
Identification of SARS-CoV-2 Main Protease (Mpro) Cleavage Sites Using Two-Dimensional Electrophoresis and In Silico Cleavage Site Prediction

 , ,
, ,  ,
,  , ,
, ,  and
and
Abstract
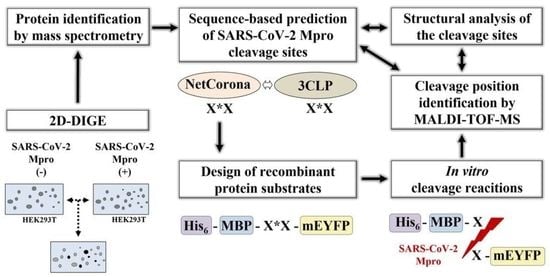
:
1. Introduction
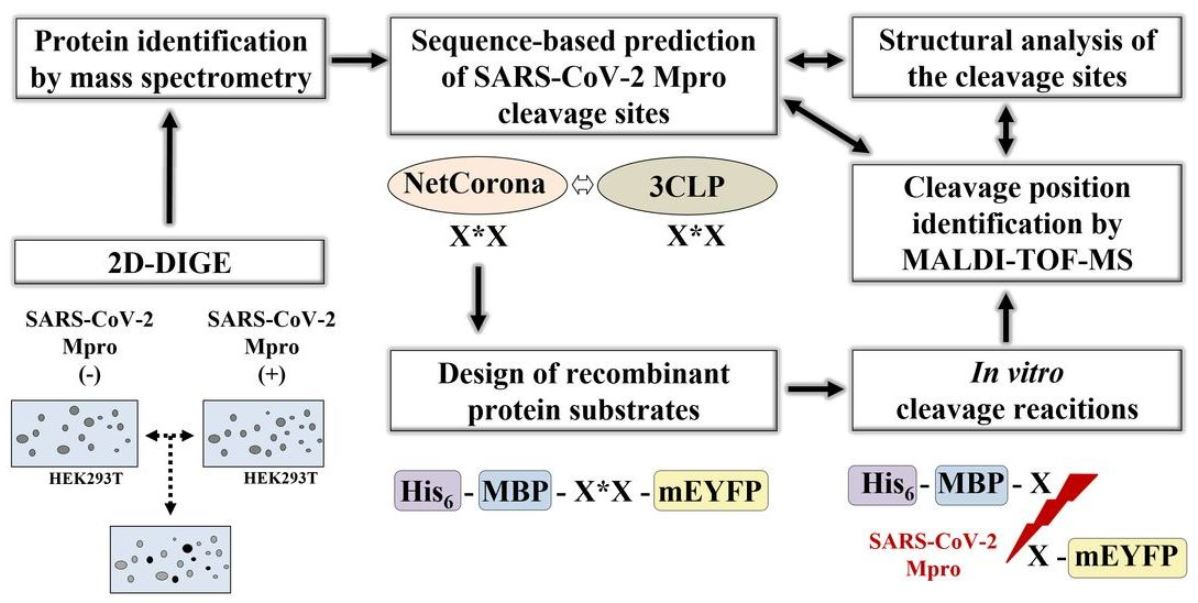
2. Results
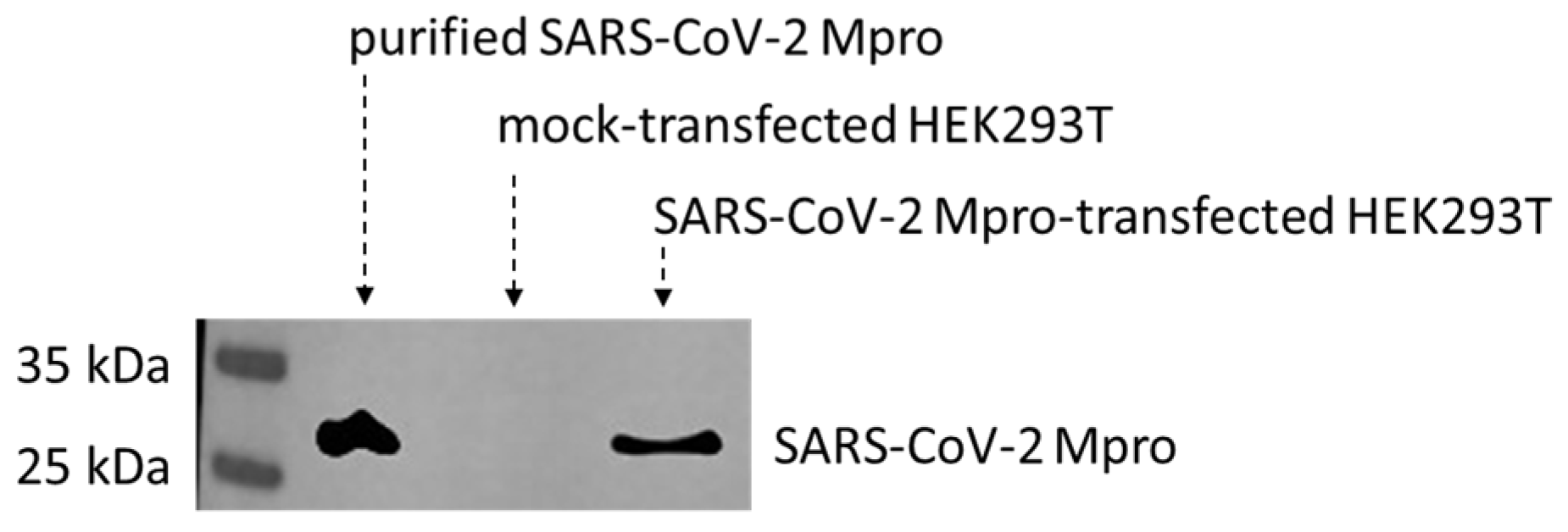
2.1. Cell Culturing and Transfection
2.2. Two-Dimensional Difference Gel Electrophoresis and Protein Identification
2.3. SARS-CoV-2 Mpro Cleavage Site Prediction
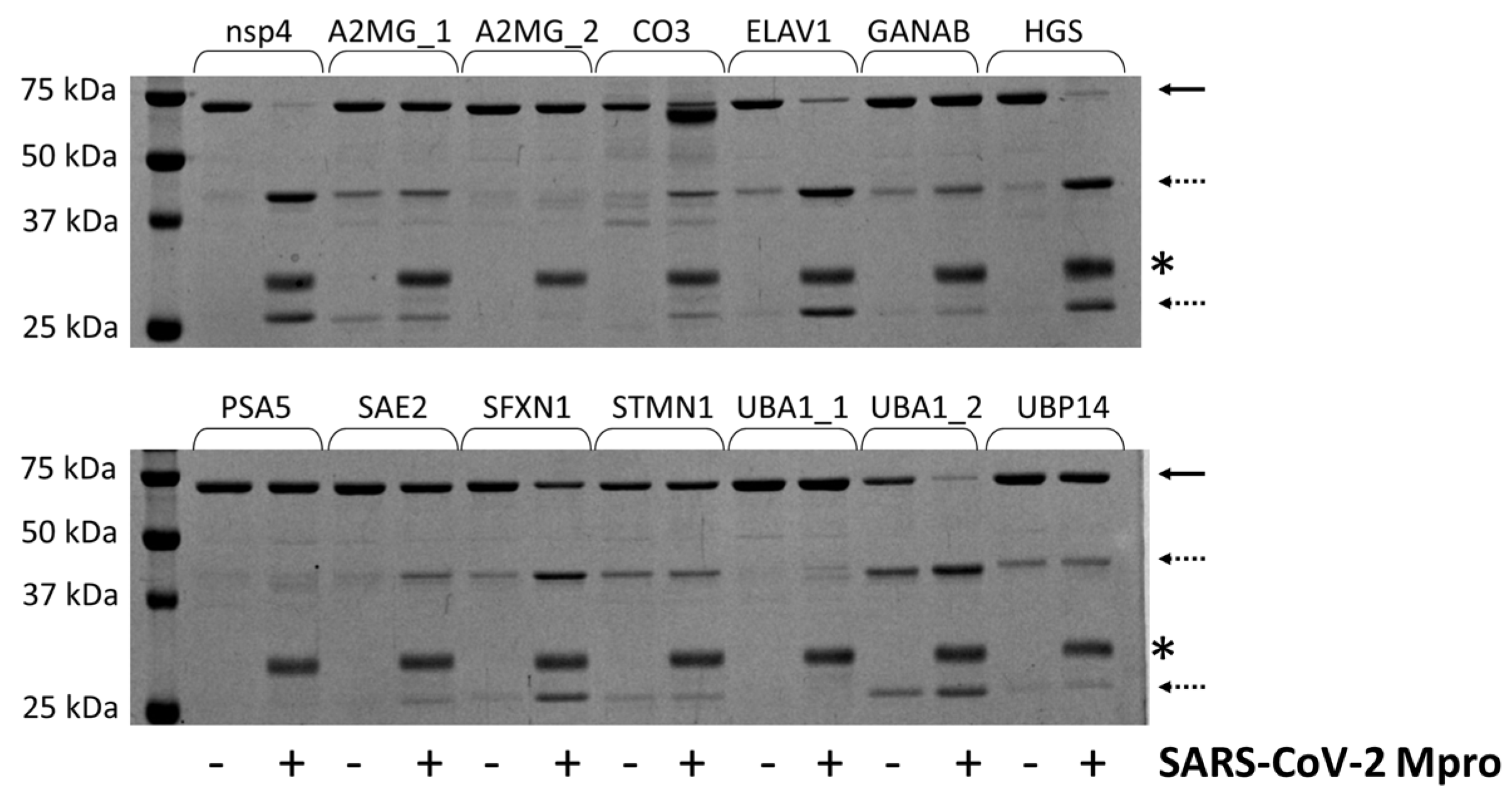
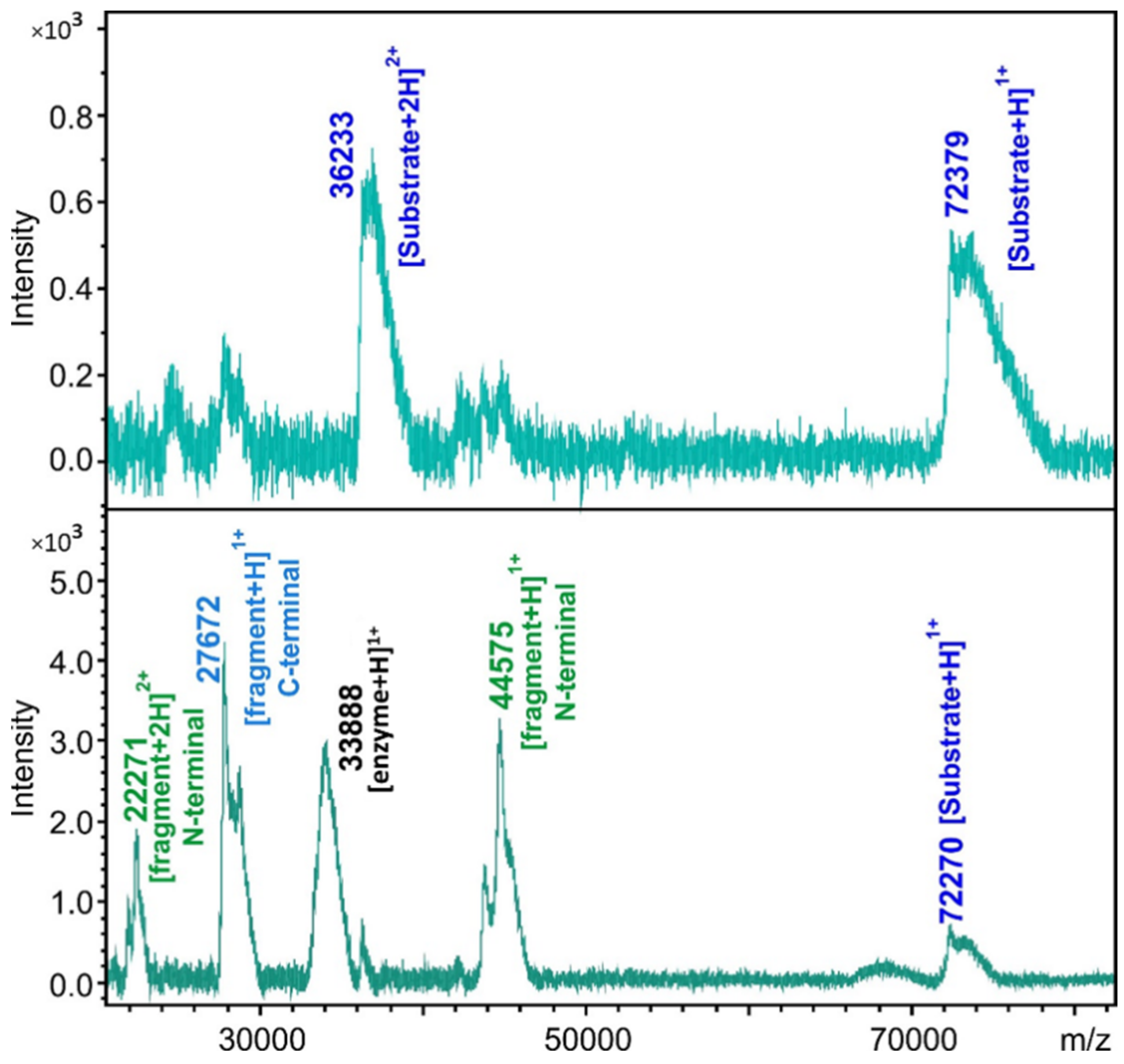
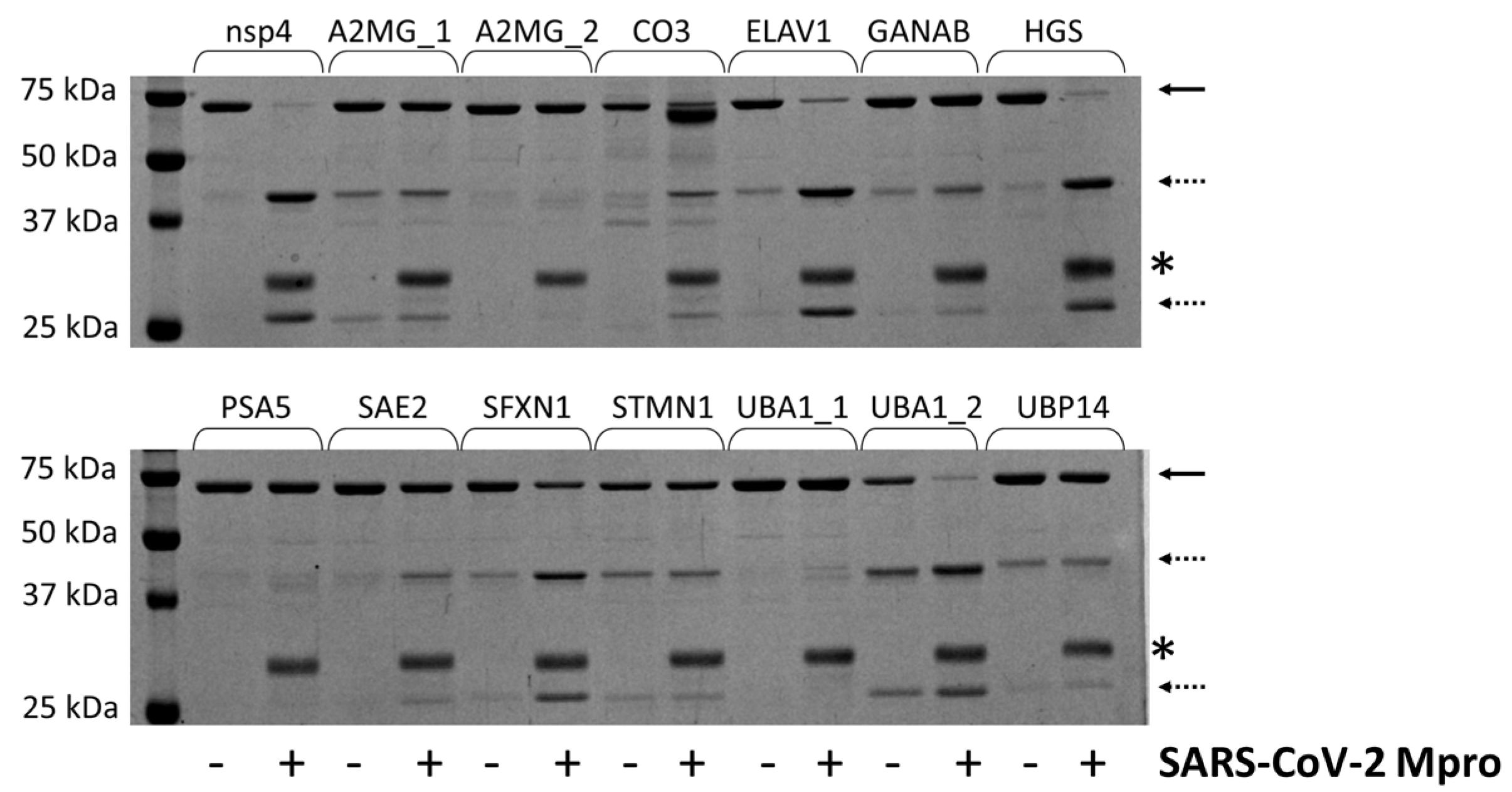
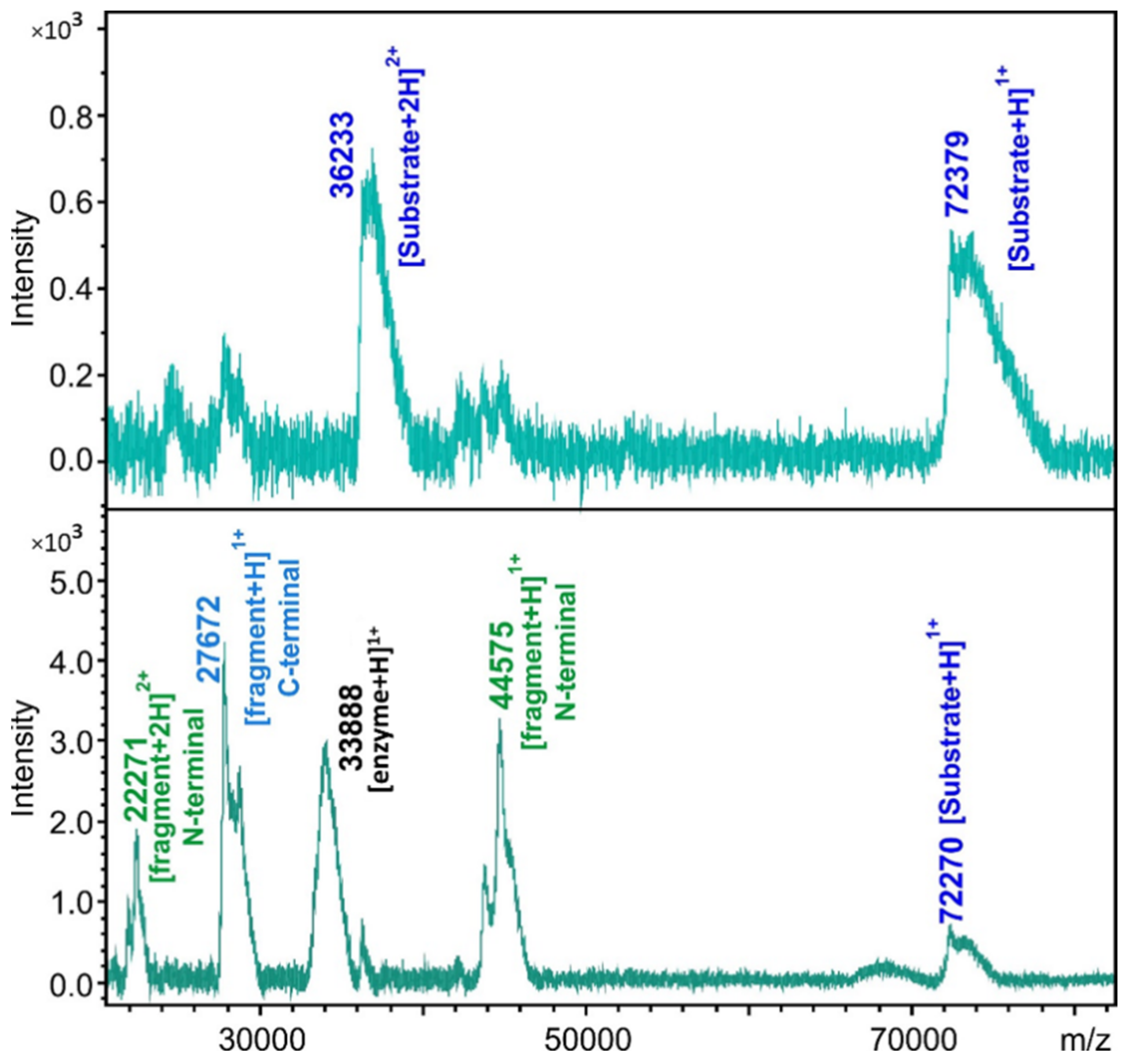
2.4. Cleavage Reactions and Cleavage Site Identification
2.5. Structural Coordinates
2.6. Comparison of the Results of NetCorona and 3CLP Predictions
3. Materials and Methods
3.1. Cell Culturing and Transfection
3.2. Sample Preparation and Protein Labelling
3.3. Two-Dimensional Difference Gel Electrophoresis (2D-DIGE)
3.4. Quantitative Analysis and Statistics
3.5. In-Gel Digestion of Proteins
3.6. Liquid Chromatography-Mass Spectrometry Analysis
3.7. Protein Identification
3.8. Western-Blot
3.9. Structural Coordinates and Cleavage Site Prediction
3.10. Preparation of Recombinant Protein Substrates
3.11. Cleavage Reactions
3.12. Cleavage Site Identification by MALDI-TOF MS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Courouble, V.V.; Dey, S.K.; Harrison, J.J.E.K.; Timm, J.; Hopkins, J.B.; Slack, R.L.; Sarafianos, S.G.; Ruiz, F.X.; Griffin, P.R.; et al. Biochemical and structural insights into SARS-CoV-2 polyprotein processing by Mpro. Sci. Adv. 2022, 8, eadd2191. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Nkeze, J.; Zhao, R.Y. Effects of HIV-1 protease on cellular functions and their potential applications in antiretroviral therapy. Cell Biosci. 2012, 2, 32. [Google Scholar] [CrossRef]
- Morazzani, E.M.; Compton, J.R.; Leary, D.H.; Berry, A.V.; Hu, X.; Marugan, J.J.; Glass, P.J.; Legler, P.M. Proteolytic cleavage of host proteins by the Group IV viral proteases of Venezuelan equine encephalitis virus and Zika virus. Antiviral Res. 2019, 164, 106–122. [Google Scholar] [CrossRef]
- Nelemans, T.; Kikkert, M. Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections. Viruses 2019, 11, 961. [Google Scholar] [CrossRef]
