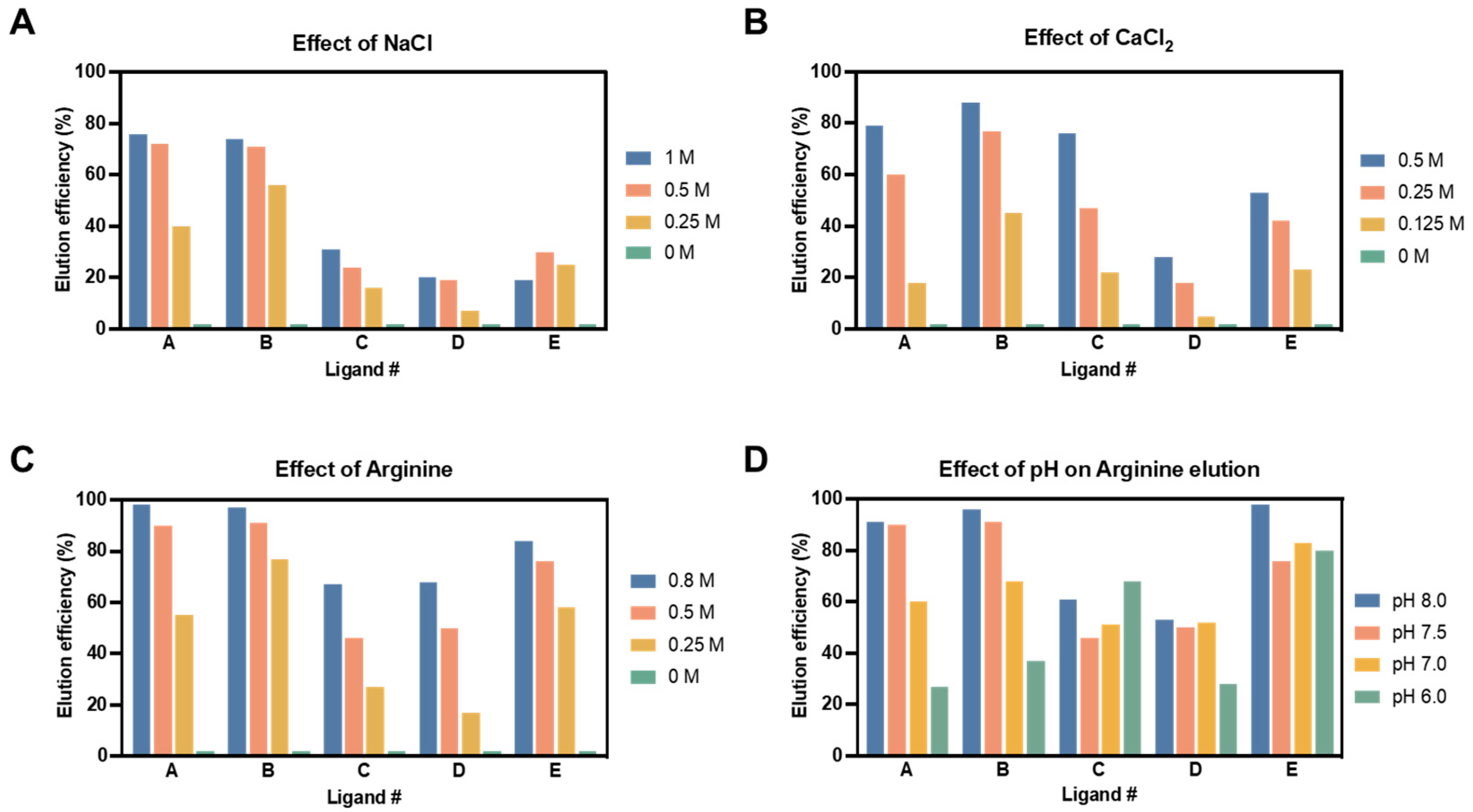
3.3.2. Elution Efficiency
3.4. VSVG-LVs Viral Capture Using Streptavidin Agarose Beads
3.5. Generation of Prototype Affinity Resins
3.6. Affinity Chromatography Studies
3.6.1. Residence Time Optimization
3.6.2. Dynamic Binding Capacity Determination
3.7. Analytics
3.7.1. VSVG-LVs Transducing Particle Quantification
3.7.2. Total Particle Quantification
3.7.3. Total Protein, Host Cell Protein, and DNA Quantification
3.7.4. Transmission Electron Microscopy
3.7.5. Quality Analysis of VSVG-LVs Particles by CE-SDS with LIF Detector
3.7.6. Western Blot Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Segura, M.M.; Mangion, M.; Gaillet, B.; Garnier, A. New developments in lentiviral vector design, production and purification. Expert Opin. Biol. Ther. 2013, 13, 987–1011. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.V.; Cabral, E.T.; Coroadinha, A.S. Progress and Perspectives in the Development of Lentiviral Vector Producer Cells. Biotechnol. J. 2021, 16, 2000017. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.; Rayat, A.C.M.E. Lentiviral vector bioprocessing. Viruses 2021, 13, 268. [Google Scholar] [CrossRef] [PubMed]
- McCarron, A.; Donnelley, M.; McIntyre, C.; Parsons, D. Challenges of up-scaling lentivirus production and processing. J. Biotechnol. 2016, 240, 23–30. [Google Scholar] [CrossRef]
- Kumar, S.R.; Markusic, D.M.; Biswas, M.; High, K.A.; Herzog, R.W. Clinical development of gene therapy: Results and lessons from recent successes. Mol. Ther. Methods Clin. Dev. 2016, 3, 16034. [Google Scholar] [CrossRef]
- Cordes, N.; Winter, N.; Kolbe, C.; Kotter, B.; Mittelstaet, J.; Assenmacher, M.; Cathomen, T.; Kaiser, A.; Schaser, T. Adapter-Mediated Transduction with Lentiviral Vectors: A Novel Tool for Cell-Type-Specific Gene Transfer. Viruses 2022, 14, 2157. [Google Scholar] [CrossRef]
- Dufait, I.; Liechtenstein, T.; Lanna, A.; Bricogne, C.; Laranga, R.; Padella, A.; Breckpot, K.; Escors, D. Retroviral and Lentiviral Vectors for the Induction of Immunological Tolerance. Scientifica 2012, 2012, 694137. [Google Scholar] [CrossRef]
- Oldham, R.A.A.; Berinstein, E.M.; Medin, J.A. Lentiviral vectors in cancer immunotherapy. Immunotherapy 2015, 7, 271–284. [Google Scholar] [CrossRef]
