
Inhibitory Effects of a Novel μ-Opioid Receptor Nonpeptide Antagonist, UD-030, on Morphine-Induced Conditioned Place Preference

Abstract
1. Introduction
2. Results
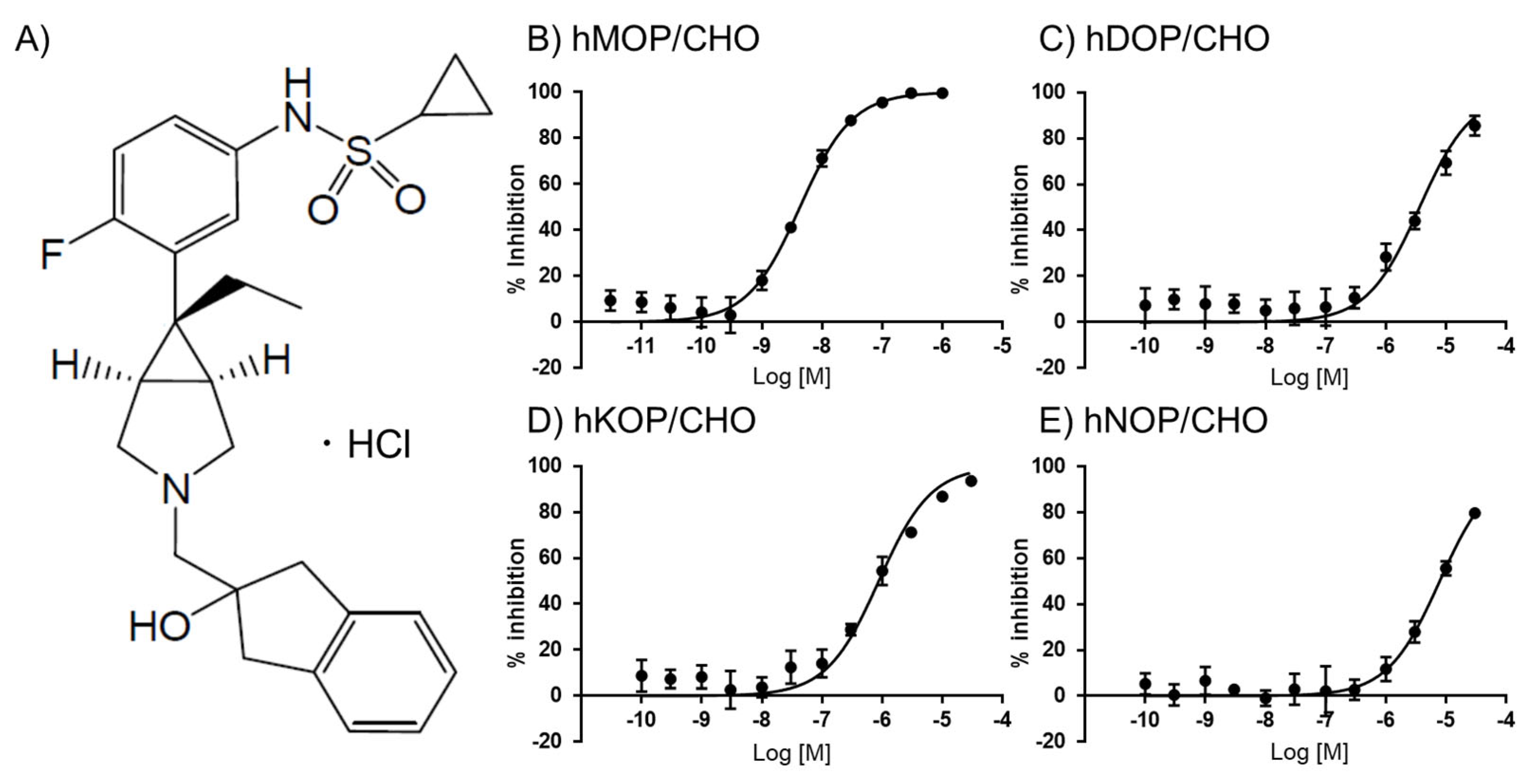
2.1. In Vitro Assays of UD-030
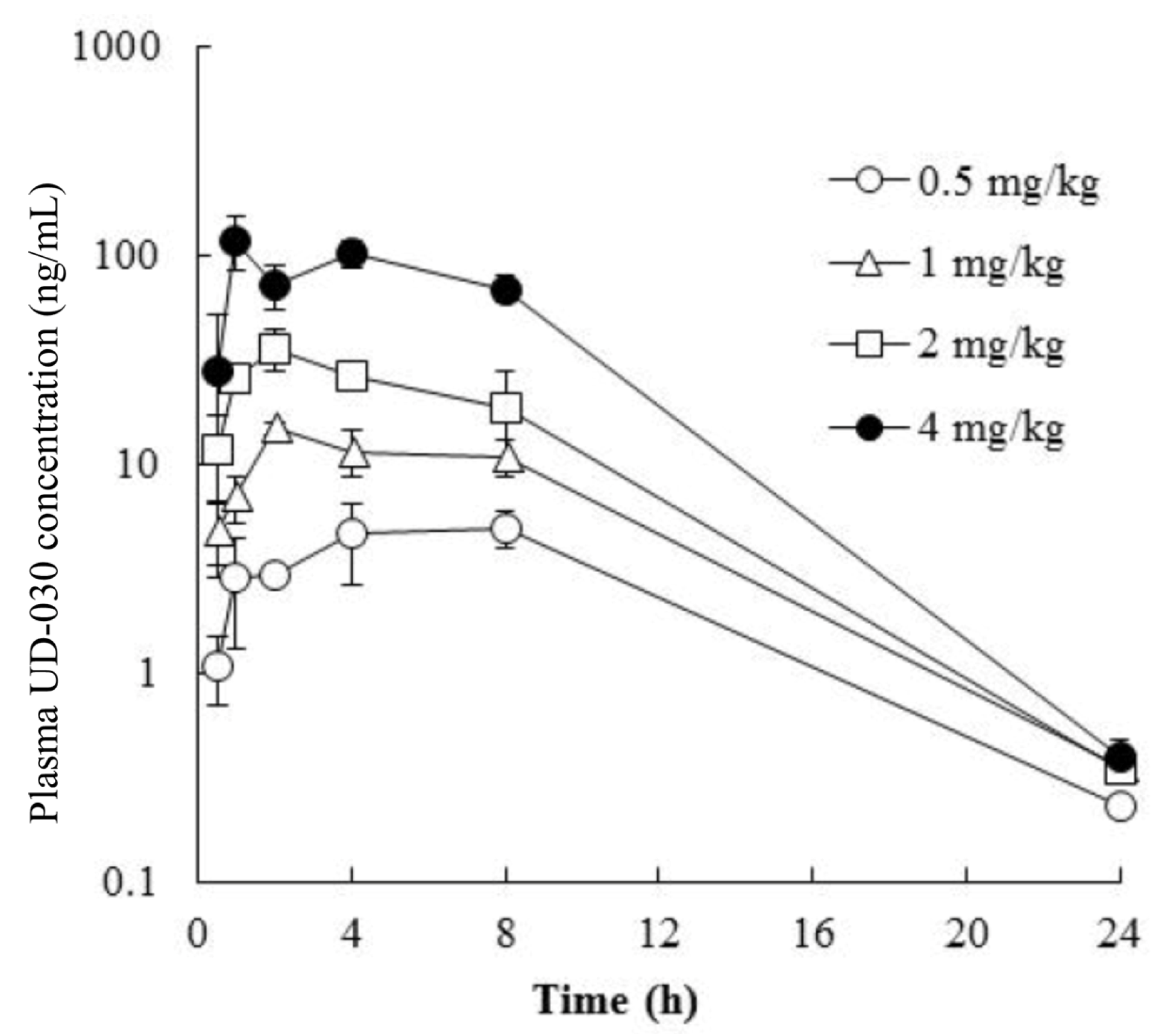
2.2. Pharmacokinetics of Orally Administered UD-030
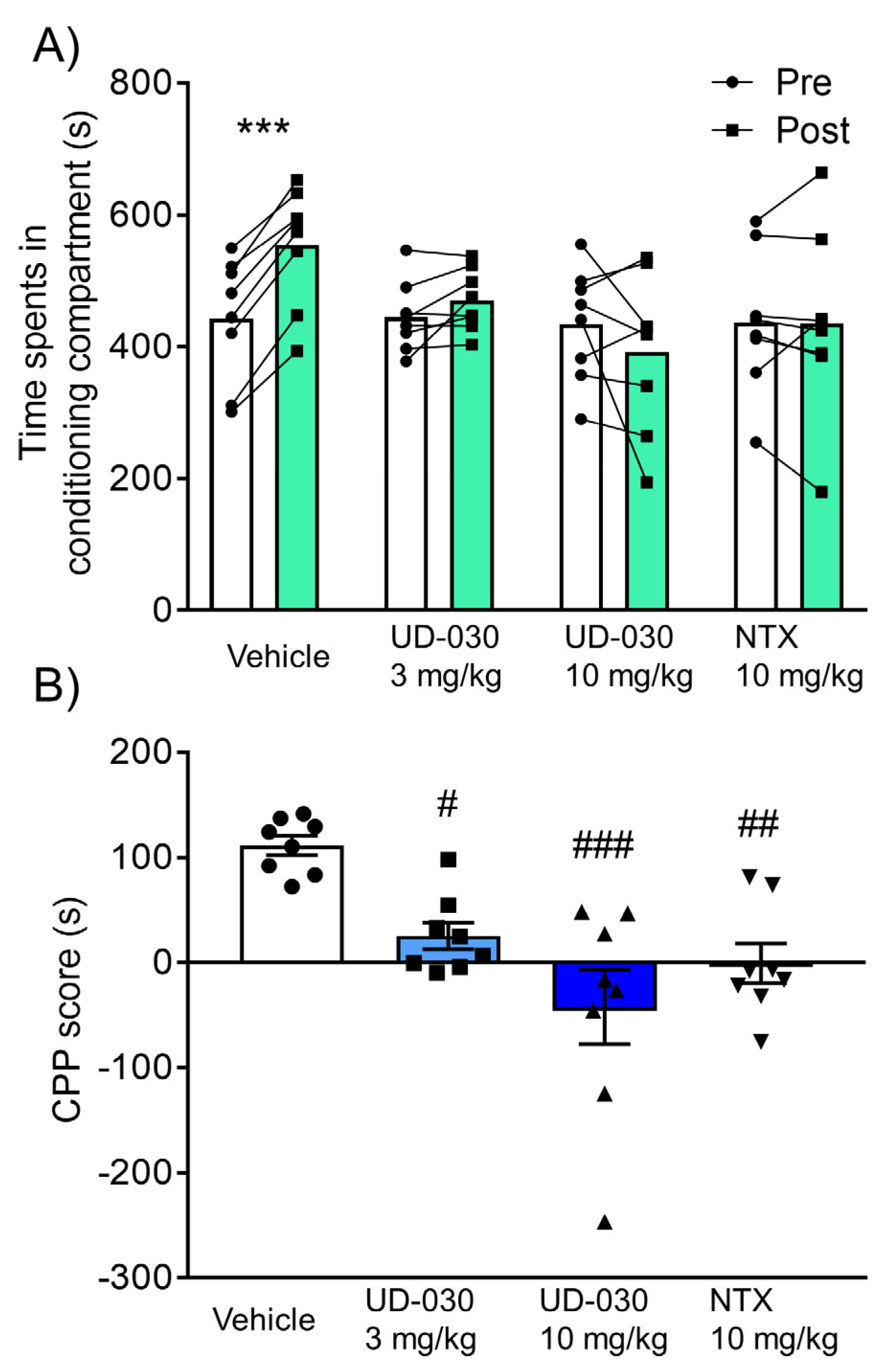
2.3. Inhibitory Effect of UD-030 on the Acquisition of Morphine-Induced Conditioned Place Preference
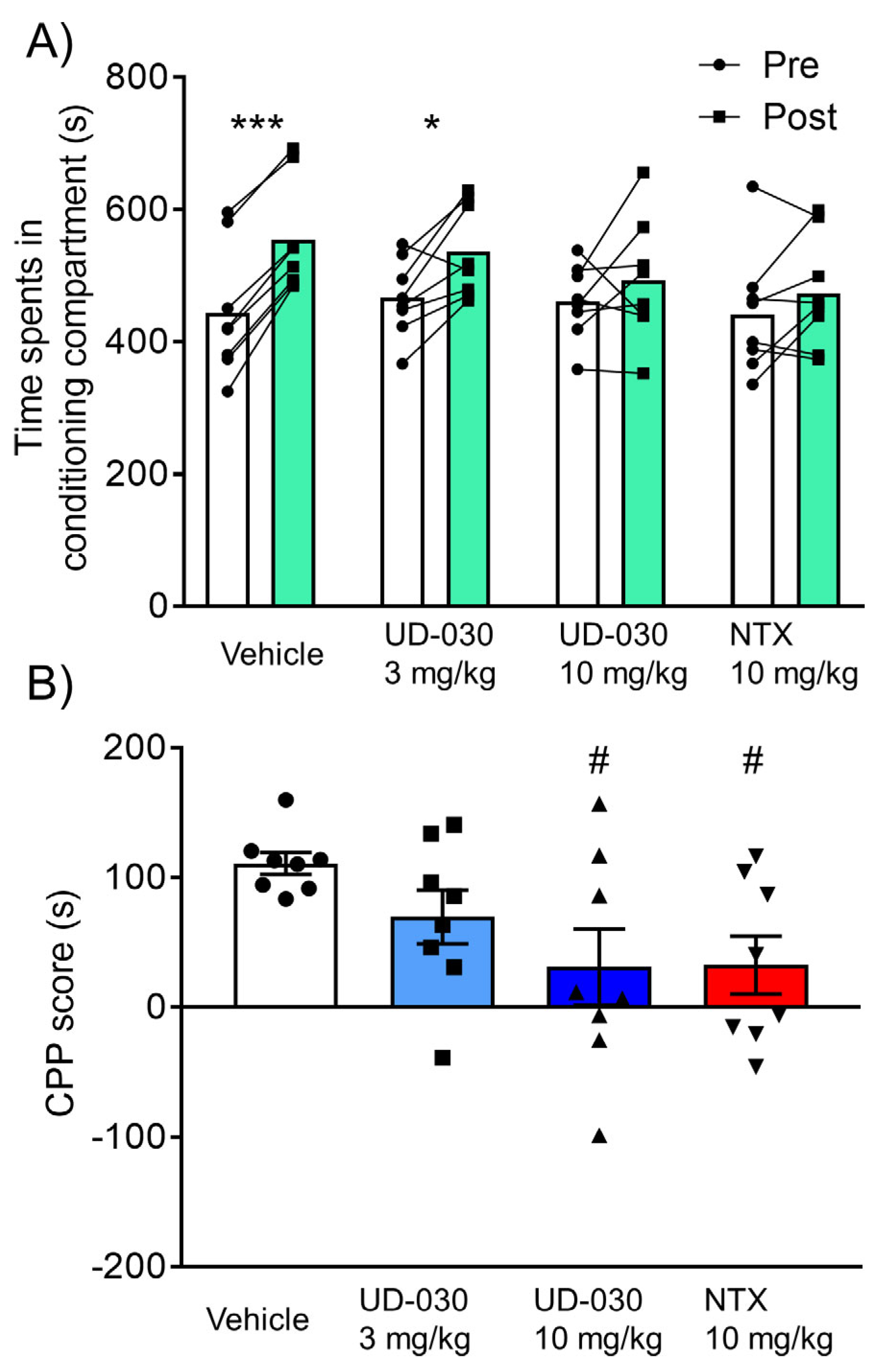
2.4. Inhibitory Effect of UD-030 on the Expression of Morphine-Induced CPP
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Radioligand Binding Assay
4.3. [35S]-GTPγS Binding
4.4. Animals
4.5. Pharmacokinetic Analysis
4.6. Conditioned Place Preference Test
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. World Drug Report 2021; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Dietis, N.; Rowbotham, D.J.; Lambert, D.G. Opioid receptor subtypes: Fact or artifact? Br. J. Anaesth. 2011, 107, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.E.; Eubanks, L.M.; Janda, K.D. Chemical Interventions for the Opioid Crisis: Key Advances and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 1798–1806. [Google Scholar] [CrossRef]
- Lutfy, K.; Cowan, A. Buprenorphine: A unique drug with complex pharmacology. Curr. Neuropharmacol. 2004, 2, 395–402. [Google Scholar] [CrossRef]
- Sora, I.; Takahashi, N.; Funada, M.; Ujike, H.; Revay, R.S.; Donovan, D.M.; Miner, L.L.; Uhl, G.R. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc. Natl. Acad. Sci. USA 1997, 94, 1544–1549. [Google Scholar] [CrossRef]
