
BTEAC Catalyzed Ultrasonic-Assisted Synthesis of Bromobenzofuran-Oxadiazoles: Unravelling Anti-HepG-2 Cancer Therapeutic Potential through In Vitro and In Silico Studies

 , ,
, ,  ,
,  ,
,  , and
, and
Abstract
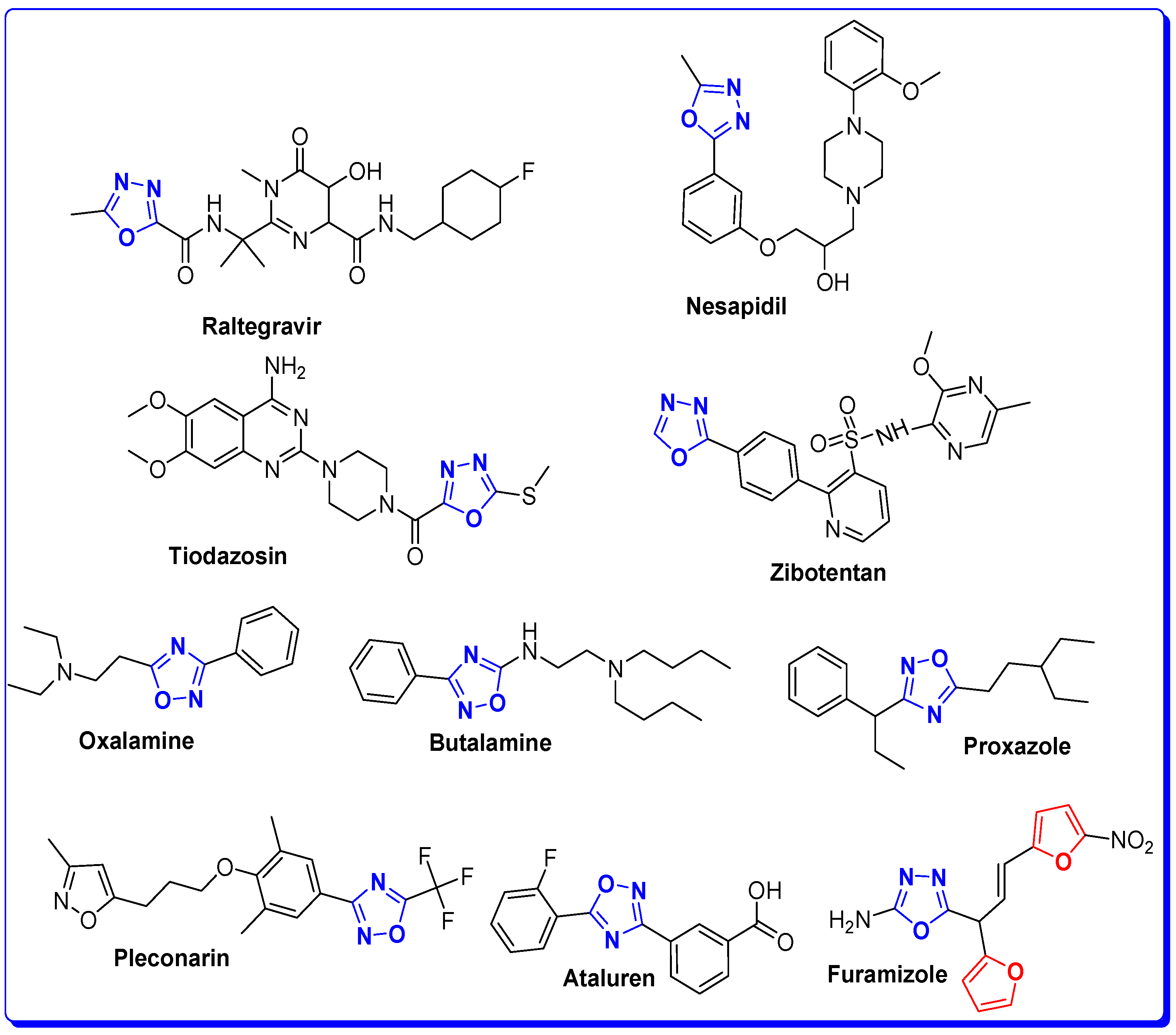
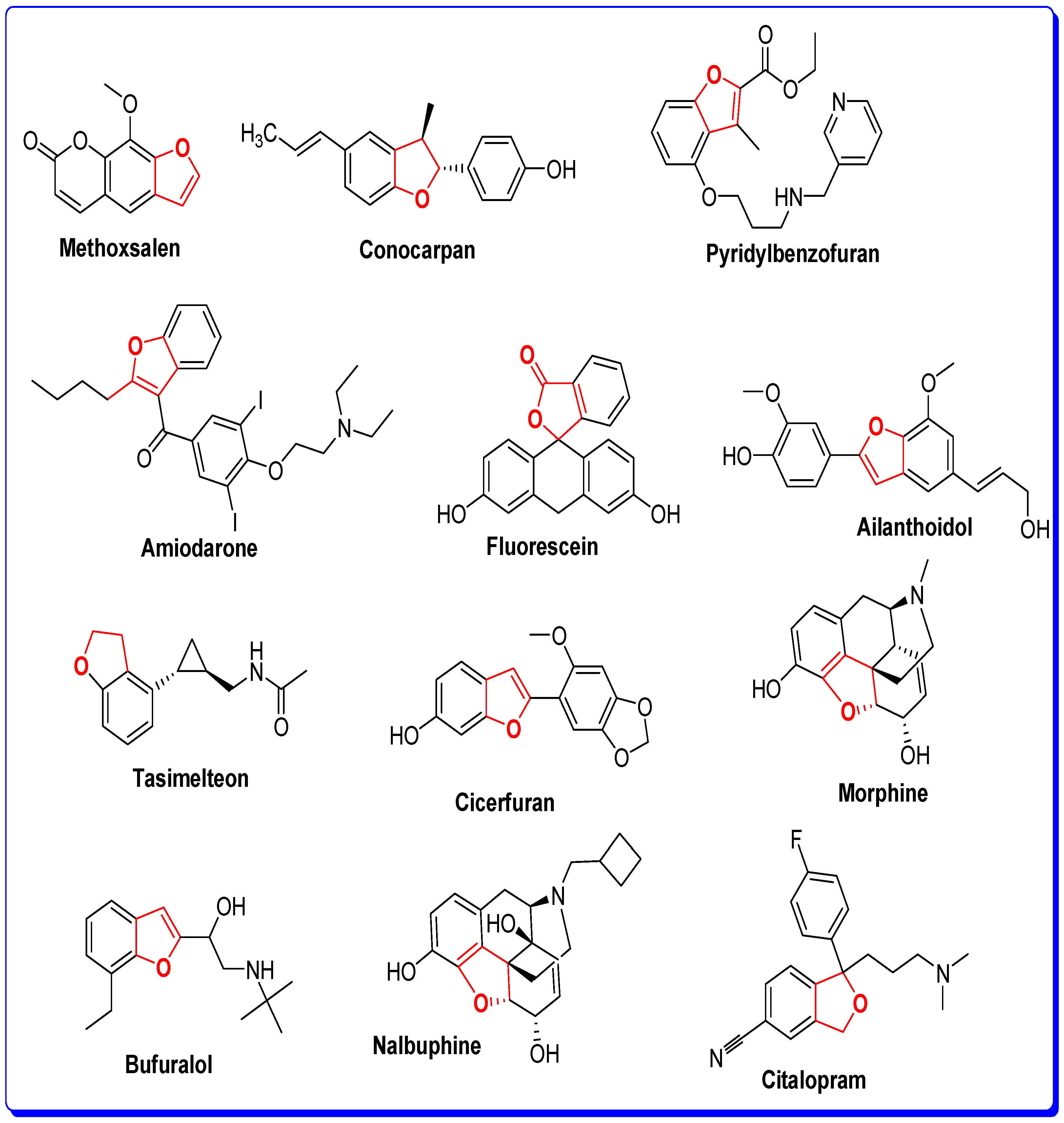
1. Introduction
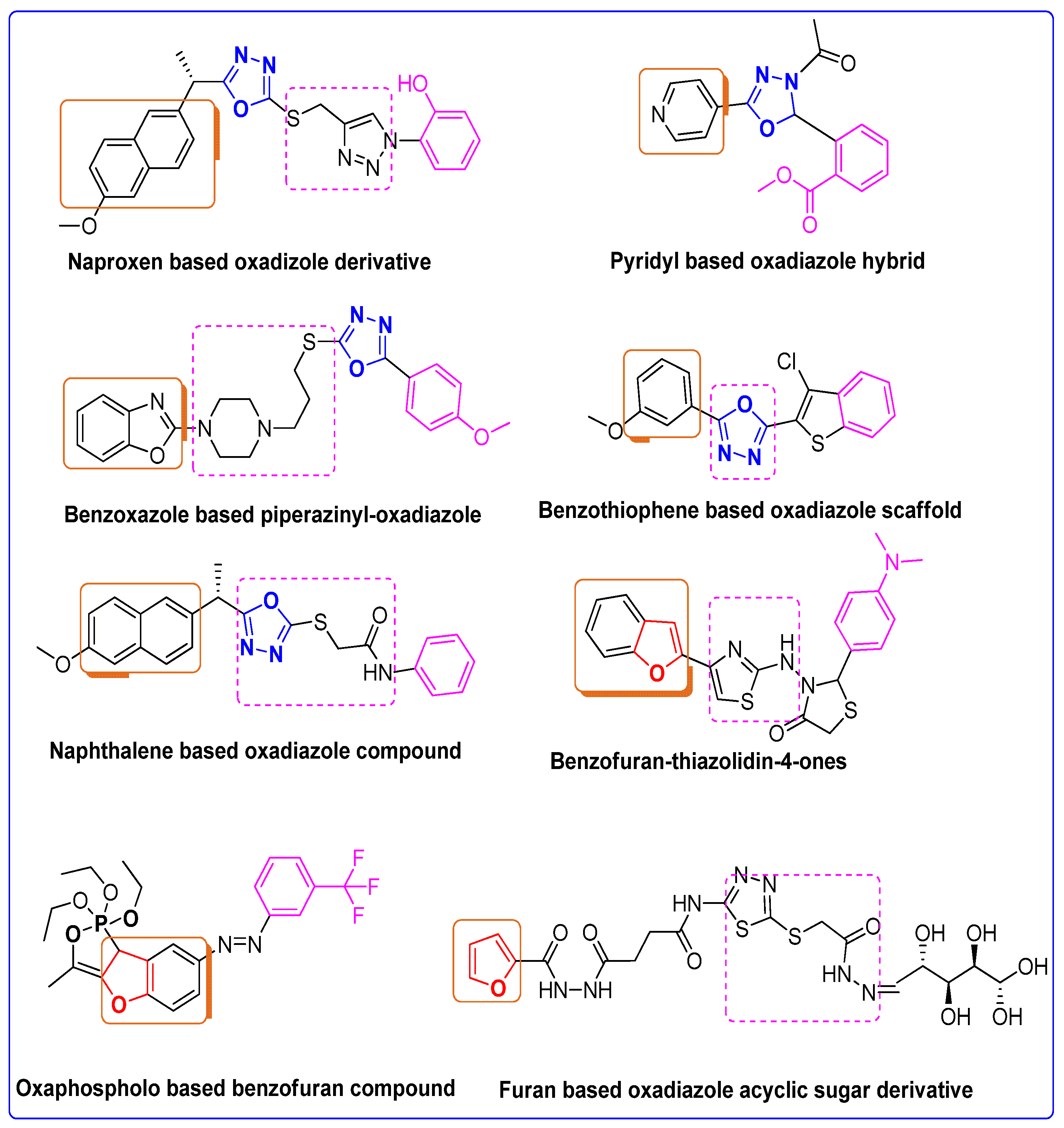
The Rationale of Molecular Design of Bromobenzofuran-oxadiazoles BF1-9 against HepG2 Cancer Cell Line
2. Materials and Methods
2.1. Materials for the Synthesis Bromobenzofuran-Oxadiazole Derivatives BF1-9
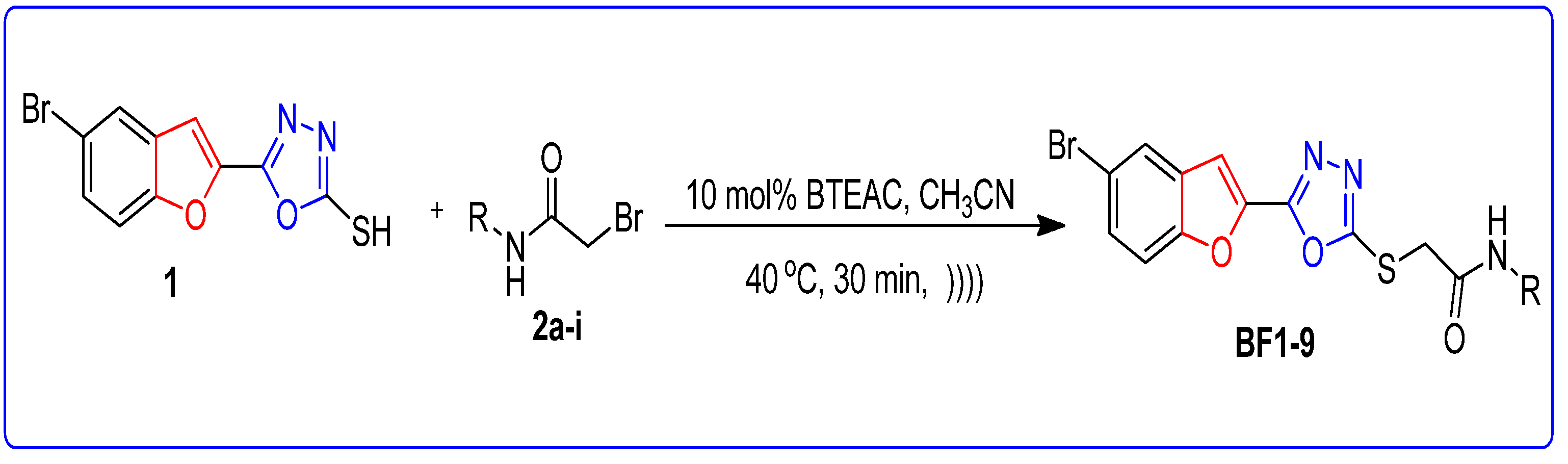
2.2. BTEAC Catalyzed Synthesis of Bromobenzofuran-Oxadiazole Structural Hybrids BF1-9 by Ultrasonic Irradiated Synthetic Approach
2.3. Anti-hepatocellular Carcinoma MTT Assay
2.4. Computational Approach of Bromobenzofuran-Oxadiazoles BF1-9
2.4.1. Retrieval of EGFR, PI3K, mTOR, AKT, Tubulin Polymerization, and GSK-3β Protein PDB Structures
2.4.2. Designing of Ligands and Molecular Docking of Bromobenzofuran-Oxadiazoles BF1-9
2.4.3. ADMET and Drug-Likeness Studies of Bromobenzofuran-Oxadiazoles BF1-9
2.4.4. Molecular Dynamic Simulation of Bromobenzofuran-oxadiazoles BF-2, BF-5, and BF-6
2.4.5. DFT Studies of Bromobenzofuran-Oxadiazoles BF-2, BF-5, and BF-6
2.5. Statistical Data
3. Results and Discussion
3.1. Chemistry
Synthesis of Bromobenzofuran-Oxadiazole Structural Hybrids BF1-9
3.2. Biological Evaluation of Bromobenzofuran-Oxadiazoles
3.2.1. Anti-Hepatocellular Carcinoma Activity of Bromobenzofuran-Oxadiazoles BF1-9
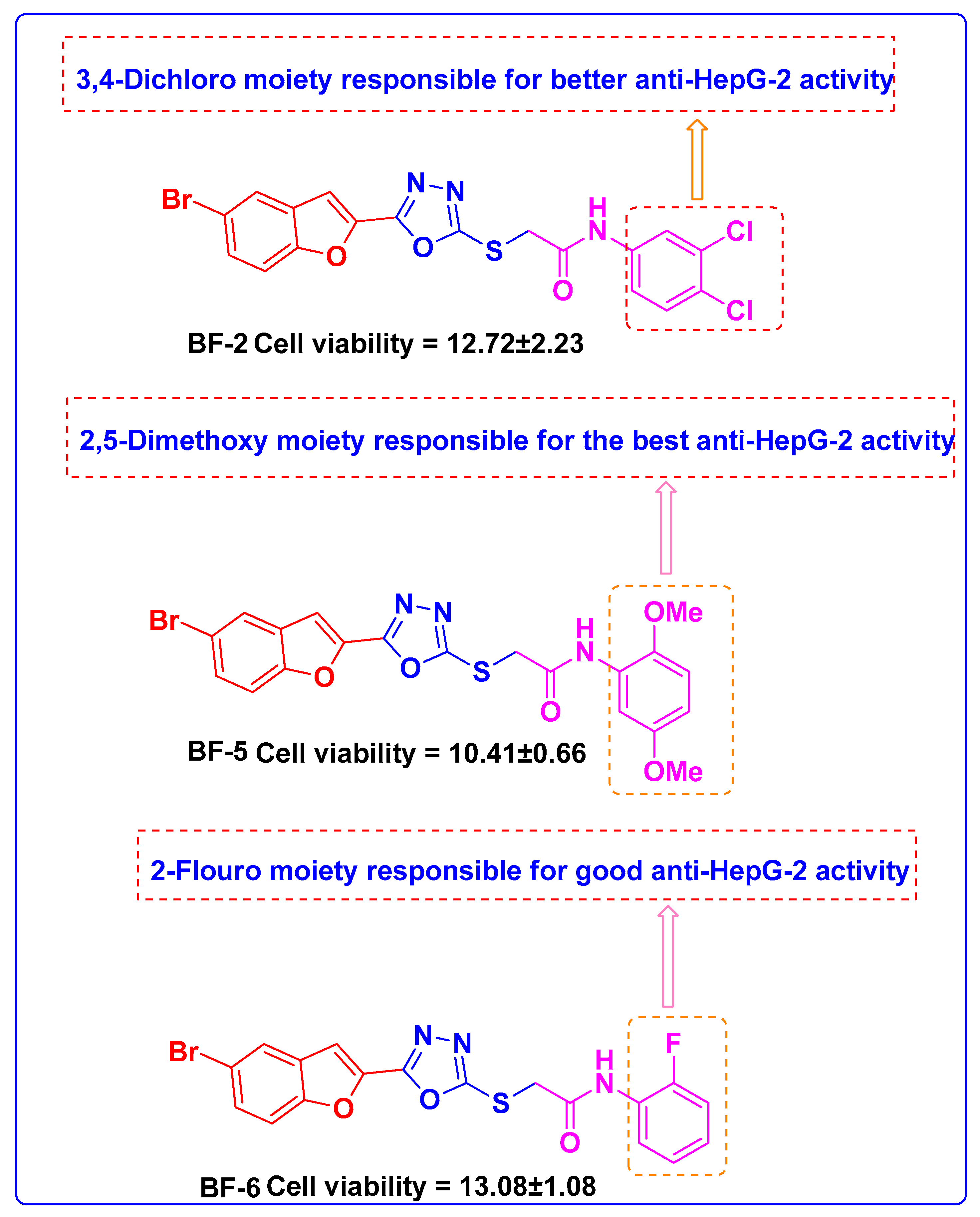
3.2.2. Structure-Activity Relationship of Bromobenzofuran-Oxadiazoles BF1-9
3.3. Computational Investigations
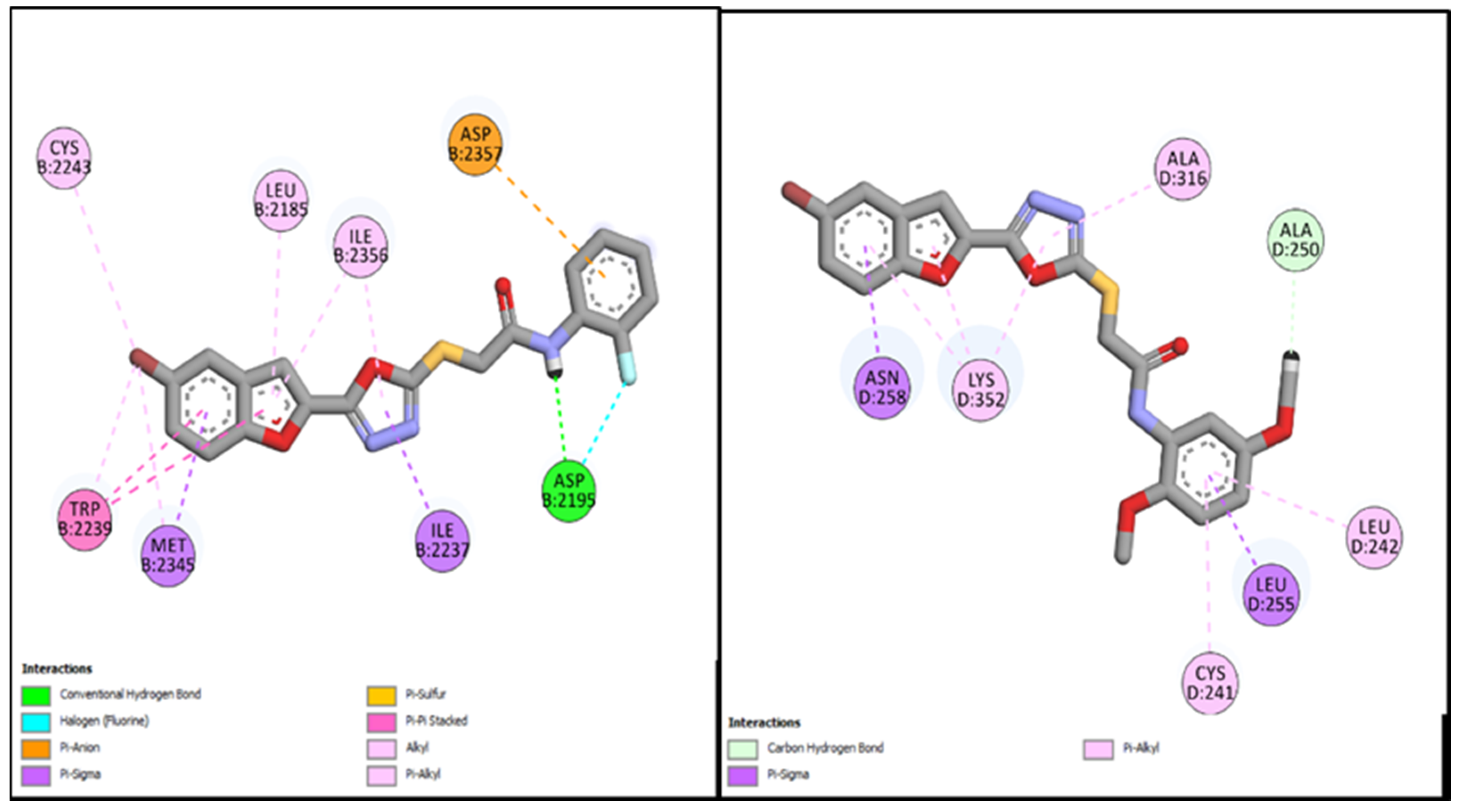
3.3.1. Molecular Docking Studies of Bromobenzofuran-Oxadiazoles BF1-9
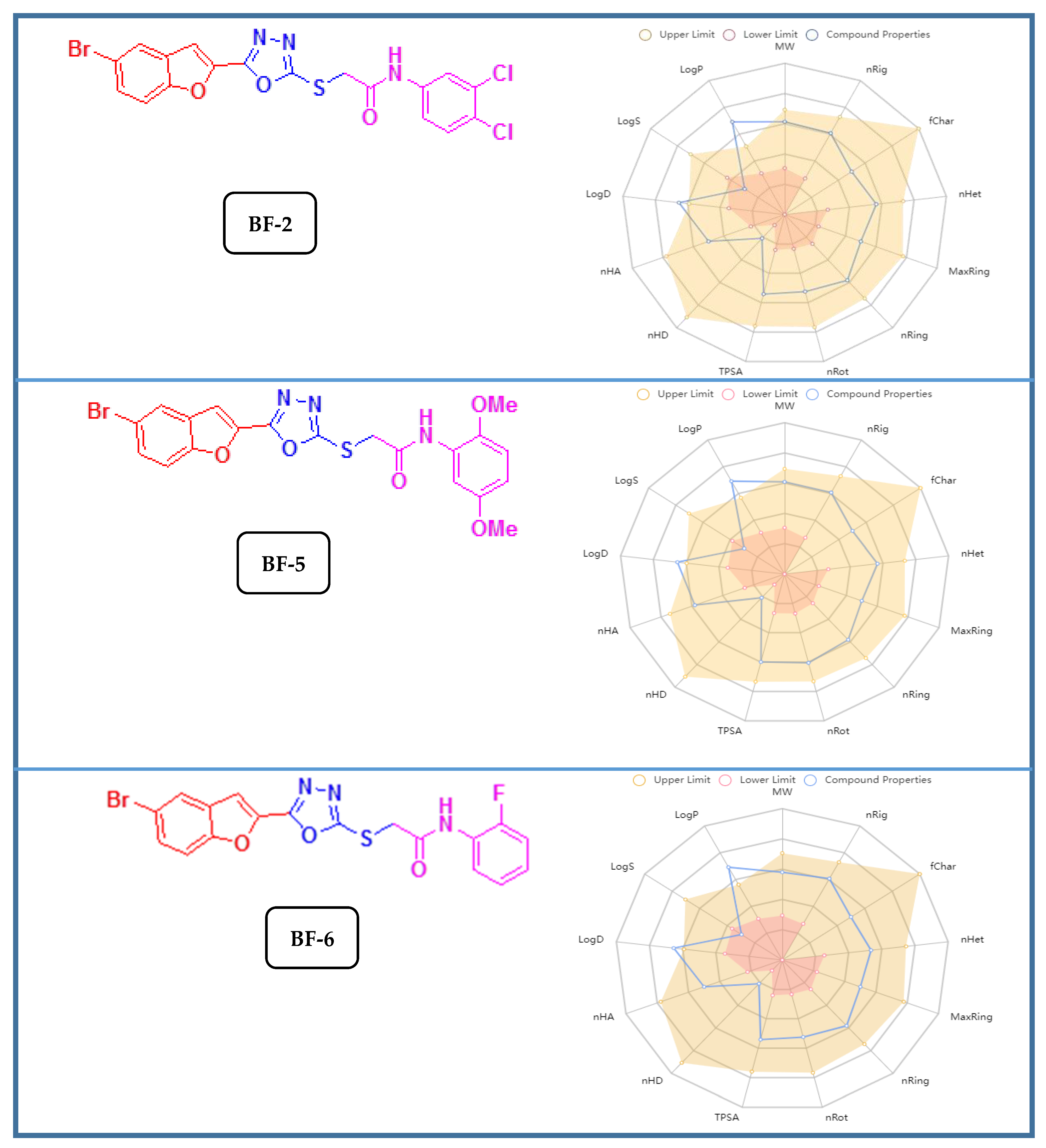
3.3.2. ADMET and Drug-Likeness Studies of Bromobenzofuran-Oxadiazoles BF1-9
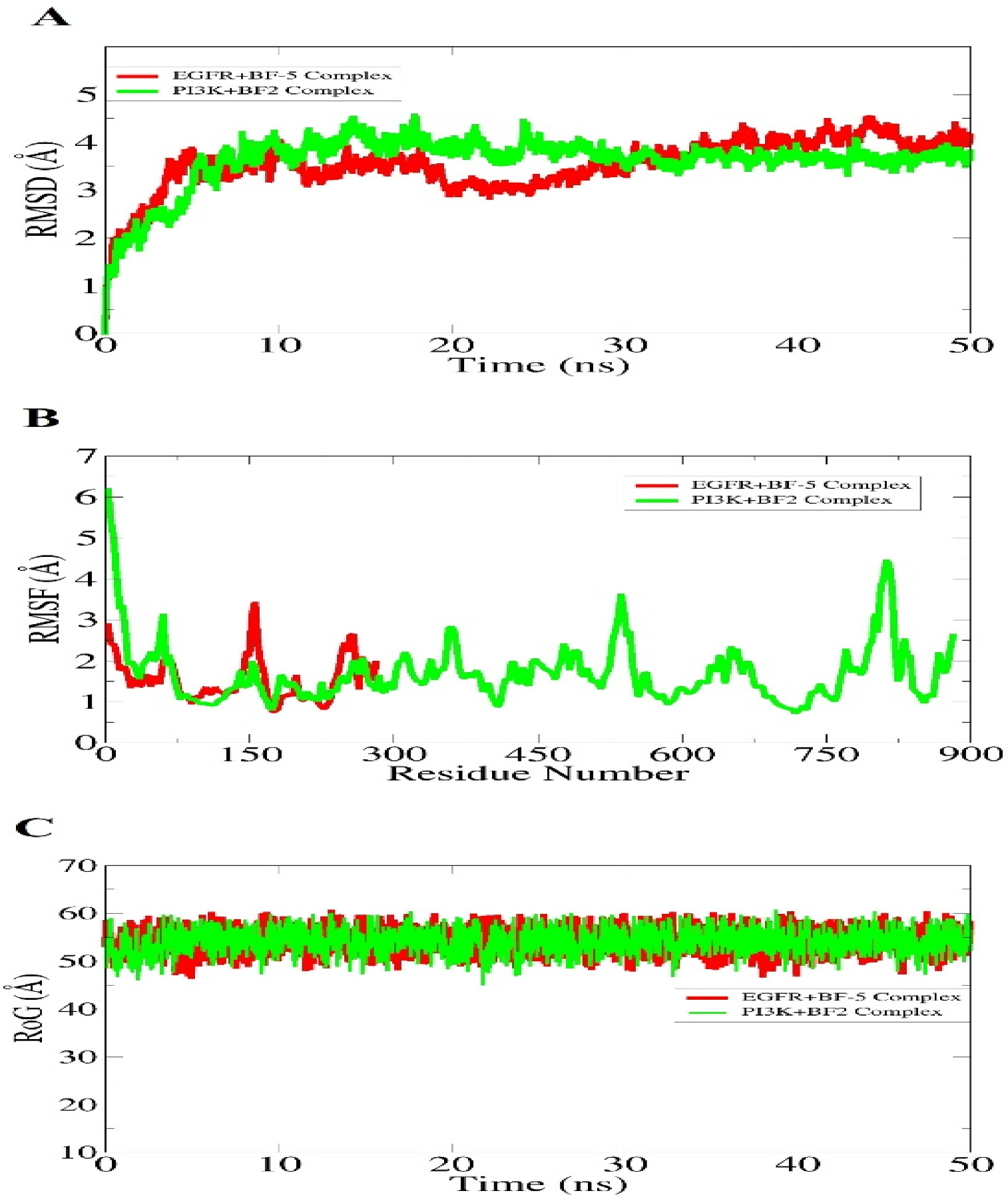
3.3.3. Molecular Dynamic Simulations of Bromobenzofuran-Oxadiazoles BF1-9
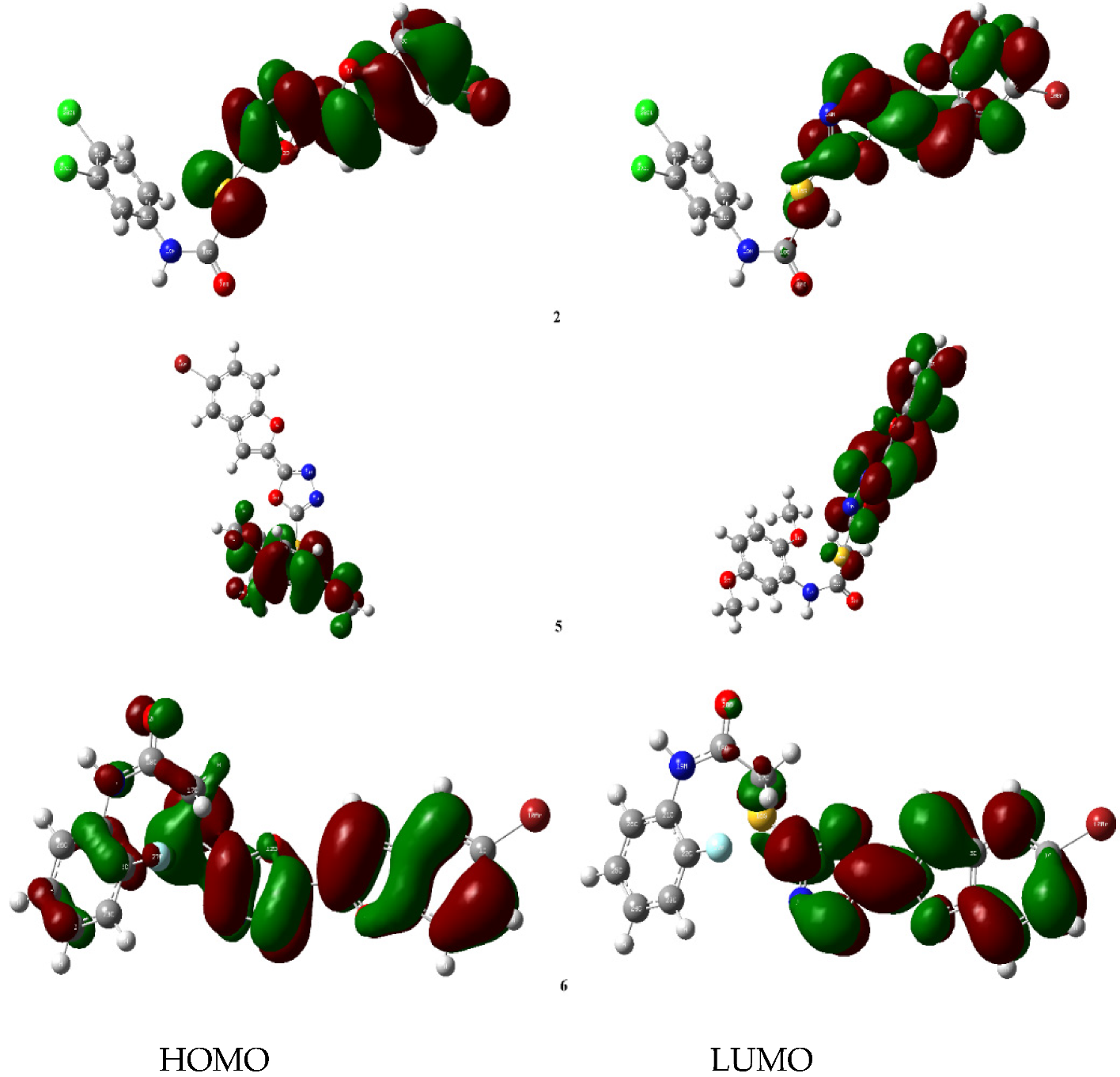
3.3.4. DFT Studies of Bromobenzofuran-Oxadiazoles BF1-9
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer Statistics, 2013. CA-Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K. The pursuit of optimal outcomes in cancer therapy in a new age of rationally designed target-based anticancer agents. Drugs 2000, 60, 1–14. [Google Scholar] [CrossRef]
- Weinberg, R.A. How Cancer Arises. Sci Am. 1996, 275, 62–70. [Google Scholar] [CrossRef]
- Johnson, D.S.; Li, J.J. The Art of Drug Synthesis; Wiley-Interscience: Hoboken, NJ, USA, 2007. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Philips, C.A.; Rajesh, S.; Nair, D.C.; Ahamed, R.; Abduljaleel, J.K.; Augustine, P. Hepatocellular Carcinoma in 2021: An Exhaustive Update. Cureus 2021, 13, e19274. [Google Scholar] [CrossRef]