- Reynolds, N.D.; Aceves, N.M.; Liu, J.L.; Compton, J.R.; Leary, D.H.; Freitas, B.T.; Pegan, S.D.; Doctor, K.Z.; Wu, F.Y.; Hu, X.; et al. The SARS-CoV-2 SSHHPS Recognized by the Papain-like Protease. ACS Infect Dis. 2021, 7, 1483–1502. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, C.; Rao, Y.; Ngo, C.; Feng, J.J.; Zhao, J.; Zhang, S.; Wang, T.Y.; Carriere, J.; Savas, A.C.; et al. SARS-CoV-2 Nsp5 Demonstrates Two Distinct Mechanisms Targeting RIG-I and MAVS To Evade the Innate Immune Response. mBio 2021, 12, e0233521. [Google Scholar] [CrossRef]
- Hu, X.; Compton, J.R.; Legler, P.M. Analysis of Group IV Viral SSHHPS Using In Vitro and In Silico Methods. J. Vis. Exp. 2019, e60421. [Google Scholar] [CrossRef]
- Miczi, M.; Golda, M.; Kunkli, B.; Nagy, T.; Tőzsér, J.; Mótyán, J.A. Identification of Host Cellular Protein Substrates of SARS-CoV-2 Main Protease. Int. J. Mol. Sci. 2020, 21, 9523. [Google Scholar] [CrossRef] [PubMed]
- Kiemer, L.; Lund, O.; Brunak, S.; Blom, N. Coronavirus 3CLpro proteinase cleavage sites: Possible relevance to SARS virus pathology. BMC Bioinform. 2004, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhu, Z.; Qiu, Y.; Ge, X.; Zheng, H.; Peng, Y. Prediction of coronavirus 3C-like protease cleavage sites using machine-learning algorithms. Virol. Sin. 2022, 37, 437–444. [Google Scholar] [CrossRef]
- Moustaqil, M.; Ollivier, E.; Chiu, H.P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.N.; Jacques, D.; et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg. Microbes Infect. 2021, 10, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, J.; Cheng, G. Protease cleavage of RNF20 facilitates coronavirus replication via stabilization of SREBP1. Proc. Natl. Acad. Sci. USA 2021, 118, e2107108118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, S.; Xu, Q.; Li, H.; Lu, K. Cleavage of the selective autophagy receptor SQSTM1/p62 by the SARS-CoV-2 main protease NSP5 prevents the autophagic degradation of viral membrane proteins. Mol. Biomed. 2022, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Chiaravalli, J.; Gellenoncourt, S.; Brownridge, P.; Bryne, D.P.; Daly, L.A.; Grauslys, A.; Walter, M.; Agou, F.; Chakrabarti, L.A.; et al. Characterising proteolysis during SARS-CoV-2 infection identifies viral cleavage sites and cellular targets with therapeutic potential. Nat. Commun. 2021, 12, 5553. [Google Scholar] [CrossRef]