- Kieffer, B.L.; Gaveriaux-Ruff, C. Exploring the opioid system by gene knockout. Prog. Neurobiol. 2002, 66, 285–306. [Google Scholar] [CrossRef] [PubMed]
- Ide, S.; Minami, M.; Ishihara, K.; Uhl, G.R.; Satoh, M.; Sora, I.; Ikeda, K. Abolished thermal and mechanical antinociception but retained visceral chemical antinociception induced by butorphanol in mu-opioid receptor knockout mice. Neuropharmacology 2008, 54, 1182–1188. [Google Scholar] [CrossRef]
- Ide, S.; Minami, M.; Uhl, G.R.; Satoh, M.; Sora, I.; Ikeda, K. (-)-Pentazocine induces visceral chemical antinociception, but not thermal, mechanical, or somatic chemical antinociception, in mu-opioid receptor knockout mice. Mol. Pain 2011, 7, 23. [Google Scholar] [CrossRef]
- Ide, S.; Minami, M.; Satoh, M.; Uhl, G.R.; Sora, I.; Ikeda, K. Buprenorphine antinociception is abolished, but naloxone-sensitive reward is retained, in mu-opioid receptor knockout mice. Neuropsychopharmacology 2004, 29, 1656–1663. [Google Scholar] [CrossRef]
- Spreen, L.A.; Dittmar, E.N.; Quirk, K.C.; Smith, M.A. Buprenorphine initiation strategies for opioid use disorder and pain management: A systematic review. Pharmacotherapy 2022, 42, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Bonner, G.G.; Davis, P.; Stropova, D.; Edsall, S.; Yamamura, H.I.; Porreca, F.; Hruby, V.J. Opiate aromatic pharmacophore structure-activity relationships in CTAP analogues determined by topographical bias, two-dimensional NMR, and biological activity assays. J. Med. Chem. 2000, 43, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.H.; Shook, J.E.; Kazmierski, W.; Ayres, E.A.; Wire, W.S.; Hruby, V.J.; Burks, T.F. Novel peptidic mu opioid antagonists: Pharmacologic characterization in vitro and in vivo. J. Pharmacol. Exp. Ther. 1989, 249, 544–551. [Google Scholar] [PubMed]