- Suresh, D.; Srinivas, A.N.; Kumar, D.P. Etiology of Hepatocellular Carcinoma: Special Focus on Fatty Liver Disease. Front. Oncol. 2020, 30, 601710. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer. 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Zahoor, A.F.; Parveen, B.; Rasul, A.; Raza, Z.; Ahmad, S.; Irfan, I.; El-Hiti, G.A. Acefylline Derivatives as a New Class of Anticancer Agents: Synthesis, Molecular Docking, and Anticancer, Hemolytic, and Thrombolytic Activities of Acefylline-Triazole Hybrids. J. Chem. 2022, 2022, 3502872. [Google Scholar] [CrossRef]
- Sharma, R.A.; Plummer, R.; Stock, J.K.; Greenhalgh, T.A.; Ataman, O.; Kelly, S.; Clay, R.; Adams, R.A.; Baird, R.D.; Billingham, L.; et al. Clinical development of new drug-radiotherapy combinations. Nat. Rev. Clin. Oncol. 2016, 13, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Arruebo, M.; Vilaboa, N.; Sáez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; González-Fernández, A. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, V.; Pingaew, R.; Anuwongcharoen, N.; Worachartcheewan, A.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Discovery of novel 1,2,3-triazole derivatives as anticancer agents using QSAR and in silico structural modification. SpringerPlus 2015, 4, 571. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, G.; Bhutani, R.; Pathak, P.D.; Chauhan, G.; Kant, R.; Grover, P.; Nagarajan, K.; Siddiqui, S.A. Current Advancement in the Oxadiazole-Based Scaffolds as Anticancer Agents. Polycycl. Aromat. Compd. 2022, 42, 4183–4215. [Google Scholar] [CrossRef]
- Aziz, H.; Zahoor, A.F.; Shahzadi, I.; Irfan, A. Recent Synthetic Methodologies Towards the Synthesis of Pyrazoles. Polycycl. Aromat. Compd. 2019, 41, 698–720. [Google Scholar] [CrossRef]
- Sudeesh, K.; Gururaja, R. Facile synthesis of some novel derivatives of 1,3,4-oxadiazole derivatives associated with quinolone moiety as cytotoxic and antibacterial agents. Org. Chem. Curr. Res. 2017, 6, 2–5. [Google Scholar]
- Nayak, S.; Gaonkar, S.L.; Musad, E.A.; Dawsar, A.M.A. 1,3,4-Oxadiazole-containing hybrids as potential anticancer agents: Recent developments, mechanism of action and structure-activity relationships. J. Saudi Chem. Soc. 2021, 25, 101284. [Google Scholar] [CrossRef]
- Lelyukh, M.; Martynets, M.; Kalytovska, M.; Drapak, I.; Harkov, S.; Chaban, T.; Chaban, I.; Matiychuk, V. Approaches for synthesis and chemical modification of non-condensed heterocyclic systems based on 1,3,4-oxadiazole ring and their biological activity: A review. J. Appl. Pharm. Sci. 2020, 10, 151–165. [Google Scholar]
- Luczynski, M.; Kudelko, A. Synthesis and Biological Activity of 1,3,4-Oxadiazoles Used in Medicine and Agriculture. Appl. Sci. 2022, 12, 3756. [Google Scholar] [CrossRef]