- Koudelka, T.; Boger, J.; Henkel, A.; Schönherr, R.; Krantz, S.; Fuchs, S.; Rodríguez, E.; Redecke, L.; Tholey, A. N-Terminomics for the Identification of In Vitro Substrates and Cleavage Site Specificity of the SARS-CoV-2 Main Protease. Proteomics 2021, 21, e2000246. [Google Scholar] [CrossRef]
- Pablos, I.; Machado, Y.; de Jesus, H.C.R.; Mohamud, Y.; Kappelhoff, R.; Lindskog, C.; Vlok, M.; Bell, P.A.; Butler, G.S.; Grin, P.M.; et al. Mechanistic insights into COVID-19 by global analysis of the SARS-CoV-2 3CLpro substrate degradome. Cell Rep. 2021, 37, 109892. [Google Scholar] [CrossRef]
- Minden, J.S. Two-Dimensional Difference Gel Electrophoresis (2D DIGE). Methods Cell Biol. 2012, 112, 111–141. [Google Scholar]
- Marrero, A.; Duquerroy, S.; Trapani, S.; Goulas, T.; Guevara, T.; Andersen, G.R.; Navaza, J.; Sottrup-Jensen, L.; Gomis-Rüth, F.X. The crystal structure of human α2-macroglobulin reveals a unique molecular cage. Angew. Chem. Int. Ed. Engl. 2012, 51, 3340–3344. [Google Scholar] [CrossRef] [PubMed]
- Forneris, F.; Wu, J.; Xue, X.; Ricklin, D.; Lin, Z.; Sfyroera, G.; Tzekou, A.; Volokhina, E.; Granneman, J.C.; Hauhart, R.; et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode. EMBO J. 2016, 35, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zeng, F.; Liu, Q.; Liu, H.; Liu, Z.; Niu, L.; Teng, M.; Li, X. The structure of the ARE-binding domains of Hu antigen R (HuR) undergoes conformational changes during RNA binding. Acta Crystallogr. D Biol. Crystallogr. 2013, 69 Pt 3, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Harshbarger, W.; Miller, C.; Diedrich, C.; Sacchettini, J. Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 2015, 23, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Lois, L.M.; Lima, C.D. Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. EMBO J. 2005, 24, 439–451. [Google Scholar] [CrossRef]
- Lv, Z.; Williams, K.M.; Yuan, L.; Atkison, J.H.; Olsen, S.K. Crystal structure of a human ubiquitin E1-ubiquitin complex reveals conserved functional elements essential for activity. J. Biol. Chem. 2018, 293, 18337–18352. [Google Scholar] [CrossRef]