- Cronin, J.; Zhang, X.-Y.; Reiser, J. Altering the Tropism of Lentiviral Vectors through Pseudotyping. Curr. Gene Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef]
- Joglekar, A.V.; Sandoval, S. Pseudotyped Lentiviral Vectors: One Vector, Many Guises. Hum. Gene Ther. Methods 2017, 28, 291–301. [Google Scholar] [CrossRef]
- Arabi, F.; Mansouri, V.; Ahmadbeigi, N. Gene therapy clinical trials, where do we go? An overview. Biomed. Pharmacother. 2022, 153, 113324. [Google Scholar] [CrossRef]
- Boudeffa, D.; Bertin, B.; Biek, A.; Mormin, M.; Leseigneur, F.; Galy, A.; Merten, O.-W. Toward a Scalable Purification Protocol of GaLV-TR-Pseudotyped Lentiviral Vectors. Hum. Gene Ther. Methods 2019, 30, 153–171. [Google Scholar] [CrossRef]
- Comisel, R.M.; Kara, B.; Fiesser, F.H.; Farid, S.S. Lentiviral vector bioprocess economics for cell and gene therapy commercialization. Biochem. Eng. J. 2021, 167, 107868. [Google Scholar] [CrossRef]
- Higashikawa, F.; Chang, L.J. Kinetic analyses of stability of simple and complex retroviral vectors. Virology 2001, 280, 124–131. [Google Scholar] [CrossRef]
- Carmo, M.; Dias, J.; Panet, A.; Coroadinha, A.; Carrondo, M.; Alves, P.; Cruz, P. Thermosensitivity of the Reverse Transcription Process as an Inactivation Mechanism of Lentiviral Vectors. Hum. Gene Ther. 2009, 20, 1168–1176. [Google Scholar] [CrossRef]
- de las Mercedes Segura, M.; Kamen, A.; Trudel, P.; Garnier, A. A novel purification strategy for retrovirus gene therapy vectors using heparin affinity chromatography. Biotechnol. Bioeng. 2005, 90, 391–404. [Google Scholar] [CrossRef]
- Bandeira, V.; Peixoto, C.; Rodrigues, A.F.; Cruz, P.; Alves, P.; Coroadinha, A.; Carrondo, M. Downstream processing of lentiviral vectors: Releasing bottlenecks. Hum. Gene Ther. Methods 2012, 23, 255–263. [Google Scholar] [CrossRef]
- Kumru, O.S.; Wang, Y.; Gombotz, C.W.R.; Kelley-Clarke, B.; Cieplak, W.; Kim, T.; Joshi, S.B.; Volkin, D.B. Physical Characterization and Stabilization of a Lentiviral Vector Against Adsorption and Freeze-Thaw. J. Pharm. Sci. 2018, 107, 2764–2774. [Google Scholar] [CrossRef]
- Rodrigues, T.; Carrondo, M.J.T.T.; Alves, P.M.; Cruz, P.E. Purification of retroviral vectors for clinical application: Biological implications and technological challenges. J. Biotechnol. 2007, 127, 520–541. [Google Scholar] [CrossRef]
- Schweizer, M.; Merten, O.-W. Large-Scale Production Means for the Manufacturing of Lentiviral Vectors. Curr. Gene Ther. 2010, 10, 474–486. [Google Scholar] [CrossRef]