- Napiórkowska, M.; Cieślak, M.; Barańska, K.J.; Golińska, K.K.; Nawrot, B. Synthesis of new derivatives of benzofuran aspotential anticancer agents. Molecules 2019, 24, 1529. [Google Scholar] [CrossRef]
- Radadiya, A.; Shah, A. Bioactive benzofuran derivatives: An insight on lead developments, radioligands and advances of the last decade. Eur. J. Med. Chem. 2015, 97, 356–376. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Dighe, S.N. Biological and medicinal significance of benzofuran. Eur. J. Med. Chem. 2014, 97, 561–581. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.-H.; Hu, Y.-H.; Yang, J.; Liu, T.; Sun, J.; Wang, X.-J. Natural source, bioactivity and synthesis of benzofuran derivatives. RSC Adv. 2019, 9, 27510. [Google Scholar] [CrossRef]
- Othman, D.I.; Abdelal, A.M.M.; El-Sayed, M.A.; El Bialy, S.A.A. Novel Benzofuran Derivatives: Synthesis and Antitumor Activity. Heterocycl. Commun. 2013, 19, 29–35. [Google Scholar] [CrossRef]
- Abdelfatah, S.; Berg, A.; Huang, Q.; Yang, L.J.; Hamdoun, S.; Klinger, A.; Greten, H.J.; Fleischer, E.; Berg, T.; Wong, V.K.W.; et al. MCC1019, a Selective Inhibitor of the Polo-Box Domain of Polo-like Kinase 1 as Novel, Potent Anticancer Candidate. Acta Pharm. Sin. B 2019, 9, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Law, P.-Y.; Loh, H. Inhibition of PI3K/Akt Signaling: An Emerging Paradigm for Targeted Cancer Therapy. Curr. Med. Chem. Agents. 2005, 5, 575–589. [Google Scholar] [CrossRef]
- Abdelhafez, O.M.; Amin, K.M.; Ali, H.I.; Abdalla, M.M.; Ahmed, E.Y. Design, Synthesis and Anticancer Activity of Benzofuran Derivatives Targeting VEGFR-2 Tyrosine Kinase. RSC Adv. 2014, 4, 11569–11579. [Google Scholar] [CrossRef]
- El-Zahabi, M.A.; Sakr, H.; El-Adl, K.; Zayed, M.; Abdelraheem, A.S.; Eissa, S.I.; Elkady, H.; Eissa, I.H. Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents. Bioorg. Chem. 2020, 104, 104218. [Google Scholar] [CrossRef]
- Vaidya, A.; Pathak, D.; Shah, K. 1,3,4-oxadiazole and its derivatives: A review on recent progress in anticancer activities. Chem. Biol. Drug Des. 2021, 97, 572–591. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Nazreen, S.; Almalki, A.S.A.; Elhenawy, A.A.; Alsenani, N.I.; Elbehairi, S.E.I.; Malebari, A.M.; Alfaifi, M.Y.; Alsharif, M.A.; Alfaifi, S.Y.M. Naproxen Based 1,3,4-Oxadiazole Derivatives as EGFR Inhibitors: Design, Synthesis, Anticancer, and Computational Studies. Pharmaceuticals 2021, 14, 870. [Google Scholar] [CrossRef]
- Sankhe, N.M.; Durgashivaprasad, E.; Kutty, N.G.; Rao, J.V.; Narayanan, K.; Kumar, N.; Jain, P.; Udupa, N.; Palanimuthu, V.R. Novel 2,5-disubstituted-1,3,4-oxadiazole derivatives induce apoptosis in HepG2 cells through p53 mediated intrinsic pathway. Arab. J. Chem. 2019, 12, 2548–2555. [Google Scholar] [CrossRef]