- Bozóki, B.; Gazda, L.; Tóth, F.; Miczi, M.; Mótyán, J.A.; Tőzsér, J. A recombinant fusion protein-based, fluorescent protease assay for high throughput-compatible substrate screening. Anal. Biochem. 2018, 540, 52–63. [Google Scholar] [CrossRef]
- Mótyán, J.A.; Miczi, M.; Bozóki, B.; Tőzsér, J. Data supporting Ni-NTA magnetic bead-based fluorescent protease assay using recombinant fusion prostein substrates. Data Brief. 2018, 18, 203–208. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, M.; Mótyán, J.A.; Szojka, Z.I.; Golda, M.; Miczi, M.; Tőzsér, J. Analysis of the efficacy of HIV protease inhibitors against SARS-CoV-2’s main protease. Virol. J. 2020, 17, 190. [Google Scholar] [CrossRef] [PubMed]
- Márkus, B.; Szabó, K.; Pfliegler, W.P.; Petrényi, K.; Boros, E.; Pócsi, I.; Tőzsér, J.; Csősz, É.; Dombrádi, V. Proteomic analysis of protein phosphatase Z1 from Candida albicans. PLoS ONE 2017, 12, e0183176. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Strub, J.-M.; Luche, S.; Dorsselaer, A.; van Lunardi, J. A comparison between Sypro Ruby and ruthenium II tris (bathophenanthroline disulfonate) as fluorescent stains for protein detection in gels. Proteomics 2001, 1, 699–704. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and pro-teome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Bozóki, B.; Mótyán, J.A.; Miczi, M.; Gazda, L.D.; Tőzsér, J. Use of Recombinant Fusion Proteins in a Fluorescent Protease Assay Platform and Their In-gel Renaturation. J. Vis. Exp. 2019, 16, e58824. [Google Scholar]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat. Commun. 2021, 12, 250645. [Google Scholar] [CrossRef]
- Chuck, C.P.; Chong, L.T.; Chen, C.; Chow, H.F.; Wan, D.C.; Wong, K.B. Profiling of substrate specificity of SARS-CoV 3CLpro. PLoS ONE 2010, 5, e13197. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.M.; Ramanathan, M.; Shanderson, R.L.; Beck, A.; Donohue, L.; Ferguson, I.; Guo, M.G.; Rao, D.S.; Miao, W.; Reynolds, D.; et al. The proximal proteome of 17 SARS-CoV-2 proteins links to disrupted antiviral signaling and host translation. PLoS Pathog. 2021, 17, e1009412. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.W.; Radding, W. Understanding Selenium and Glutathione as Antiviral Factors in COVID-19: Does the Viral Mpro Protease Target Host Selenoproteins and Glutathione Synthesis? Front. Nutr. 2020, 7, 143. [Google Scholar] [CrossRef]