- Pelton, J.T.; Kazmierski, W.; Gulya, K.; Yamamura, H.I.; Hruby, V.J. Design and synthesis of conformationally constrained somatostatin analogues with high potency and specificity for mu opioid receptors. J. Med. Chem. 1986, 29, 2370–2375. [Google Scholar] [CrossRef]
- Peng, X.; Knapp, B.I.; Bidlack, J.M.; Neumeyer, J.L. Pharmacological properties of bivalent ligands containing butorphan linked to nalbuphine, naltrexone, and naloxone at mu, delta, and kappa opioid receptors. J. Med. Chem. 2007, 50, 2254–2258. [Google Scholar] [CrossRef]
- Bespalov, A.Y.; Tokarz, M.E.; Bowen, S.E.; Balster, R.L.; Beardsley, P.M. Effects of test conditions on the outcome of place conditioning with morphine and naltrexone in mice. Psychopharmacology 1999, 141, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Shoblock, J.R.; Maidment, N.T. Constitutively active mu opioid receptors mediate the enhanced conditioned aversive effect of naloxone in morphine-dependent mice. Neuropsychopharmacology 2006, 31, 171–177. [Google Scholar] [CrossRef]
- Anton, R.F. Naltrexone for the management of alcohol dependence. N. Engl. J. Med. 2008, 359, 715–721. [Google Scholar] [CrossRef]