- Mohan, C.D.; Anilkumar, N.C.; Rangappa, S.; Shanmugam, M.K.; Mishra, S.; Chinnathambi, A.; Alharbi, S.A.; Bhattacharjee, A.; Sethi, G.; Kumar, A.P.; et al. Novel1,3,4-Oxadiazole Induces Anticancer Activity by Targeting NF-κB in Hepatocellular Carcinoma Cells. Front. Oncol. 2018, 8, 42. [Google Scholar] [CrossRef]
- Hagras, M.; Saleh, M.A.; Eldin, R.R.E.; Abuelkhir, A.A.; Khidr, E.G.; El-Husseiny, A.A.; El-Mahdy, H.A.; Elkaeed, E.B.; Eissa, I.H. 1,3,4-Oxadiazole-naphthalene hybrids as potential VEGFR-2 inhibitors:design, synthesis, antiproliferative activity, apoptotic effect, and in-silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 386–402. [Google Scholar] [CrossRef]
- Kassem, A.F.; Nassar, I.F.; Abdel-Aal, M.T.; Awad, H.M.; El-Sayed, W.A. Synthesis and Anticancer Activity of New ((Furan-2-yl)-1,3,4-thiadiazolyl)-1,3,4-oxadiazole Acyclic Sugar Derivatives. Chem. Pharm. Bull. 2019, 67, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Faiz, S.; Zahoor, A.F.; Ajmal, M.; Kamal, S.; Ahmad, S.; Abdelgawad, A.M.; Elnaggar, M.E. Design, Synthesis, Antimicrobial Evaluation, and Laccase Catalysis Effect of Novel Benzofuran–Oxadiazole and Benzofuran–Triazole Hybrids. J. Heterocycl. Chem. 2019, 56, 2839–2852. [Google Scholar] [CrossRef]
- Irfan, A.; Faiz, S.; Rasul, A.; Zafar, R.; Zahoor, A.F.; Kotwica-Mojzych, K.; Mojzych, M. Exploring the Synergistic Anticancer Potential of Benzofuran–Oxadiazoles and Triazoles: Improved Ultrasound-and Microwave-Assisted Synthesis, Molecular Docking, Hemolytic, Thrombolytic and Anticancer Evaluation of Furan-Based Molecules. Molecules 2022, 27, 1023. [Google Scholar] [CrossRef]
- Majumdar, C.K.; Biswas, A.; Mukhopadhyay, P.P. Carbon–Carbon Bond Formation by Radical Cyclisation: Regioselective Synthesis of Coumarin-Annulated Sulfur Heterocycles. Synthesis 2003, 15, 2385–2389. [Google Scholar] [CrossRef]
- Majumdar, C.K.; Biswas, A. Regioselective Synthesis of Thieno[3,2-c][1]benzopyran-4-ones by Thio-Claisen Rearrangement. Mon. Für Chem. 2004, 135, 1001–1007. [Google Scholar] [CrossRef]
- Irfan, A.; Zahoor, A.F.; Kamal, S.; Hassan, M.; Kloczkowski, A. Ultrasonic-Assisted Synthesis of Benzofuran Appended Oxadiazole Molecules as Tyrosinase Inhibitors: Mechanistic Approach through Enzyme Inhibition, Molecular Docking, Chemoinformatics, ADMET and Drug-Likeness Studies. Int. J. Mol. Sci. 2022, 23, 10979. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Zahoor, A.F.; Rasul, A.; Rasool, N.; Raza, Z.; Faisal, S.; Parveen, B.; Kamal, S.; Zia-ur-Rehman, M.; Zahid, F.M. Synthesis, anticancer, and computational studies of 1, 3, 4-oxadiazole-purine derivatives. Heterocycl. Chem. 2020, 57, 2782–2794. [Google Scholar] [CrossRef]