- Martínez-Molina, E.; Chocarro-Wrona, C.; Martínez-Moreno, D.; Marchal, J.A.; Boulaiz, H. Large-scale production of lentiviral vectors: Current perspectives and challenges. Pharmaceutics 2020, 12, 1051. [Google Scholar] [CrossRef] [PubMed]
- Merten, O.-W.; Charrier, S.; Laroudie, N.; Fauchille, S.; Dugué, C.; Jenny, C.; Audit, M.; Zanta-Boussif, M.-A.; Chautard, H.; Radrizzani, M.; et al. Large-Scale Manufacture and Characterization of a Lentiviral Vector Produced for Clinical Ex Vivo Gene Therapy Application. Hum. Gene Ther. 2011, 22, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.; Faria, T.; Oliveira, J.; Kavara, A.; Schofield, M.; Sanderson, T.; Collins, M.; Gantier, R.; Alves, P.; Carrondo, M.; et al. Enhancing the purification of Lentiviral vectors for clinical applications. Sep. Purif. Technol. 2021, 274, 118598. [Google Scholar] [CrossRef]
- Cheeks, M.C.; Kamal, N.; Sorrell, A.; Darling, D.; Farzaneh, F.; Slater, N.K.H. Immobilized metal affinity chromatography of histidine-tagged lentiviral vectors using monolithic adsorbents. J. Chromatogr. A 2009, 1216, 2705–2711. [Google Scholar] [CrossRef]
- Ayyar, B.V.; Arora, S.; Murphy, C.; O’Kennedy, R. Affinity chromatography as a tool for antibody purification. Methods 2012, 56, 116–129. [Google Scholar] [CrossRef]
- Zhao, M.; Vandersluis, M.; Stout, J.; Haupts, U.; Sanders, M.; Jacquemart, R. Affinity chromatography for vaccines manufacturing: Finally ready for prime time? Vaccine 2019, 37, 5491–5503. [Google Scholar] [CrossRef]
- Terova, O.; Hermans, P.; Soltys, S.; Detmers, F. Affinity Chromatography Accelerates Viral Vector Purification for Gene Therapies. Dev. Strateg. Emerg. Ther. 2017, 30, 27–30. [Google Scholar]
- Łącki, K.M.; Riske, F.J. Affinity Chromatography: An Enabling Technology for Large-Scale Bioprocessing. Biotechnol. J. 2020, 15, e1800397. [Google Scholar] [CrossRef]
- Block, H.; Maertens, B.; Spriestersbach, A.; Brinker, N.; Kubicek, J.; Fabis, R.; Labahn, J.; Schäfer, F. Immobilized-Metal Affinity Chromatography (IMAC): A Review. Methods Enzymol. 2009, 463, 439–473. [Google Scholar] [CrossRef]
- Cheung, R.C.F.; Wong, J.H.; Ng, T.B. Immobilized metal ion affinity chromatography: A review on its applications. Appl. Microbiol. Biotechnol. 2012, 96, 1411–1420. [Google Scholar] [CrossRef]
