

The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives


Abstract
1. Introduction
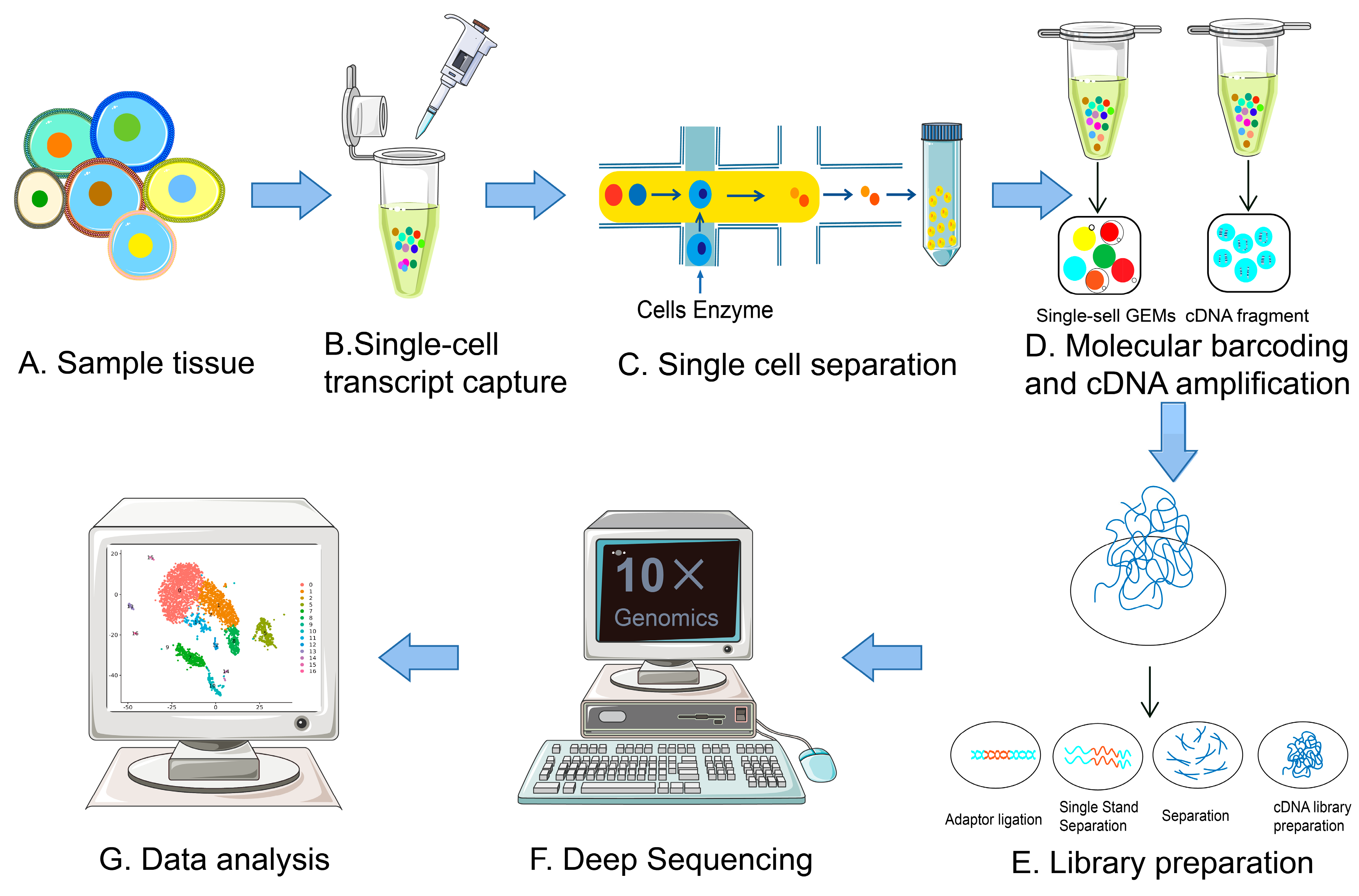
2. Evolution of scRNA-Seq Technologies
3. Application Progress of scRNA-Seq
3.1. Application of scRNA-Seq in Embryonic, Tissue and, Organ Development Research
3.1.1. Embryonic Research
3.1.2. Tissue and Organ Development Research
3.2. Application of scRNA-Seq in Tumor Research
3.2.1. Research on the Tumor Microenvironment (TME)
3.2.2. Research on Metabolic Heterogeneity
3.3. Application of scRNA-Seq in Immune System Research
3.3.1. Research on Immune Cell Differentiation
3.3.2. Research on the Mechanism of Immune Disease
3.3.3. Research on the Regulatory Processes of the Immune System
4. Application and Prospects of scRNA-Seq in TCM
4.1. Research on TCM Syndrome Differentiation
4.2. Research on the Interaction Mechanisms of TCM
4.3. Research on Pharmacodynamic Substances of TCM
4.4. Research on the Toxicity of TCM
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nassar, S.F.; Raddassi, K.; Wu, T. Single-Cell Multiomics Analysis for Drug Discovery. Metabolites 2021, 11, 729. [Google Scholar] [CrossRef] [PubMed]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, Y. Single cell sequencing: A distinct new field. Clin. Transl. Med. 2017, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Memczak, S.; Qu, J.; Belmonte, J.C.I.; Liu, G.-H. Single-cell omics in ageing: A young and growing field. Nat. Metab. 2020, 2, 293–302. [Google Scholar] [CrossRef]
- Lodato, M.A.; Rodin, R.E.; Bohrson, C.L.; Coulter, M.E.; Barton, A.R.; Kwon, M.; Sherman, M.A.; Vitzthum, C.M.; Luquette, L.J.; Yandava, C.N.; et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 2018, 359, 555–559. [Google Scholar] [CrossRef]
- Enge, M.; Arda, H.E.; Mignardi, M.; Beausang, J.; Bottino, R.; Kim, S.K.; Quake, S.R. Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell 2017, 171, 321–330.e14. [Google Scholar] [CrossRef]
- Liu, N.; Liu, L.; Pan, X. Single-cell analysis of the transcriptome and its application in the characterization of stem cells and early embryos. Cell. Mol. Life Sci. 2014, 71, 2707–2715. [Google Scholar] [CrossRef]
- De Klerk, E.; t Hoen, P.A. Alternative mRNA transcription, processing, and translation: Insights from RNA sequencing. Trends Genet. TIG 2015, 31, 128–139. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Salmen, F.; De Jonghe, J.; Kaminski, T.S.; Alemany, A.; Parada, G.E.; Verity-Legg, J.; Yanagida, A.; Kohler, T.N.; Battich, N.; Brekel, F.V.D.; et al. High-throughput total RNA sequencing in single cells using VASA-seq. Nat. Biotechnol. 2022, 40, 1780–1793. [Google Scholar] [CrossRef]
- Hagemann-Jensen, M.; Ziegenhain, C.; Sandberg, R. Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nat. Biotechnol. 2022, 40, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramsköld, D.; Hendriks, G.-J.; Larsson, A.J.M.; Faridani, O.R.; Sandberg, R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 2020, 38, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhong, Y.; Zhuang, Z.; Xie, J.; Lu, Y.; Huang, C.; Sun, Y.; Wu, L.; Yin, J.; Yu, H.; et al. Multiregion single-cell sequencing reveals the transcriptional landscape of the immune microenvironment of colorectal cancer. Clin. Transl. Med. 2021, 11, e253. [Google Scholar] [CrossRef] [PubMed]