- Akhtar, R.; Zahoor, A.F.; Rasul, A.; Khan, S.G.; Ali., K.G. In-vitro cytotoxic evaluation of newly designed ciprofloxacin-oxadiazole hybrids against human liver tumor cell line (Huh7). Pak. J. Pharm. Sci. 2021, 34, 1143–1148. [Google Scholar]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological Macromolecular Structures Enabling Research and Education in Fundamental Biology, Biomedicine, Biotechnology and Energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib Binds Both Inactive and Active Conformations of the EGFR Tyrosine Kinase Domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Li, C.; Bailey, S.; Baxi, S.M.; Goulet, L.; Guo, L.; Hoffman, J.; Jiang, Y.; Johnson, T.O.; Johnson, T.W.; et al. Discovery of the Highly Potent PI3K/MTOR Dual Inhibitor PF-04979064 through Structure-Based Drug Design. ACS Med. Chem. Lett. 2013, 4, 91–97. [Google Scholar] [CrossRef]
- Buonfiglio, R.; Prati, F.; Bischetti, M.; Cavarischia, C.; Furlotti, G.; Ombrato, R. Discovery of Novel Imidazopyridine GSK-3β Inhibitors Supported by Computational Approaches. Molecules 2020, 25, 2163. [Google Scholar] [CrossRef]
- Kallan, N.C.; Spencer, K.L.; Blake, J.F.; Xu, R.; Heizer, J.; Bencsik, J.R.; Mitchell, I.S.; Gloor, S.L.; Martinson, M.; Risom, T.; et al. Discovery and SAR of spirochromane Akt inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2410–2414. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Faisal, S.; Lal Badshah, S.; Kubra, B.; Sharaf, M.; Emwas, A.H.; Jaremko, M.; Abdalla, M. Computational Study of SARS-CoV-2 Rna Dependent Rna Polymerase Allosteric Site Inhibition. Molecules 2022, 27, 223. [Google Scholar] [CrossRef]
- Studio, A.D. Discovery Studio Modeling Environment, Release 3.5; Accelrys Software Inc.: San Diego, CA, USA, 2012. [Google Scholar]
- Mills, N. ChemDraw Ultra 10.0 CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. www.cambridgesoft.com. Commercial Price: $1910 for Download, $2150 for CD-ROM; Academic Price: $710 for Download, $800 for CD-ROM. J. Am. Chem. Soc. 2006, 128, 13649–13650. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001, 222, 1–41. [Google Scholar]
- Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar]
- Sprenger, K.G.; Jaeger, V.W.; Pfaendtner, J. The general AMBER force field (GAFF) can accurately predict thermodynamic and transport properties of many ionic liquids. J. Phys. Chem. B 2015, 119, 5882–5895. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian. J. Nat. Sci. Ed. 2009, 09, 53–54. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Perdew, J.P.; Kurth, S. Accurate density functional with correct formal properties: A step beyond the generalized gradient approximation. Phys. Rev. Lett. 1999, 82, 2544–2547. [Google Scholar] [CrossRef]
- Dennington, J.M.; Keith, R.D.; Millam, T.A. GaussView 5.0, Semichem Inc.: Shawnee Mission, KS, USA, 2008.