- Chan, L.; Nesbeth, D.; MacKey, T.; Galea-Lauri, J.; Gäken, J.; Martin, F.; Collins, M.; Mufti, G.; Farzaneh, F.; Darling, D. Conjugation of lentivirus to paramagnetic particles via nonviral proteins allows efficient concentration and infection of primary acute myeloid leukemia cells. J. Virol. 2005, 79, 13190–13194. [Google Scholar] [CrossRef]
- Nesbeth, D.; Williams, S.L.; Chan, L.; Brain, T.; Slater, N.K.; Farzaneh, F.; Darling, D. Metabolic biotinylation of lentiviral pseudotypes for scalable paramagnetic microparticle-dependent manipulation. Mol. Ther. 2006, 13, 814–822. [Google Scholar] [CrossRef]
- Mekkaoui, L.; Parekh, F.; Kotsopoulou, E.; Darling, D.; Dickson, G.; Cheung, G.W.; Chan, L.; MacLellan-Gibson, K.; Mattiuzzo, G.; Farzaneh, F.; et al. Lentiviral Vector Purification Using Genetically Encoded Biotin Mimic in Packaging Cell. Mol. Ther. Methods Clin. Dev. 2018, 11, 155–165. [Google Scholar] [CrossRef]
- Moser, A.C.; Hage, D.S. Immunoaffinity chromatography: An introduction to applications and recent developments. Bioanalysis 2010, 2, 769–790. [Google Scholar] [CrossRef]
- Segura, M.M.; Garnier, A.; Durocher, Y.; Coelho, H.; Kamen, A. Production of lentiviral vectors by large-scale transient transfection of suspension cultures and affinity chromatography purification. Biotechnol. Bioeng. 2007, 98, 789–799. [Google Scholar] [CrossRef]
- Pabst, T.M.; Wendeler, M.; Wang, X.; Bezemer, S.; Hermans, P.; Hunter, A.K. Camelid VHH affinity ligands enable separation of closely related biopharmaceuticals. Biotechnol. J. 2017, 12. [Google Scholar] [CrossRef]
- Terova, O.; Soltys, S.; Hermans, P.; de Rooij, J.; Detmers, F. Overcoming Downstream Purification Challenges for Viral Vector Manufacturing: Enabling Advancement of Gene Therapies in the Clinic. Cell Gene Ther. Insights 2018, 4, 101–111. [Google Scholar] [CrossRef]
- Moleirinho, M.G.; Fernandes, R.P.; Carvalho, S.B.; Bezemer, S.; Detmers, F.; Hermans, P.; Silva, R.J.; Alves, P.M.; Carrondo, M.J.; Peixoto, C. Baculovirus affinity removal in viral-based bioprocesses. Sep. Purif. Technol. 2020, 241. [Google Scholar] [CrossRef]
- Lange, C.; Rudolph, R. Suppression of Protein Aggregation by L-Arginine. 2009. Available online: http://www.emea.europa.eu/humandocs/PDFs/EPAR/Rapilysin/H-105-PI-en.pdf (accessed on 19 September 2022).