- Scott, B.M.; Lacasse, V.; Blom, D.G.; Tonner, P.D.; Blom, N.S. Predicted coronavirus Nsp5 protease cleavage sites in the human proteome. BMC Genom. Data. 2022, 23, 25. [Google Scholar] [CrossRef] [PubMed]
- Goetz, D.H.; Choe, Y.; Hansell, E.; Chen, Y.T.; McDowell, M.; Jonsson, C.B.; Roush, W.R.; McKerrow, J.; Craik, C.S. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry 2007, 46, 8744–8752. [Google Scholar] [CrossRef] [PubMed]
- Chuck, C.P.; Chow, H.F.; Wan, D.C.; Wong, K.B. Profiling of substrate specificities of 3C-like proteases from group 1, 2a, 2b, and 3 coronaviruses. PLoS ONE 2011, 6, e27228. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Mahajan, R.; Delphin, C.; Guan, T.; Gerace, L.; Melchior, F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 1997, 88, 97–107. [Google Scholar] [CrossRef]
- Lowrey, A.J.; Cramblet, W.; Bentz, G.L. Viral manipulation of the cellular sumoylation machinery. Cell Commun. Signal 2017, 15, 2750. [Google Scholar] [CrossRef]
- Imbert, F.; Langford, D. Viruses, SUMO, and immunity: The interplay between viruses and the host SUMOylation system. J. Neurovirol. 2021, 27, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qiao, J.; You, Q.; Zong, S.; Peng, Q.; Liu, Y.; Hu, S.; Liu, W.; Li, S.; Shu, X.; et al. SARS-CoV-2 Nsp5 Activates NF-κB Pathway by Upregulating SUMOylation of MAVS. Front. Immunol. 2021, 12, 750969. [Google Scholar] [CrossRef] [PubMed]
- Colombo, R.; Boggio, R.; Seiser, C.; Draetta, G.F.; Chiocca, S. The adenovirus protein Gam1 interferes with sumoylation of histone deacetylase 1. EMBO Rep. 2002, 3, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio 2018, 9, e01753-18. [Google Scholar] [CrossRef]
- Sinkovits, G.; Mező, B.; Réti, M.; Müller, V.; Iványi, Z.; Gál, J.; Gopcsa, L.; Reményi, P.; Szathmáry, B.; Lakatos, B.; et al. Complement Overactivation and Consumption Predicts In-Hospital Mortality in SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 663187. [Google Scholar] [CrossRef]
- Zinellu, A.; Mangoni, A.A. Serum Complement C3 and C4 and COVID-19 Severity and Mortality: A Systematic Review and Meta-Analysis With Meta-Regression. Front. Immunol. 2021, 12, 696085. [Google Scholar] [CrossRef]