- Chiu, C.T.; Ma, T.; Ho, I.K. Attenuation of methamphetamine-induced behavioral sensitization in mice by systemic administration of naltrexone. Brain Res. Bull. 2005, 67, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Ray, L.A.; Bujarski, S.; Courtney, K.E.; Moallem, N.R.; Lunny, K.; Roche, D.; Leventhal, A.M.; Shoptaw, S.; Heinzerling, K.; London, E.D.; et al. The effects of naltrexone on subjective response to methamphetamine in a clinical sample: A double-blind, placebo-controlled laboratory study. Neuropsychopharmacology 2015, 40, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Al-Eitan, L.N.; Rababa’h, D.M.; Alghamdi, M.A. Genetic susceptibility of opioid receptor genes polymorphism to drug addiction: A candidate-gene association study. BMC Psychiatry 2021, 21, 5. [Google Scholar] [CrossRef]
- Bart, G.; Kreek, M.J.; Ott, J.; LaForge, K.S.; Proudnikov, D.; Pollak, L.; Heilig, M. Increased attributable risk related to a functional mu-opioid receptor gene polymorphism in association with alcohol dependence in central Sweden. Neuropsychopharmacology 2005, 30, 417–422. [Google Scholar] [CrossRef]
- Ide, S.; Kobayashi, H.; Ujike, H.; Ozaki, N.; Sekine, Y.; Inada, T.; Harano, M.; Komiyama, T.; Yamada, M.; Iyo, M.; et al. Linkage disequilibrium and association with methamphetamine dependence/psychosis of mu-opioid receptor gene polymorphisms. Pharm. J. 2006, 6, 179–188. [Google Scholar] [CrossRef]
- Umbricht, A.; Montoya, I.D.; Hoover, D.R.; Demuth, K.L.; Chiang, C.T.; Preston, K.L. Naltrexone shortened opioid detoxification with buprenorphine. Drug Alcohol Depend. 1999, 56, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Wentland, M.P.; Lu, Q.; Lou, R.; Bu, Y.; Knapp, B.I.; Bidlack, J.M. Synthesis and opioid receptor binding properties of a highly potent 4-hydroxy analogue of naltrexone. Bioorg. Med. Chem. Lett. 2005, 15, 2107–2110. [Google Scholar] [CrossRef] [PubMed]