- Arakawa, T.; Ejima, D.; Tsumoto, K.; Obeyama, N.; Tanaka, Y.; Kita, Y.; Timasheff, S.N. Suppression of protein interactions by arginine: A proposed mechanism of the arginine effects. Biophys. Chem. 2007, 127, 1–8. [Google Scholar] [CrossRef]
- Stärtzel, P. Arginine as an Excipient for Protein Freeze-Drying: A Mini Review. J. Pharm. Sci. 2018, 107, 960–967. [Google Scholar] [CrossRef]
- Arakawa, T.; Philo, J.S.; Tsumoto, K.; Yumioka, R.; Ejima, D. Elution of antibodies from a Protein-A column by aqueous arginine solutions. Protein Expr. Purif. 2004, 36, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Ejima, D.; Yumioka, R.; Tsumoto, K.; Arakawa, T. Effective elution of antibodies by arginine and arginine derivatives in affinity column chromatography. Anal. Biochem. 2005, 345, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Trovão, F.; Gonçalves, J.; Rothbauer, U.; Freire, M.; Barbosa, A.; Pina, A.; Roque, A. A purification platform for antibodies and derived fragments using a de novo designed affinity adsorbent. Sep. Purif. Technol. 2021, 265, 118476. [Google Scholar] [CrossRef]
- Moreira, A.S.; Cavaco, D.G.; Faria, T.Q.; Alves, P.M.; Carrondo, M.J.T.; Peixoto, C. Advances in Lentivirus Purification. Biotechnol. J. 2021, 16, e2000019. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Trout, B.L. Understanding the role of arginine and citrate as eluents in affinity chromatography. ACS Symp. Ser. 2013, 1125, 67–86. [Google Scholar] [CrossRef]
- Mendes, J.P.; Bergman, M.; Solbrand, A.; Peixoto, C.; Carrondo, M.J.T.; Silva, R.J.S. Continuous Affinity Purification of Adeno-Associated Virus Using Periodic Counter-Current Chromatography. Pharmaceutics 2022, 14, 1346. [Google Scholar] [CrossRef]
- Ye, K.; Dhiman, H.K.; Suhan, J.; Schultz, J.S. Effect of pH on infectivity and morphology of ecotropic moloney murine leukemia virus. Biotechnol. Prog. 2003, 19, 538–543. [Google Scholar] [CrossRef]
- Xue, A.; Fan, S. Matrices and Affinity Ligands for Antibody Purification and Corresponding Applications in Radiotherapy. Biomolecules 2022, 12, 821. [Google Scholar] [CrossRef]
- Müller, E.; Vajda, J. Routes to improve binding capacities of affinity resins demonstrated for Protein A chromatography. J. Chromatogr. B 2016, 1021, 159–168. [Google Scholar] [CrossRef]
- Gao, Z.-Y.; Zhang, Q.-L.; Shi, C.; Gou, J.-X.; Gao, D.; Wang, H.-B.; Yao, S.-J.; Lin, D.-Q. Antibody capture with twin-column continuous chromatography: Effects of residence time, protein concentration and resin. Sep. Purif. Technol. 2020, 253, 117554. [Google Scholar] [CrossRef]
- Eslami, T.; Jakob, L.A.; Satzer, P.; Ebner, G.; Jungbauer, A.; Lingg, N. Productivity for free: Residence time gradients during loading increase dynamic binding capacity and productivity. Sep. Purif. Technol. 2022, 281, 119985. [Google Scholar] [CrossRef]
- Scherr, M.; Battmer, K.; Eder, M.; Schüle, S.; Hohenberg, H.; Ganser, A.; Grez, M.; Blömer, U. Efficient gene transfer into the CNS by lentiviral vectors purified by anion exchange chromatography. Gene Ther. 2002, 9, 1708–1714. [Google Scholar] [CrossRef]
- Ghosh, R.; Koley, S.; Gopal, S.; Rodrigues, A.L.; Dordick, J.S.; Cramer, S.M. Evaluation of lentiviral vector stability and development of ion exchange purification processes. Biotechnol. Prog. 2022, 38, e3286. [Google Scholar] [CrossRef] [PubMed]
- Valkama, A.J.; Oruetxebarria, I.; Lipponen, E.M.; Leinonen, H.M.; Käyhty, P.; Hynynen, H.; Turkki, V.; Malinen, J.; Miinalainen, T.; Heikura, T.; et al. Development of Large-Scale Downstream Processing for Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2020, 17, 717–730. [Google Scholar] [CrossRef]