- Zhang, C.; Peng, L.; Zhang, Y.; Liu, Z.; Li, W.; Chen, S.; Li, G. The Identification of Key Genes and Pathways in Hepatocellular Carcinoma by Bioinformatics Analysis of High-Throughput Data. Med. Oncol. 2017, 34, 101. [Google Scholar] [CrossRef] [PubMed]
- Juaid, N.; Amin, A.; Abdalla, A.; Reese, K.; Alamri, Z.; Moulay, M.; Abdu, S.; Miled, N. Anti-Hepatocellular Carcinoma Biomolecules: Molecular Targets Insights. Int. J. Mol. Sci. 2021, 22, 10774. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Du, Y.; Kong, L.; Zhang, X.; Chen, Q. Identification of Molecular Target Genes and Key Pathways in Hepatocellular Carcinoma by Bioinformatics Analysis. Onco. Targets. Ther. 2018, 11, 1861–1869. [Google Scholar] [CrossRef]
- Zhu, M.; Li, W.; Lu, Y.; Dong, X.; Lin, B.; Chen, Y.; Zhang, X.; Guo, J.; Li, M. HBx Drives Alpha Fetoprotein Expression to Promote Initiation of Liver Cancer Stem Cells through Activating PI3K/AKT Signal Pathway. Int. J. Cancer 2017, 140, 1346–1355. [Google Scholar] [CrossRef]
- Tsai, K.H.; Hsien, H.H.; Chen, L.M.; Ting, W.J.; Yang, Y.S.; Kuo, C.H.; Tsai, C.H.; Tsai, F.J.; Tsai, H.J.; Huang, C.Y. Rhubarb Inhibits Hepatocellular Carcinoma Cell Metastasis via GSK-3-β Activation to Enhance Protein Degradation and Attenuate Nuclear Translocation of β-Catenin. Food Chem. 2013, 138, 278–285. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 4629495. [Google Scholar]
- Hindson, J. Lenvatinib plus EGFR Inhibition for Liver Cancer. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 675. [Google Scholar] [CrossRef]
- Abbas, H.A.S.; Abd El-Karim, S.S. Design, Synthesis and Anticervical Cancer Activity of New Benzofuran–Pyrazol-Hydrazono- Thiazolidin-4-One Hybrids as Potential EGFR Inhibitors and Apoptosis Inducing Agents. Bioorg. Chem. 2019, 89, 103035. [Google Scholar] [CrossRef]
- Mphahlele, M.J.; Maluleka, M.M.; Parbhoo, N.; Malindisa, S.T. Synthesis, Evaluation for Cytotoxicity and Molecular Docking Studies of Benzo[c]Furan-Chalcones for Potential to Inhibit Tubulin Polymerization and/or EGFR-Tyrosine Kinase Phosphorylation. Int. J. Mol. Sci. 2018, 19, 2552. [Google Scholar] [CrossRef]
- Mervai, Z.; Reszegi, A.; Miklya, I.; Knoll, J.; Schaff, Z.; Kovalszky, I.; Baghy, K. Inhibitory Effect of (2R)-1-(1-Benzofuran-2-Yl)-N-Propylpentan-2-Amine on Lung Adenocarcinoma. Pathol. Oncol. Res. 2020, 26, 727–734. [Google Scholar] [CrossRef]
- Saitoh, M.; Kunitomo, J.; Kimura, E.; Iwashita, H.; Uno, Y.; Onishi, T.; Uchiyama, N.; Kawamoto, T.; Tanaka, T.; Mol, C.D.; et al. 2-{3-[4-(Alkylsulfinyl)Phenyl]-1-Benzofuran-5-Yl}-5-Methyl-1,3,4-Oxadiazole Derivatives as Novel Inhibitors of Glycogen Synthase Kinase-3β with Good Brain Permeability. J. Med. Chem. 2009, 52, 6270–6286. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Al-Rashood, S.T.; Al-Warhi, T.; Eskandrani, R.O.; Alharbi, A.; El Kerdawy, A.M. Novel Oxindole/Benzofuran Hybrids as Potential Dual CDK2/GSK-3β Inhibitors Targeting Breast Cancer: Design, Synthesis, Biological Evaluation, and in Silico Studies. J. Enzyme Inhib. Med. Chem. 2021, 36, 270–285. [Google Scholar] [CrossRef]
- Gaisina, I.N.; Gallier, F.; Ougolkov, A.V.; Kim, K.H.; Kurome, T.; Guo, S.; Holzle, D.; Luchini, D.N.; Blond, S.Y.; Billadeau, D.D.; et al. From a Natural Product Lead to the Identification of Potent and Selective Benzofuran-3-Yl-(Indol-3-Yl)Maleimides as Glycogen Synthase Kinase 3βinhibitors That Suppress Proliferation and Survival of Pancreatic Cancer Cells. J. Med. Chem. 2009, 52, 1853–1863. [Google Scholar] [CrossRef]
- El-Khouly, O.A.; Henen, M.A.; El-Sayed, M.A.A.; Shabaan, M.I.; El-Messery, S.M. Synthesis, Anticancer and Antimicrobial Evaluation of New Benzofuran Based Derivatives: PI3K Inhibition, Quorum Sensing and Molecular Modeling Study. Bioorganic Med. Chem. 2021, 31, 115976. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ma, C.; Feng, X.; Liu, Y.; Haimiti, X. BF12, a Novel Benzofuran, Exhibits Antitumor Activity by Inhibiting Microtubules and the PI3K/Akt/MTOR Signaling Pathway in Human Cervical Cancer Cells. Chem. Biodivers. 2020, 17, e1900622. [Google Scholar] [CrossRef] [PubMed]