- Yu, J.; Gerber, G.F.; Chen, H.; Yuan, X.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Complement dysregulation is associated with severe COVID-19 illness. Haematologica 2022, 107, 1095–1105. [Google Scholar] [CrossRef]
- Afzali, B.; Noris, M.; Lambrecht, B.N.; Kemper, C. The state of complement in COVID-19. Nat. Rev. Immunol. 2022, 22, 77–84. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef]
- Mintoo, M.; Chakravarty, A.; Tilvawala, R. N-Terminomics Strategies for Protease Substrates Profiling. Molecules 2021, 26, 4699. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | NetCorona Prediction Output | 3CLP Prediction Output | ||||||
|---|---|---|---|---|---|---|---|---|
| Spot | (UniProt ID; Name) | Cleavage Position | Cleavage | Score | Cleavage Site | Cleavage | Score | Cleavage Site |
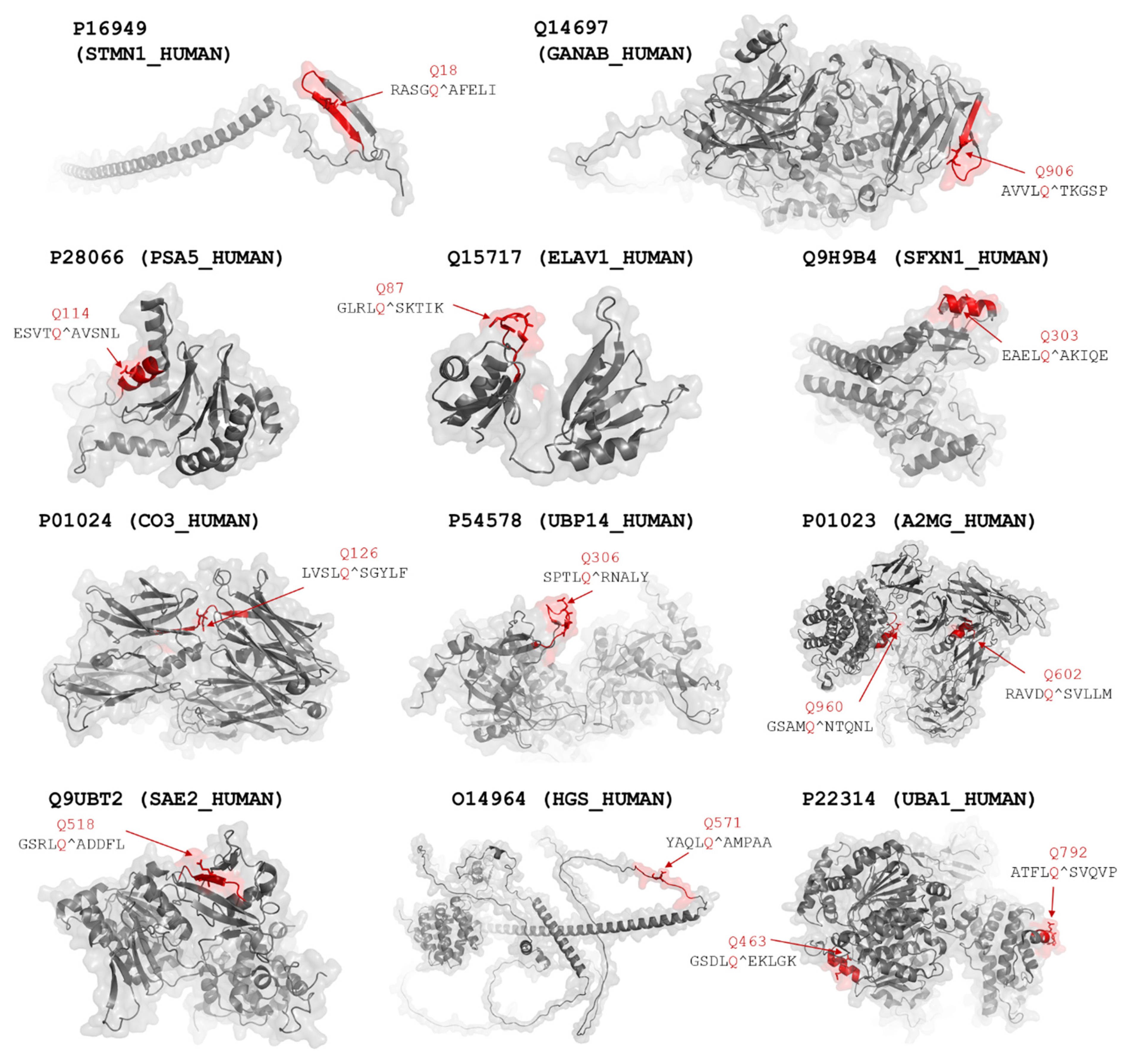
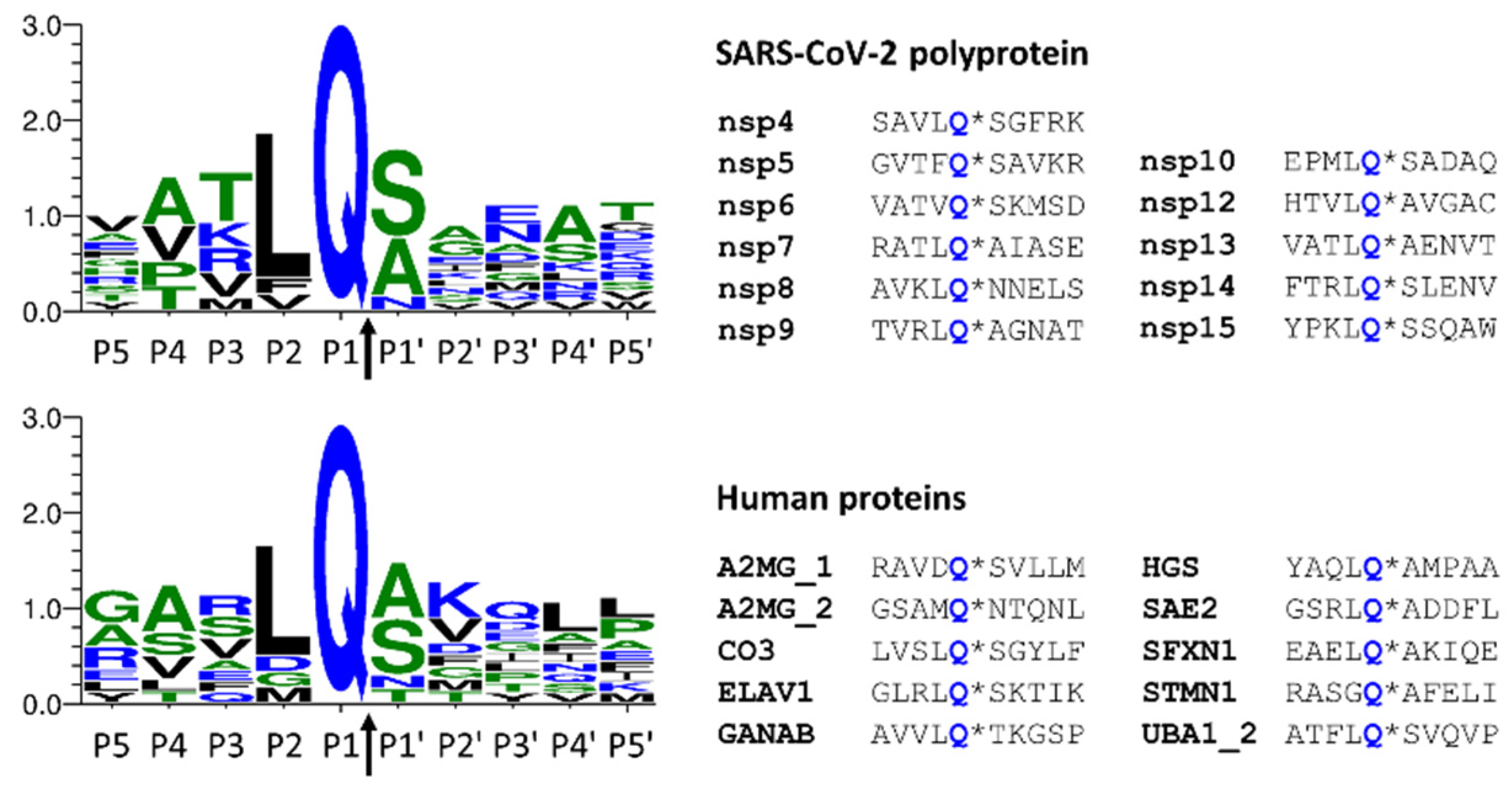
| 3, 4 | P11021; BIP | Q401 | No | 0.276 | - | Yes | 0.95 | AAVQ*AG |
| 9, 10 | P09651; ROA1 | - | No | - | - | No | - | - |
| 2 | P55072; TERA | Q421 | No | 0.368 | - | Yes | 0.98 | AALQ*AI |
| 11 | P18669; PGAM1 | - | No | - | - | No | - | - |
| 1 | Q14697; GANAB | Q92 | No | 0.142 | - | Yes | 0.79 | LELQ*GL |
| Q343 # | No | 0.143 | - | No | 0.20 | - | ||
| Q780 | No | 0.122 | - | Yes | 0.66 | YDIQ*SY | ||
| Q906 | Yes | 0.574 | AVVLQ*TKGSP | No | 0.46 | - | ||
| 15 | P16949; STMN1 | Q18 # | Yes | 0.546 | RASGQ*AFELI | No | 0.29 | - |
| 9, 10 | Q9H9B4; SFXN1 | Q303 | Yes | 0.772 | EAELQ*AKIQE | Yes | 0.98 | AELQ*AK |
| 9, 10 | Q15717; ELAV1 | Q87 # | Yes | 0.888 | GLRLQ*SKTIK | Yes | 0.95 | LRLQ*SK |
| 9, 10 | Q99623; PHB2 | - | No | - | - | No | - | - |
| 1 | O95757; HS74L | Q470 | No | 0.150 | - | Yes | 0.66 | VFPQ*SD |
| Q585 | No | 0.102 | - | Yes | 0.67 | LPIQ*SS | ||
| 12 | P28066; PSA5 | Q114 | Yes | 0.509 | ESVTQ*AVSNL | No | 0.26 | - |
| Q164 | No | 0.184 | - | Yes | 0.69 | TFVQ*CD | ||
| 3 | P01024; CO3 | Q109 | No | 0.292 | - | Yes | 1.00 | VTVQ*AT |
| Q126 | Yes | 0.724 | LVSLQ*SGYLF | Yes | 1.00 | VSLQ*SG | ||
| Q392 | No | 0.157 | - | Yes | 0.97 | VAVQ*GE | ||
| Q398 | No | 0.283 | - | Yes | 0.75 | DTVQ*SL | ||
| Q661 | No | 0.171 | - | Yes | 0.72 | AELQ*CP | ||
| Q1277 | No | 0.124 | - | Yes | 0.56 | MVFQ*AL | ||
| Q1299 | No | 0.112 | - | Yes | 0.57 | VSLQ*LP | ||
| Q1553 | No | 0.076 | - | Yes | 0.67 | VKVQ*LS | ||