- Greenwood-Van Meerveld, B.; Standifer, K.M. Methylnaltrexone in the treatment of opioid-induced constipation. Clin. Exp. Gastroenterol. 2008, 1, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Karver, S.; Cooney, G.A.; Chamberlain, B.H.; Watt, C.K.; Slatkin, N.E.; Stambler, N.; Kremer, A.B.; Israel, R.J. Methylnaltrexone for opioid-induced constipation in advanced illness. N. Engl. J. Med. 2008, 358, 2332–2343. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.; Torup, L.; Sorensen, P.; Gual, A.; Swift, R.; Walker, B.; van den Brink, W. Nalmefene for the management of alcohol dependence: Review on its pharmacology, mechanism of action and meta-analysis on its clinical efficacy. Eur. Neuropsychopharmacol. 2016, 26, 1941–1949. [Google Scholar] [CrossRef]
- Takemori, A.E.; Larson, D.L.; Portoghese, P.S. The irreversible narcotic antagonistic and reversible agonistic properties of the fumaramate methyl ester derivative of naltrexone. Eur. J. Pharmacol. 1981, 70, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Madariaga-Mazon, A.; Marmolejo-Valencia, A.F.; Li, Y.; Toll, L.; Houghten, R.A.; Martinez-Mayorga, K. Mu-Opioid receptor biased ligands: A safer and painless discovery of analgesics? Drug Discov. Today 2017, 22, 1719–1729. [Google Scholar] [CrossRef]
- Karkhanis, A.; Holleran, K.M.; Jones, S.R. Dynorphin/Kappa Opioid Receptor Signaling in Preclinical Models of Alcohol, Drug, and Food Addiction. Int. Rev. Neurobiol. 2017, 136, 53–88. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Fernandez, B.; Calomarde-Gomez, C.; Lopez-Lazcano, A.; Lligona, A.; Gual, A.; Lopez-Pelayo, H. Systematic review on the clinical management of chronic pain and comorbid opioid use disorder. Adicciones 2022, 1680. [Google Scholar] [CrossRef]