- Farhat, J.; Alzyoud, L.; Alwahsh, M.; Al-Omari, B. Structure–Activity Relationship of Benzofuran Derivatives with Potential Anticancer Activity. Cancers 2022, 14, 2196. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef]
- Koopmans, T. About the assignment of wave functions and eigenvalues to the individual electrons of an atom. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Miar, M.; Shiroudi, A.; Pourshamsian, K.; Oliaey, A.R.; Hatamjafari, F. Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo[d]thiazole-2(3H)-imine and its para-substituted derivatives: Solvent and subs. J. Chem. Res. 2021, 45, 147–158. [Google Scholar] [CrossRef]
- Akkoc, S.; Karatas, H.; Muhammed, M.T.; Ceylan, A.; Almalki, F.; Laaroussi, H.; Ben, T.; Laaroussi, H.; Ben Hadda, T. Drug design of new therapeutic agents: Molecular docking, molecular dynamics simulation, DFT and POM analyses of new Schiff base ligands and impact of substituents on bioactivity of their potential antifungal pharmacophore site. J. Biomol. Struct. Dyn. 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Morales, Y. HOMO-LUMO gap as an index of molecular size and structure for polycyclic aromatic hydrocarbons (PAHs) and asphaltenes: A theoretical study. I. J. Phys. Chem. A 2002, 106, 11283–11308. [Google Scholar] [CrossRef]
- Han, M.İ.; Dengiz, C.; Doğan, Ş.D.; Gündüz, M.G.; Köprü, S.; Özkul, C. Isoquinolinedione-urea hybrids: Synthesis, antibacterial evaluation, drug-likeness, molecular docking and DFT studies. J. Mol. Struct. 2022, 1252, 132007. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Products | Percentage Yields | Melting Points (MP) oC | |
|---|---|---|---|---|
| This Work (Using BTEAC Catalyst) | Reported [39] | Found | ||
| BF-1 |  | 82 | 204–205 | 203–204 |
| BF-2 |  | 86 | 227–228 | 226–227 |
| BF-3 |  | 80 | 238–240 | 239–240 |
| BF-4 |  | 88 | 177–179 | 177–178 |
| BF-5 |  | 83 | 188–190 | 189–190 |
| BF-6 |  | 76 | 187–188 | 186–187 |
| BF-7 |  | 75 | 197–199 | 197–198 |
| BF-8 |  | 77 | 192–193 | 191–193 |
| BF-9 |  | 80 | 185–186 | 185–186 |
| Compounds | Products Structure | HepG2 % Cell Viability ± SD |
|---|---|---|
| BF-1 |  | 26.29 ± 17.54 |
| BF-2 |  | 12.72 ± 2.23 |
| BF-3 |  | 33.12 ± 6.15 |
| BF-4 |  | 13.88 ± 0.6 |
| BF-5 |  | 10.41 ± 0.66 |
| BF-6 |  | 13.08 ± 1.08 |
| BF-7 |  | 21.47 ± 8.55 |
| BF-8 |  | 13.85 ± 1.08 |
| BF-9 |  | 44.69 ± 6.85 |
| Control | DMSO | 100 ± 0 |
| Compound | Binding Affinity | Interacting Residues | Types of Interactions Made |
|---|---|---|---|
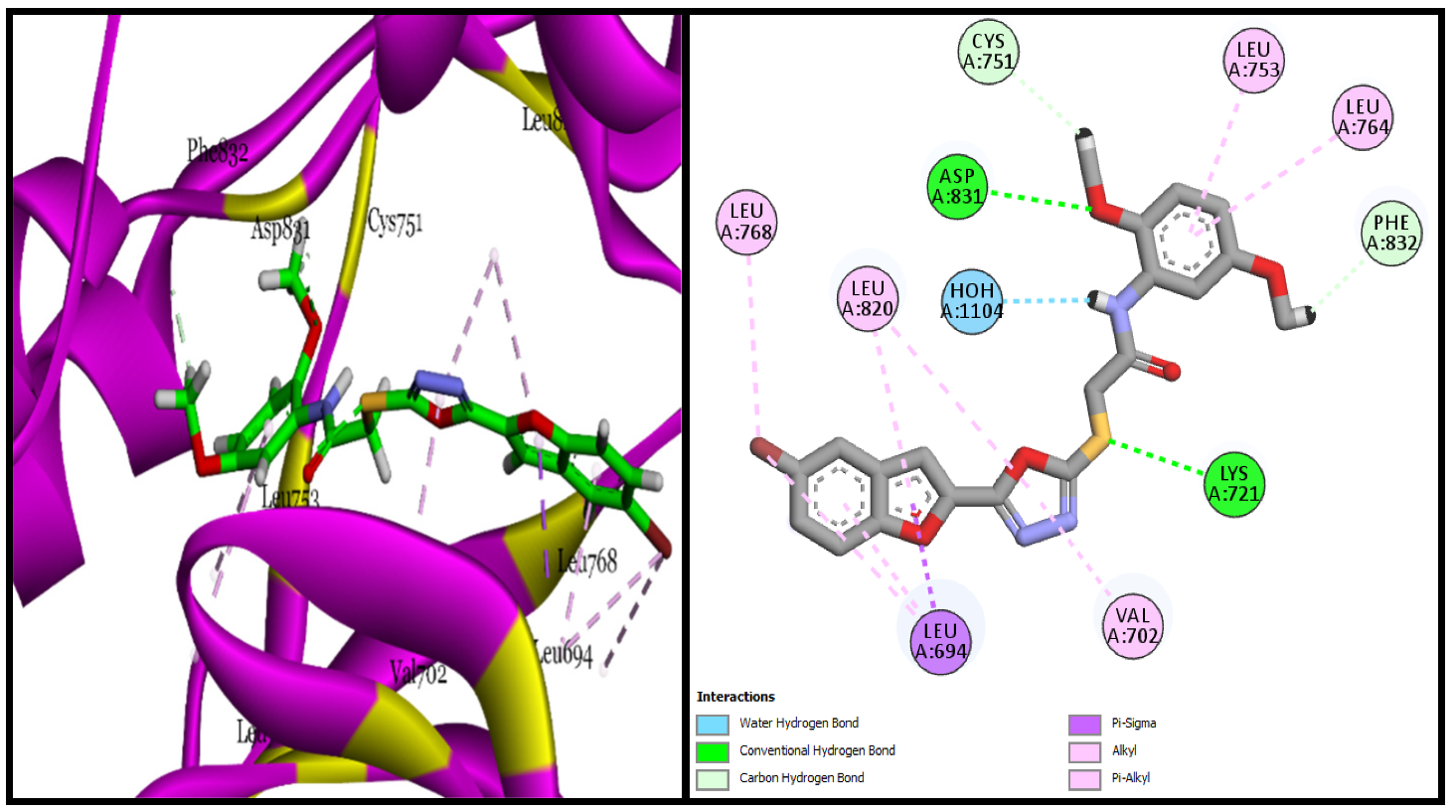
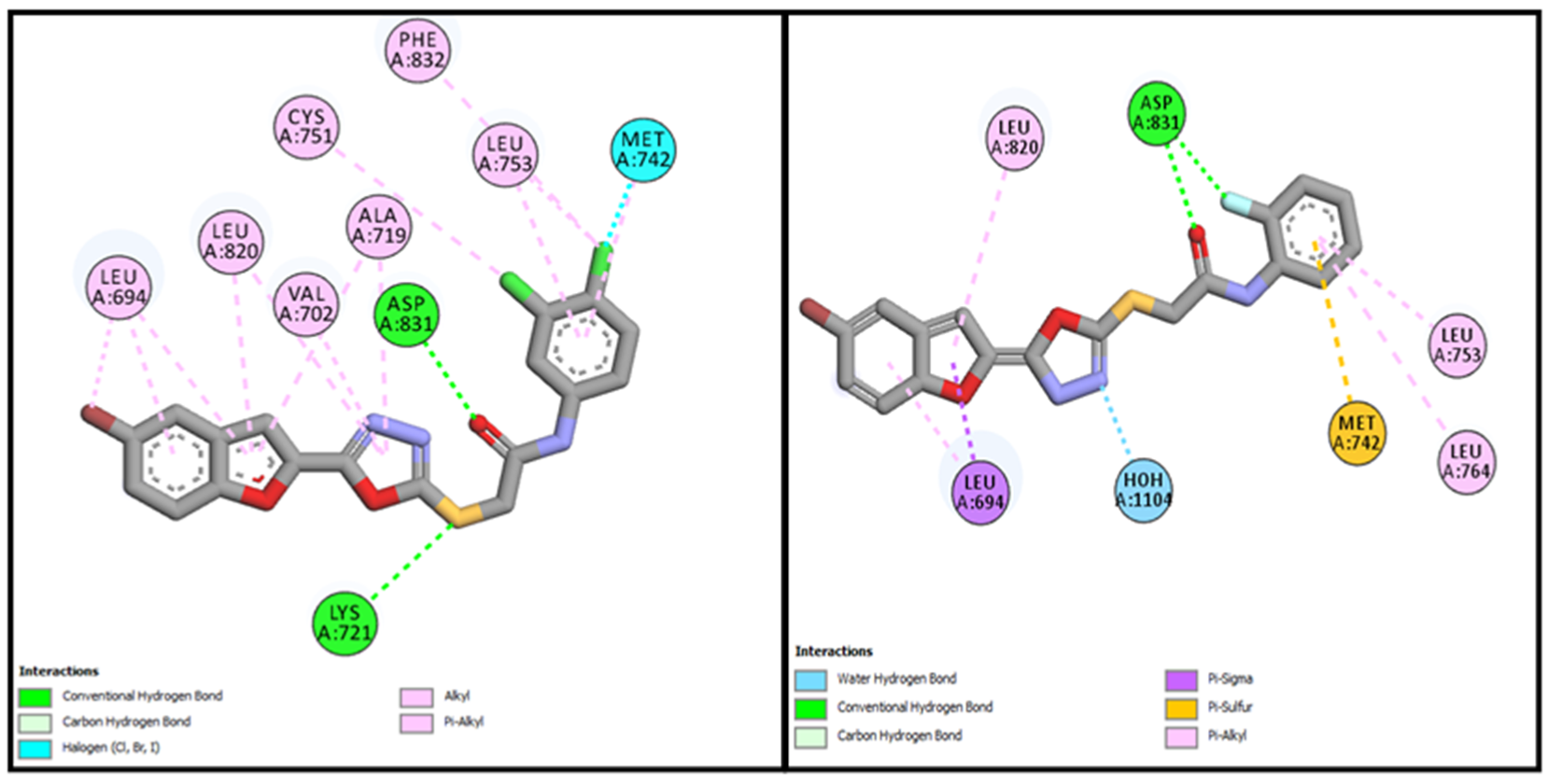
| BF-2 | −14.17 Kcal/mol | LEU694, VAL702, ALA719, LYS721, MET742, LEU753ASP831, CYS751, PHE832 | Conventional and Carbon type H-bonds, Halogen interactions, Pi-Alkyl & Alkyl |
| BF-5 | −15.17 Kcal/mol | LEU694, VAL702, LYS721, ASP831, PHE732, CYS751, LEU820, ASP831, PHE832 etc. | H2O-Assisted, Conventional and Carbon type H-bonds, Pi-Sigma, Pi-Alkyl & Alkyl |
| BF-6 | −12.59 Kcal/mol | MET742, LEU694, LEU764, LEU753, LEU820 | Conventional and Carbon type H-bonds, Pi-Sigma, Pi-Alkyl, Pi-Sulfur |
| Erlotinib | −11.67 Kcal/mol | LEU694, LYS704, VAL702, ALA719, LYS721, MET769, LEU820 | Conventional and Carbon type H-bonds, Pi-Alkyl, Alkyl |
| Compound | Binding Affinity | Interacting Residues | Types of Interactions Made |
|---|---|---|---|
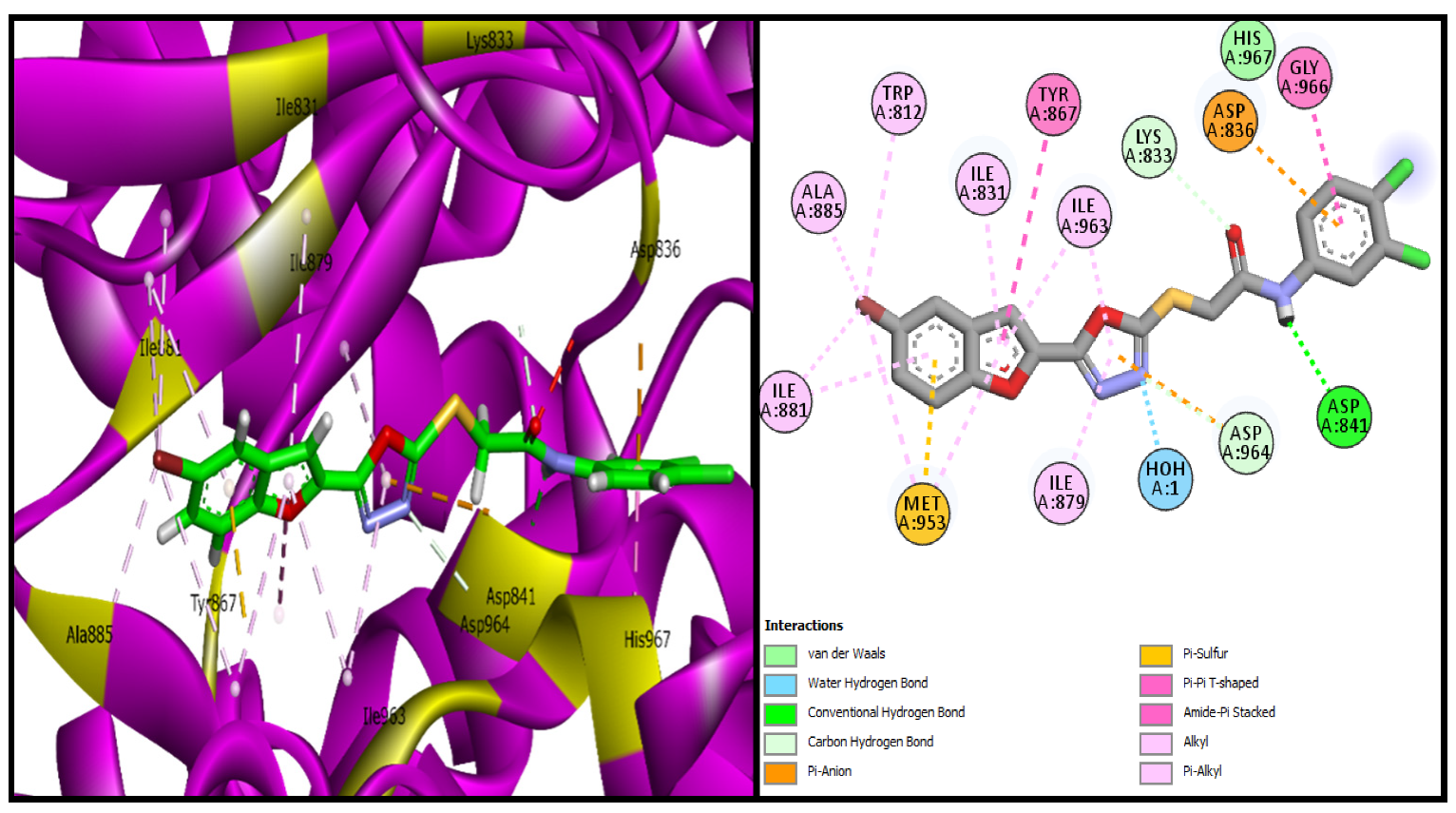
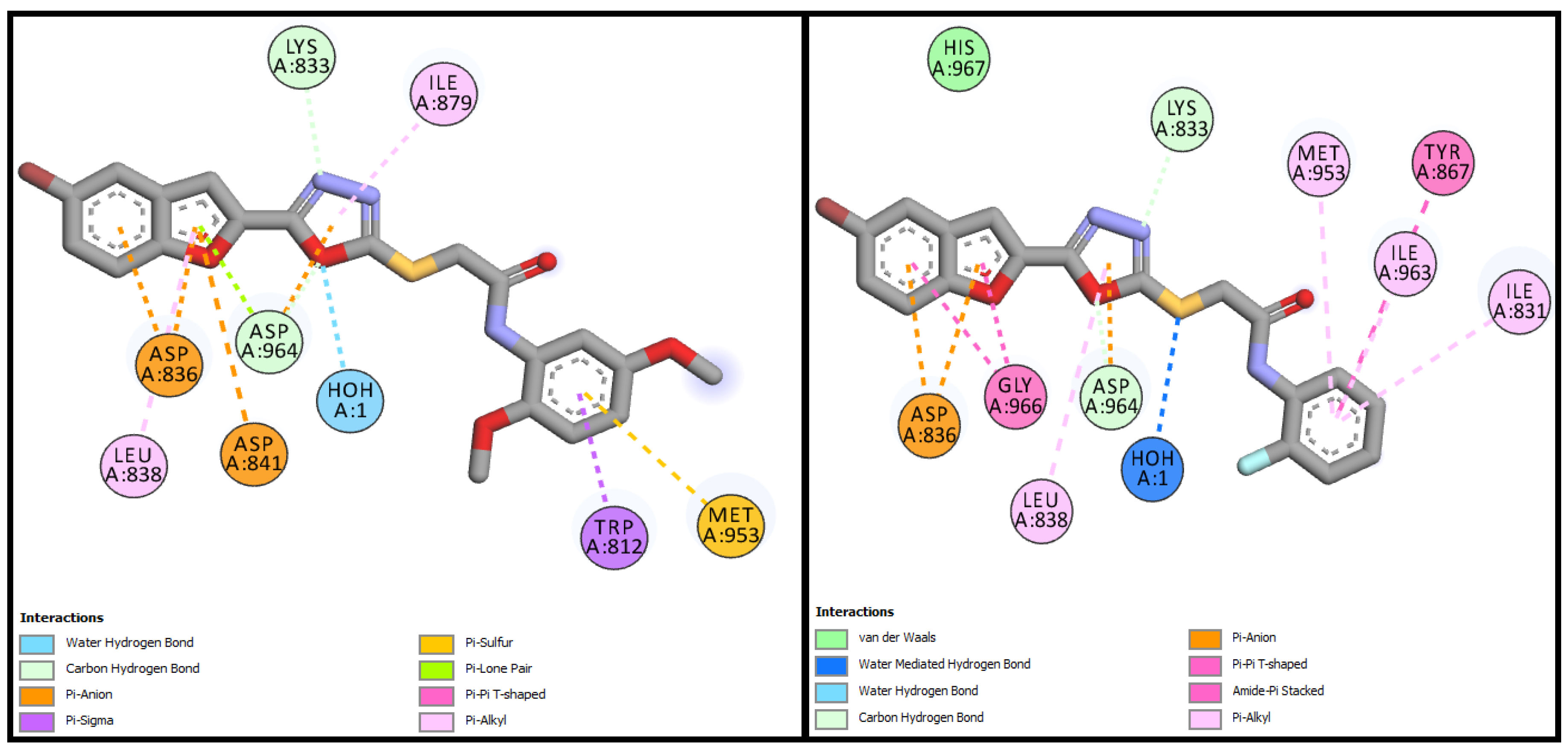
| BF-2 | −15.17 Kcal/mol | MET953, ASP836, LYS833, ASP964, ALA885, ILE881, ILE879, ILE963, TYR867, GLY966 | H2O-Assisted, Conventional and Carbon-H type H-bonds, Halogen interactions, Pi-Alkyl, Alkyl, Pi-Anion, Amide-Pi Stacked, Pi-Sulfur |
| BF-5 | −13.17 Kcal/mol | TRP812, LYS833, ASP836, LEU838, ASP841, MET953, ASP964 | H2O-Assisted, Conventional and Carbon-H type H-bonds, Halogen interactions, Pi-Alkyl, Alkyl, Pi-Anion, Pi-Lone Pair, Amide-Pi Stacked, Pi-Sulfur, etc. |
| BF-6 | −12.90 Kcal/mol | MET953, ASP836, LYS833, ASP964, ILE831, ILE963, TYR867, ASP964, GLY966, LEU838 | H2O-Assisted, Conventional and Carbon-H type H-bonds, Halogen interactions, Pi-Alkyl, Alkyl, Pi-Anion, Amide-Pi Stacked, etc |