| 1 | P19338; NUCL | Q558 | No | 0.095 | - | Yes | 0.80 | LELQ*GP |
| 7 | P05455; LA | Q358 | No | 0.462 | - | Yes | 0.97 | VQFQ*GK |
| 5 | P54578; UBP14 | Q40 | No | 0.083 | - | Yes | 0.67 | TGVQ*PA |
| Q121 | No | 0.106 | - | Yes | 0.69 | ATVQ*CI | ||
| Q306 | Yes | 0.541 | SPTLQ*RNALY | No | 0.35 | - | ||
| Q421 | No | 0.424 | Yes | 0.71 | YDLQ*AV | |||
| 3 | P13667; PDIA4 | Q377 | No | 0.111 | - | Yes | 0.64 | MDVQ*GS |
| 3 | Q9NY33; DPP3 | - | No | - | - | No | - | - |
| 1 | P36776; LONM | - | No | - | - | No | - | - |
| 14 | P30048; PRDX3 | - | No | - | - | No | - | - |
| 3 | P00738; HPT | - | No | - | - | No | - | - |
| 3 | P01023; A2MG | Q592 | No | 0.230 | - | Yes | 0.62 | AAPQ*SV |
| Q602 | Yes | 0.550 | RAVDQ*SVLLM | No | 0.16 | - | ||
| Q827 | No | 0.152 | - | Yes | 0.54 | VSVQ*LE | ||
| Q960 | Yes | 0.641 | GSAMQ*NTQNL | Yes | 0.76 | SAMQ*NT | ||
| Q1281 | No | 0.386 | - | Yes | 0.93 | VTIQ*SS | ||
| 13 | P15374; UCHL3 | - | No | - | - | No | - | - |
| 2 | Q9UBT2; SAE2 | Q82 # | No | 0.302 | - | No | 0.28 | - |
| Q288 | No | 0.310 | - | Yes | 0.97 | AEVQ*SQ | ||
| Q518 | Yes | 0.920 | GSRLQ*ADDFL | Yes | 0.98 | SRLQ*AD | ||
| 1 | O14964; HGS | Q444 | No | 0.192 | - | Yes | 0.75 | SLFQ*SI |
| Q571 | Yes | 0.721 | YAQLQ*AMPAA | Yes | 0.88 | AQLQ*AM | ||
| Q649 | No | 0.109 | - | Yes | 0.61 | AAPQ*AQ | ||
| 3 | P10909; CLUS | - | No | - | - | No | - | - |
| 1, 7 | P22314; UBA1 | Q254 | No | 0.097 | - | Yes | 0.89 | SEVQ*GM |
| Q334 | No | 0.138 | - | Yes | 0.55 | IGFQ*AL | ||
| Q463 | Yes | 0.506 | GSDLQ*EKLGK | No | 0.11 | - | ||
| Q792 # | Yes | 0.796 | ATFLQ*SVQVP | Yes | 0.97 | TFLQ*SV | ||
| Q815 | No | 0.322 | - | Yes | 0.57 | QELQ*SA | ||
| Q961 | No | 0.110 | - | Yes | 0.60 | FEVQ*GL | ||
| Spot Number | Protein (UniProt ID and Name) | Cleavage Site | Calculated Mw (kDa) | NetCorona | In Vitro Cleavage ** | Coordinate File | |||
|---|---|---|---|---|---|---|---|---|---|
| Sequence | Identified as Substrate/Cleavage Site * | Full- Length Protein | Cleavage Products | Cleavage Probability | PDB ID | Ref. | |||
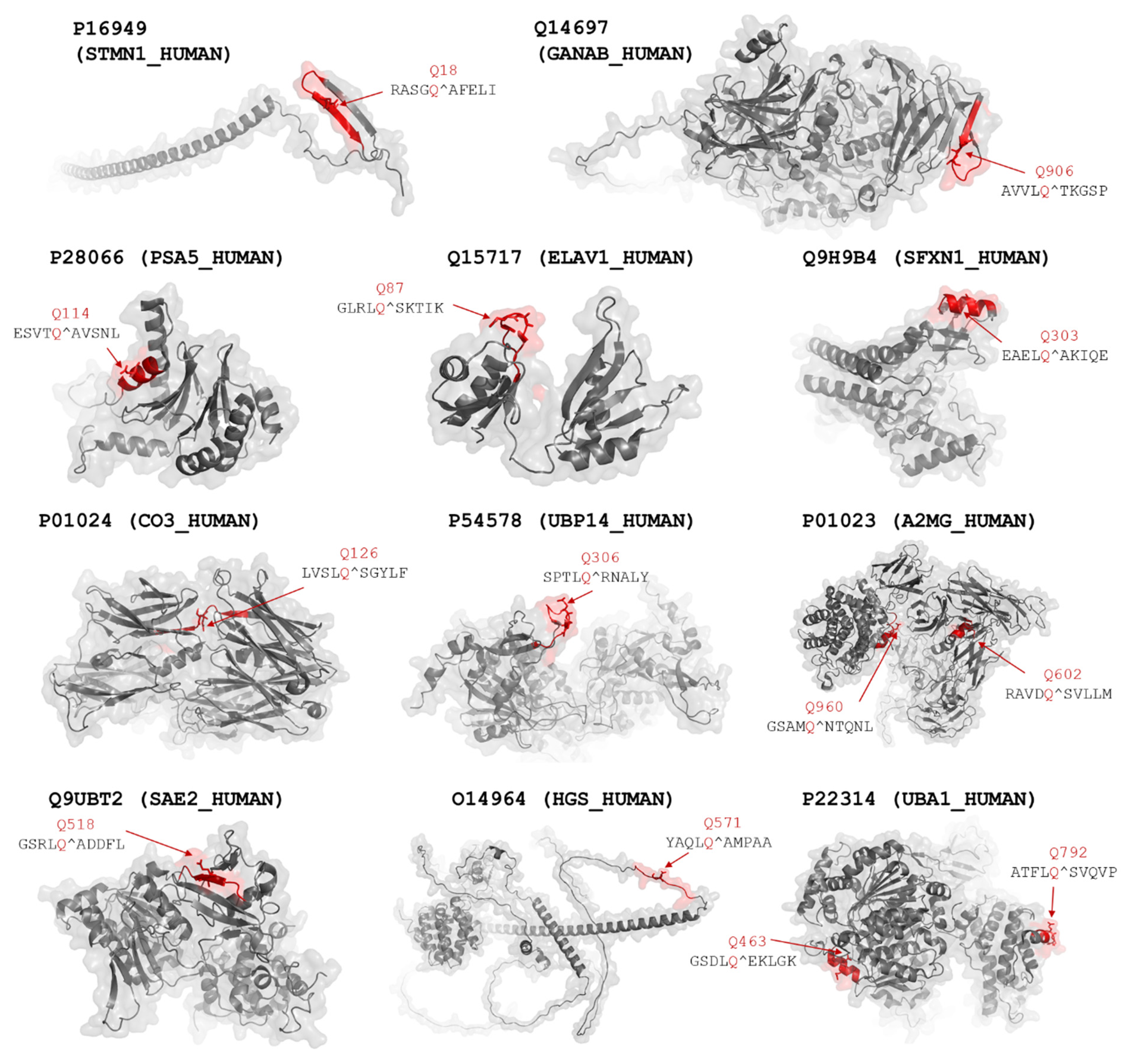
| 3 | P01023; A2MG | RAVDQ*SVLLM | No/No | 163.3 | 67.0/96.3 | + | Yes | 4ACQ | [22] |
| GSAMQ*NTQNL | No/No | 106.1/57.2 | ++ | Yes | |||||