- Wachholtz, A.; Robinson, D.; Epstein, E. Developing a novel treatment for patients with chronic pain and Opioid User Disorder. Subst. Abuse Treat. Prev. Policy 2022, 17, 35. [Google Scholar] [CrossRef]
- Tzschentke, T.M. Measuring reward with the conditioned place preference paradigm: A comprehensive review of drug effects, recent progress and new issues. Prog. Neurobiol. 1998, 56, 613–672. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hMOP/CHO | hDOP/CHO | hKOP/CHO | hNOP/CHO | |
|---|---|---|---|---|
| Competitive binding assay | ||||
| UD-030 Ki (nM) | 3.1 | 1800 | 460 | 1800 |
| [2.7–3.5] | [1500–2100] | [390–550] | [1600–2100] | |
| Selectivity (vs. μ) | 581 | 148 | 581 | |
| NTX Ki (nM) a | 0.23 | 38 | 0.25 | ND |
| Selectivity (vs. μ) | 165 | 1 | ND | |
| [35S]-GTPγS binding assay | ||||
| UD-030 EC50 (nM) | >10,000 | >10,000 | >10,000 | >10,000 |
| IC50 (nM) | 1.7 [1.4–2.0] | 850 [760–940] | 85 [76–96] | 330 [290–370] |
| DAMGO EC50 (nM) | 2.5 [2.1–2.9] | — | — | — |
| DPDPE EC50 (nM) | — | 1.8 [1.5–2.1] | — | — |
| U69593 EC50 (nM) | — | — | 14 [13–15] | — |
| Nociceptin EC50 (nM) | — | — | — | 16 [15–18] |
| Time (h) | Plasma Concentration (ng/mL) | |||
|---|---|---|---|---|
| 0.5 mg/kg | 1 mg/kg | 2 mg/kg | 4 mg/kg | |
| 0.5 | 1.09 ± 0.4 | 4.76 ± 1.9 | 11.8 ± 5.4 | 27.7 ± 24 |
| 1 | 2.86 ± 1.6 | 6.98 ± 1.7 | 25.5 ± 2.2 | 119 ± 35 |
| 2 | 2.96 ± 0.3 | 14.9 ± 0.9 | 36.1 ± 8.3 | 72.7 ± 18 |