| Idelalisib | −11.42 Kcal/mol | MET953, LYS833, ASP964, ILE831, ILE963, TYR867, ASP964, MET804, VAL882 | Conventional and Carbon-H type H-bonds and Pi-Donor H-bond, Pi-Alkyl, Alkyl, Pi-Anion, Pi-Pi T-shaped, etc. |
| Compound | Binding Affinity with mTOR | Binding Affinity with Tubulin |
|---|---|---|
| BF-2 | −11.61 kcal/mol | −13.14 kcal/mol |
| BF-5 | −11.84 kcal/mol | −13.79 kcal/mol |
| BF-6 | −11.89 kcal/mol | −10.59 kcal/mol |
| COLCHICINE Standard against Tubulin | -- | −11.85 kcal/mol |
| TORIN-2 Standard against mTOR | −11.77 kcal/mol | -- |
| Compound | iLogP | LogS | Renal OCTs | AMES Toxicity | BBB+ | Carcinogenicity | HIA+ |
|---|---|---|---|---|---|---|---|
| BF-2 | 3.76 | −7.55 | Non-inhibitor | None | 0.97 | None | 0.87 |
| BF-5 | 3.99 | −6.56 | Non-inhibitor | None | 0.97 | None | 0.87 |
| BF-6 | 3.41 | −6.34 | Non-inhibitor | None | 0.97 | None | 0.84 |
| Compounds | Bioavailability Score | PAINS Alerts | Lipinski’s Rule | Pfizer Rule | Golden Triangle Rule | TPSA |
|---|---|---|---|---|---|---|
| BF-2 | 0.55 | None | complied | complied | complied | 106.46 Å2 |
| BF-5 | 0.55 | None | complied | complied | complied | 124.92 Å2 |
| BF-6 | 0.55 | None | complied | complied | complied | 106.46 Å2 |
| Energy Parameter | EGFR+BF-5 Complex | PI3K+BF-2 Complex |
|---|---|---|
| MMGBSA | ||
| Van der Waals | −29.10 | −33.90 |
| Electrostatic | −12.20 | −10.85 |
| Polar | 12.36 | 11.75 |
| Non-polar | −9.50 | −9.55 |
| Delta G gas | −41.3 | −44.75 |
| Delta G solv | 2.86 | 2.2 |
| Delta Total | −38.44 | −42.55 |
| MMPBSA | ||
| Van der Waals | −29.10 | −33.90 |
| Electrostatic | −12.20 | −10.85 |
| Polar | 11.11 | 10.36 |
| Non-polar | −9.35 | −10.74 |
| Delta G gas | −41.3 | −44.75 |
| Delta G solv | −1.76 | −0.38 |
| Delta Total | −39.54 | −45.13 |
| Parameters | BF-2 | BF-5 | BF-6 |
|---|---|---|---|
| Etotal | −30,873.682 | −36,325.869 | −32,792.474 |
| EHOMO | −6.851 | −6.163 | −6.532 |
| ELUMO | −2.667 | −2.444 | −2.383 |
| ΔE | 4.184 | 3.719 | 4.149 |
| Ionization potential (IP= -EHOMO) | 6.851 | 6.163 | 6.532 |
| Electron affinity (A = -ELUMO) | 2.667 | 2.444 | 2.383 |
| Chemical potential (µ = -(I + A)/2) | −4.759 | −4.304 | −4.458 |
| Hardness (η = (I-A)/2) | 2.092 | 1.860 | 2.075 |
| Mulliken electronegativity (ᵡ = (I + A)/2) [84] | 4.759 | 4.304 | −4.458 |
| Softness (S = ½η) | 0.239 | 0.269 | 0.241 |
| Electrophilicity index (ꞷ = µ2/2η) [85] | 5.413 | 4.983 | 4.790 |
| Maximum charge transfer (ΔNmax = (I + A)/2(I-A)) [86] | 1.137 | 1.157 | 1.074 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irfan, A.; Zahoor, A.F.; Rasul, A.; Al-Hussain, S.A.; Faisal, S.; Ahmad, S.; Noor, R.; Muhammed, M.T.; Zaki, M.E.A. BTEAC Catalyzed Ultrasonic-Assisted Synthesis of Bromobenzofuran-Oxadiazoles: Unravelling Anti-HepG-2 Cancer Therapeutic Potential through In Vitro and In Silico Studies. Int. J. Mol. Sci. 2023, 24, 3008. https://doi.org/10.3390/ijms24033008
Irfan A, Zahoor AF, Rasul A, Al-Hussain SA, Faisal S, Ahmad S, Noor R, Muhammed MT, Zaki MEA. BTEAC Catalyzed Ultrasonic-Assisted Synthesis of Bromobenzofuran-Oxadiazoles: Unravelling Anti-HepG-2 Cancer Therapeutic Potential through In Vitro and In Silico Studies. International Journal of Molecular Sciences. 2023; 24(3):3008. https://doi.org/10.3390/ijms24033008
Chicago/Turabian StyleIrfan, Ali, Ameer Fawad Zahoor, Azhar Rasul, Sami A. Al-Hussain, Shah Faisal, Sajjad Ahmad, Rida Noor, Muhammed Tilahun Muhammed, and Magdi E. A. Zaki. 2023. "BTEAC Catalyzed Ultrasonic-Assisted Synthesis of Bromobenzofuran-Oxadiazoles: Unravelling Anti-HepG-2 Cancer Therapeutic Potential through In Vitro and In Silico Studies" International Journal of Molecular Sciences 24, no. 3: 3008. https://doi.org/10.3390/ijms24033008
APA StyleIrfan, A., Zahoor, A. F., Rasul, A., Al-Hussain, S. A., Faisal, S., Ahmad, S., Noor, R., Muhammed, M. T., & Zaki, M. E. A. (2023). BTEAC Catalyzed Ultrasonic-Assisted Synthesis of Bromobenzofuran-Oxadiazoles: Unravelling Anti-HepG-2 Cancer Therapeutic Potential through In Vitro and In Silico Studies. International Journal of Molecular Sciences, 24(3), 3008. https://doi.org/10.3390/ijms24033008