- Joshi, P.R.H.; Bernier, A.; Moço, P.D.; Schrag, J.; Chahal, P.S.; Kamen, A. Development of a scalable and robust AEX method for enriched rAAV preparations in genome-containing VCs of serotypes 5, 6, 8, and 9. Mol. Ther. Methods Clin. Dev. 2021, 21, 341–356. [Google Scholar] [CrossRef]
- Toueille, M.; Dejoint, L.; Attebi, E.; Cartigny, J.; Rasle, C.; Potier, S.; Rundwasser, S.; Guianvarc’H, L.; Lebec, C.; Hebben, M. Development of purification steps for several AAV serotypes using POROSTM CaptureSelectTM AAVX affinity chromatography. Cell Gene Ther. Insights 2018, 4, 637–645. [Google Scholar] [CrossRef]
- Rathore, A.S.; Kumar, D.; Kateja, N. Recent developments in chromatographic purification of biopharmaceuticals. Biotechnol. Lett. 2018, 40, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Hahn, R. Methods for characterization of biochromatography media. J. Sep. Sci. 2012, 35, 3001–3032. [Google Scholar] [CrossRef]
- Nweke, M.C.; Rathore, A.S.; Bracewell, D.G. Lifetime and Aging of Chromatography Resins during Biopharmaceutical Manufacture. Trends Biotechnol. 2018, 36, 992–995. [Google Scholar] [CrossRef]
- Suzuki, N.; Yoshida, T.; Takeuchi, H.; Sakuma, R.; Sukegawa, S.; Yamaoka, S. Robust Enhancement of Lentivirus Production by Promoter Activation. Sci. Rep. 2018, 8, 15036. [Google Scholar] [CrossRef] [PubMed]
- Doyon, L.; Croteau, G.; Thibeault, D.; Poulin, F.; Pilote, L.; Lamarre, D. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 1996, 70, 3763–3769. [Google Scholar] [CrossRef]
- Ruscic, J.; Perry, C.; Mukhopadhyay, T.; Takeuchi, Y.; Bracewell, D.G. Lentiviral vector purification using nanofiber ion-exchange chromatography. Mol. Ther.-Methods Clin. Dev. 2019, 15, 52–62. [Google Scholar] [CrossRef]
- Cooper, A.R.; Patel, S.; Senadheera, S.; Plath, K.; Kohn, D.B.; Hollis, R.P. Highly efficient large-scale lentiviral vector concentration by tandem tangential flow filtration. J. Virol. Methods 2011, 177, 1–9. [Google Scholar] [CrossRef]
- Soldi, M.; Sergi, L.S.; Unali, G.; Kerzel, T.; Cuccovillo, I.; Capasso, P.; Annoni, A.; Biffi, M.; Rancoita, P.M.V.; Cantore, A.; et al. Laboratory-Scale Lentiviral Vector Production and Purification for Enhanced Ex Vivo and In Vivo Genetic Engineering. Mol. Ther. Methods Clin. Dev. 2020, 19, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Labisch, J.J.; Kassar, M.; Bollmann, F.; Valentic, A.; Hubbuch, J.; Pflanz, K. Steric exclusion chromatography of lentiviral vectors using hydrophilic cellulose membranes. J. Chromatogr. A 2022, 1674, 463148. [Google Scholar] [CrossRef] [PubMed]
- Ausubel, L.J.; Hall, C.; Sharma, A.; Shakeley, R.; Lopez, P.; Quezada, V.; Couture, S.; Laderman, K.; McMahon, R.; Huang, P.; et al. Production of CGMP-Grade Lentiviral Vectors. Bioprocess. Int. 2012, 10, 32–43. Available online: http://www.ncbi.nlm.nih.gov/pubmed/22707919 (accessed on 19 September 2022).
- Fernandes, R.P.; Escandell, J.M.; Guerreiro, A.C.L.; Moura, F.; Faria, T.Q.; Carvalho, S.B.; Silva, R.J.S.; Gomes-Alves, P.; Peixoto, C. Assessing Multi-Attribute Characterization of Enveloped and Non-Enveloped Viral Particles by Capillary Electrophoresis. Viruses 2022, 14, 2539. [Google Scholar] [CrossRef]
- Li, T.; Guttman, A. High-Precision Lentivirus Titer Determination and Protein Profiling. Available online: https://sciex.com/content/dam/SCIEX/pdf/tech-notes/biopharma/high-precision-lentivirus-titer-determination-and-protein-profiling.pdf (accessed on 18 December 2022).