| 4 | 4.61 ± 1.9 | 11.5 ± 2.9 | 26.7 ± 3.3 | 103 ± 15 |
| 8 | 4.95 ± 0.9 | 10.9 ± 2.1 | 18.8 ± 9.2 | 69.4 ± 10 |
| 24 | 0.231 ± 0 | 0.358 ± 0 | 0.35 ± 0 | 0.395 ± 0.1 |
| Tmax (h) | 8.0 | 2.0 | 2.0 | 1.0 |
| Cmax (ng/mL) | 4.95 | 14.9 | 36.1 | 119 |
| AUC0–24h (ng·h/mL) | 72.3 | 176 | 350 | 1220 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ide, S.; Iwase, N.; Arai, K.; Kojima, M.; Ushiyama, S.; Taniko, K.; Ikeda, K. Inhibitory Effects of a Novel μ-Opioid Receptor Nonpeptide Antagonist, UD-030, on Morphine-Induced Conditioned Place Preference. Int. J. Mol. Sci. 2023, 24, 3351. https://doi.org/10.3390/ijms24043351
Ide S, Iwase N, Arai K, Kojima M, Ushiyama S, Taniko K, Ikeda K. Inhibitory Effects of a Novel μ-Opioid Receptor Nonpeptide Antagonist, UD-030, on Morphine-Induced Conditioned Place Preference. International Journal of Molecular Sciences. 2023; 24(4):3351. https://doi.org/10.3390/ijms24043351
Chicago/Turabian StyleIde, Soichiro, Noriaki Iwase, Kenichi Arai, Masahiro Kojima, Shigeru Ushiyama, Kaori Taniko, and Kazutaka Ikeda. 2023. "Inhibitory Effects of a Novel μ-Opioid Receptor Nonpeptide Antagonist, UD-030, on Morphine-Induced Conditioned Place Preference" International Journal of Molecular Sciences 24, no. 4: 3351. https://doi.org/10.3390/ijms24043351
APA StyleIde, S., Iwase, N., Arai, K., Kojima, M., Ushiyama, S., Taniko, K., & Ikeda, K. (2023). Inhibitory Effects of a Novel μ-Opioid Receptor Nonpeptide Antagonist, UD-030, on Morphine-Induced Conditioned Place Preference. International Journal of Molecular Sciences, 24(4), 3351. https://doi.org/10.3390/ijms24043351