| P01024; CO3 | LVSLQ*SGYLF | No/No | 187.2 | 13.6/173.6 | +++ | Yes | 5FOA | [23] | |
| 9, 10 | Q15717; ELAV1 | GLRLQ*SKTIK | Yes/Yes | 36.1 | 9.7/26.4 | ++++ | Yes | 4ED5 | [24] |
| 1 | Q14697; GANAB | AVVLQ*TKGSP | Yes/No | 106.9 | 102.6/4.3 | + | Yes | AF | [25] |
| O14964; HGS | YAQLQ*AMPAA | No/No | 86.2 | 65.2/21.0 | +++ | Yes | AF | [25] | |
| 12 | P28066; PSA5 | ESVTQ*AVSNL | No/No | 26.4 | 12.6/13.8 | + | No | 4R3O | [26] |
| 2 | Q9UBT2; SAE2 | GSRLQ*ADDFL | Yes/No | 71.2 | 57.7/13.5 | +++++ | Yes | 1Y8Q | [27] |
| 9, 10 | Q9H9B4; SFXN1 | EAELQ*AKIQE | No/No | 35.6 | 33.3/2.3 | +++ | Yes | AF | [25] |
| 15 | P16949; STMN1 | RASGQ*AFELI | No/Yes | 17.3 | 2.0/15.3 | + | Yes | AF | [25] |
| 1, 7 | P22314; UBA1 | GSDLQ*EKLGK | No/No | 117.9 | - | + | No | 6DC6 | [28] |
| ATFLQ*SVQVP | Yes/Yes | 87.6/30.3 | +++ | Yes | |||||
| 5 | P54578; UBP14 | SPTLQ*RNALY | No/No | 56.1 | - | + | No | 2AYN | [11] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miltner, N.; Kalló, G.; Csősz, É.; Miczi, M.; Nagy, T.; Mahdi, M.; Mótyán, J.A.; Tőzsér, J. Identification of SARS-CoV-2 Main Protease (Mpro) Cleavage Sites Using Two-Dimensional Electrophoresis and In Silico Cleavage Site Prediction. Int. J. Mol. Sci. 2023, 24, 3236. https://doi.org/10.3390/ijms24043236
Miltner N, Kalló G, Csősz É, Miczi M, Nagy T, Mahdi M, Mótyán JA, Tőzsér J. Identification of SARS-CoV-2 Main Protease (Mpro) Cleavage Sites Using Two-Dimensional Electrophoresis and In Silico Cleavage Site Prediction. International Journal of Molecular Sciences. 2023; 24(4):3236. https://doi.org/10.3390/ijms24043236
Chicago/Turabian StyleMiltner, Noémi, Gergő Kalló, Éva Csősz, Márió Miczi, Tibor Nagy, Mohamed Mahdi, János András Mótyán, and József Tőzsér. 2023. "Identification of SARS-CoV-2 Main Protease (Mpro) Cleavage Sites Using Two-Dimensional Electrophoresis and In Silico Cleavage Site Prediction" International Journal of Molecular Sciences 24, no. 4: 3236. https://doi.org/10.3390/ijms24043236
APA StyleMiltner, N., Kalló, G., Csősz, É., Miczi, M., Nagy, T., Mahdi, M., Mótyán, J. A., & Tőzsér, J. (2023). Identification of SARS-CoV-2 Main Protease (Mpro) Cleavage Sites Using Two-Dimensional Electrophoresis and In Silico Cleavage Site Prediction. International Journal of Molecular Sciences, 24(4), 3236. https://doi.org/10.3390/ijms24043236