- Vaz, T.A.; Rodrigues, A.F.; Coroadinha, A.S. Exploring nutrient supplementation and bioprocess optimization to improve the production of lentiviral vectors in serum-free suspension cultures. Biotechnol. Bioeng. 2023; Submitted. [Google Scholar]
- Adams, H.; Horrevoets, W.M.; Adema, S.M.; Carr, H.E.; van Woerden, R.E.; Koster, M.; Tommassen, J. Inhibition of biofilm formation by Camelid single-domain antibodies against the flagellum of Pseudomonas aeruginosa. J. Biotechnol. 2014, 186, 66–73. [Google Scholar] [CrossRef]
- Schatz, P.J. Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: A 13 residue consensus peptide specifies biotinylation in Escherichia coli. Biotechnology 1993, 11, 1138–1143. [Google Scholar] [CrossRef]
- Adams, H.; Brummelhuis, W.; Maassen, B.; van Egmond, N.; El Khattabi, M.; Detmers, F.; Hermans, P.; Braam, B.; Stam, J.; Verrips, T. Specific immuno capturing of the staphylococcal superantigen toxic-shock syndrome toxin-1 in plasma. Biotechnol. Bioeng. 2009, 104, 143–151. [Google Scholar] [CrossRef]
- Fisher Scientific. CaptureSelect Lenti VSVG Affinity Matrix Product Information Sheet (Pub.No. MAN0028036 A.0). Available online: www.thermofisher.com/captureselect (accessed on 19 September 2022).
- Tomás, H.A.; Rodrigues, A.F.; Carrondo, M.J.T.; Coroadinha, A.S. LentiPro26: Novel stable cell lines for constitutive lentiviral vector production. Sci. Rep. 2018, 8, 5271. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix Type | Coupling Method | Selectivity Profile | Average Particle Size (μm) |
|---|---|---|---|
| Matrix 1 | Direct | − | 50 |
| Matrix 2 | Direct | +/− | 50 |
| Matrix 3 | Direct | +++ | 65 |
| Matrix 4 | Biotin | ++ | 45–165 |
| Fraction | Transduction Units (TU/mL) | Volume (mL) | Total Particles (TP/mL) | TP/TU Ratio | Residual HCP (%) |
|---|---|---|---|---|---|
| Load | 7.3 ± 2.1 × 107 | 250 | 1.0 ± 1.5 × 1010 | 143 ± 23 | - |
| Flow-through | 8.1 ± 8.9 × 106 | 258 | 1.7 ± 6.8 × 109 | 270 ± 107 | 98.5 ± 1.3 |
| Elution | 3.7 ± 1.5 × 108 | 22.5 | 3.3 ± 1.0 × 1010 | 97 ± 40 | 1.5 ± 1.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, A.S.; Bezemer, S.; Faria, T.Q.; Detmers, F.; Hermans, P.; Sierkstra, L.; Coroadinha, A.S.; Peixoto, C. Implementation of Novel Affinity Ligand for Lentiviral Vector Purification. Int. J. Mol. Sci. 2023, 24, 3354. https://doi.org/10.3390/ijms24043354
Moreira AS, Bezemer S, Faria TQ, Detmers F, Hermans P, Sierkstra L, Coroadinha AS, Peixoto C. Implementation of Novel Affinity Ligand for Lentiviral Vector Purification. International Journal of Molecular Sciences. 2023; 24(4):3354. https://doi.org/10.3390/ijms24043354
Chicago/Turabian StyleMoreira, Ana Sofia, Sandra Bezemer, Tiago Q. Faria, Frank Detmers, Pim Hermans, Laurens Sierkstra, Ana Sofia Coroadinha, and Cristina Peixoto. 2023. "Implementation of Novel Affinity Ligand for Lentiviral Vector Purification" International Journal of Molecular Sciences 24, no. 4: 3354. https://doi.org/10.3390/ijms24043354
APA StyleMoreira, A. S., Bezemer, S., Faria, T. Q., Detmers, F., Hermans, P., Sierkstra, L., Coroadinha, A. S., & Peixoto, C. (2023). Implementation of Novel Affinity Ligand for Lentiviral Vector Purification. International Journal of Molecular Sciences, 24(4), 3354. https://doi.org/10.3390/ijms24043354