- Gierahn, T.M.; Wadsworth, M.H., II; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Love, J.C.; Shalek, A.K. Seq-Well: Portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 2017, 14, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Sheng, K.; Zong, C. Single-Cell RNA-Seq by Multiple Annealing and Tailing-Based Quantitative Single-Cell RNA-Seq (MATQ-Seq). Methods Mol. Biol. Clifton N.J. 2019, 1979, 57–71. [Google Scholar]
- Wang, X.; He, Y.; Zhang, Q.; Ren, X.; Zhang, Z. Direct Comparative Analyses of 10X Genomics Chromium and Smart-Seq2. Genom. Proteom. Bioinform. 2021, 19, 253–266. [Google Scholar] [CrossRef]
- Fan, H.C.; Fu, G.K.; Fodor, S.P.A. Combinatorial labeling of single cells for gene expression cytometry. Science 2015, 347, 1258367. [Google Scholar] [CrossRef]
- Nakamura, T.; Yabuta, Y.; Okamoto, I.; Aramaki, S.; Yokobayashi, S.; Kurimoto, K.; Sekiguchi, K.; Nakagawa, M.; Yamamoto, T.; Saitou, M. SC3-seq: A method for highly parallel and quantitative measurement of single-cell gene expression. Nucleic Acids Res. 2015, 43, e60. [Google Scholar] [CrossRef]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.N. Single-Cell Tagged Reverse Transcription (STRT-Seq). Methods Mol. Biol. Clifton N.J. 2019 1979, 133–153. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Björklund, K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Sasagawa, Y.; Nikaido, I.; Hayashi, T.; Danno, H.; Uno, K.D.; Imai, T.; Ueda, H.R. Quartz-Seq: A highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013, 14, R31. [Google Scholar] [CrossRef]
- Hosic, S.; Murthy, S.K.; Koppes, A.N. Microfluidic Sample Preparation for Single Cell Analysis. Anal. Chem. 2016, 88, 354–380. [Google Scholar] [CrossRef]
- Ramsköld, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Grün, D.; Kester, L.; Van Oudenaarden, A. Validation of noise models for single-cell transcriptomics. Nat. Methods 2014, 11, 637–640. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, Q.Y.; Liu, J.L. Deciphering mouse uterine receptivity for embryo implantation at single-cell resolution. Cell Prolif. 2021, 54, e13128. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-M.; Cui, L.-S.; Hao, H.-S.; Wang, H.-Y.; Zhao, S.-J.; Du, W.-H.; Wang, D.; Liu, Y.; Zhu, H.-B. Transcriptome analyses of inner cell mass and trophectoderm cells isolated by magnetic-activated cell sorting from bovine blastocysts using single cell RNA-seq. Reprod. Domest. Anim. 2016, 51, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Valihrach, L.; Androvic, P.; Kubista, M. Platforms for Single-Cell Collection and Analysis. Int. J. Mol. Sci. 2018, 19, 807. [Google Scholar] [CrossRef]
- Warren, L.; Bryder, D.; Weissman, I.L.; Quake, S.R. Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proc. Natl. Acad. Sci. USA 2006, 103, 17807–17812. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 172, 1091–1107.e17. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.N.; Ugolini, G.S.; Sullivan, M.R.; Yang, Y.; De Ganzó, A.; Lim, J.W.; Konry, T. Droplet microfluidics for functional temporal analysis and cell recovery on demand using microvalves: Application in immunotherapies for cancer. Lab A Chip 2022, 22, 3258–3267. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, D.S.; Tamaki, W.; Sun, Y.; Ogorodnikov, A.; Hartoularos, G.C.; Winters, A.; Yeung, B.Z.; Nazor, K.L.; Song, Y.S.; et al. SCITO-seq: Single-cell combinatorial indexed cytometry sequencing. Nat. Methods 2021, 18, 903–911. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Orabi, B.; Erhan, E.; McConeghy, B.; Volik, S.V.; Le Bihan, S.; Bell, R.; Collins, C.C.; Chauve, C.; Hach, F. Alignment-free clustering of UMI tagged DNA molecules. Bioinformatics 2019, 35, 1829–1836. [Google Scholar] [CrossRef]
- Pan, X.; Urban, A.E.; Palejev, D.; Schulz, V.; Grubert, F.; Hu, Y.; Snyder, M.; Weissman, S.M. A procedure for highly specific, sensitive, and unbiased whole-genome amplification. Proc. Natl. Acad. Sci. USA 2008, 105, 15499–15504. [Google Scholar] [CrossRef]
- Pan, X.; Durrett, R.E.; Zhu, H.; Tanaka, Y.; Li, Y.; Zi, X.; Marjani, S.L.; Euskirchen, G.; Ma, C.; LaMotte, R.H.; et al. Two methods for full-length RNA sequencing for low quantities of cells and single cells. Proc. Natl. Acad. Sci. USA 2013, 110, 594–599. [Google Scholar] [CrossRef]
- Prieto, C.; Barrios, D. RaNA-Seq: Interactive RNA-Seq analysis from FASTQ files to functional analysis. Bioinformatics 2019, 36, 1955–1956. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Yu, X.; Liu, X. Mapping RNA-seq reads to transcriptomes efficiently based on learning to hash method. Comput. Biol. Med. 2020, 116, 103539. [Google Scholar] [CrossRef]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11.14.1–11.14.19. [Google Scholar]
- De Sena Brandine, G.; Smith, A.D. Falco: High-speed fast QC emulation for quality control of sequencing data. F1000Research 2019, 8, 1874. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Thomson, J.A.; Stewart, R. Quality control of single-cell RNA-seq by SinQC. Bioinformatics 2016, 32, 2514–2516. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Campbell, K.R.; Lun, A.T.L.; Wills, Q.F. Scater: Pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 2017, 33, 1179–1186. [Google Scholar] [CrossRef]
- Lytal, N.; Ran, D.; An, L. Normalization Methods on Single-Cell RNA-Seq Data: An Empirical Survey. Front. Genet. 2020, 11, 41. [Google Scholar] [CrossRef]
- Haghverdi, L.; Lun, A.T.L.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Büttner, M.; Miao, Z.; Wolf, F.A.; Teichmann, S.A.; Theis, F.J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 2019, 16, 43–49. [Google Scholar] [CrossRef]
- Fang, Z.-Y.; Lin, C.-X.; Xu, Y.-P.; Li, H.-D.; Xu, Q.-S. REBET: A method to determine the number of cell clusters based on batch effect removal. Brief. Bioinform. 2021, 22, bbab204. [Google Scholar] [CrossRef]
- Andrews, T.S.; Hemberg, M. Identifying cell populations with scRNASeq. Mol. Asp. Med. 2018, 59, 114–122. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef]
- Andrews, T.S.; Hemberg, M. M3Drop: Dropout-based feature selection for scRNASeq. Bioinformatics 2019, 35, 2865–2867. [Google Scholar] [CrossRef]
- Soneson, C.; Robinson, M.D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 2018, 15, 255–261. [Google Scholar] [CrossRef]
- Vans, E.; Patil, A.; Sharma, A. FEATS: Feature selection-based clustering of single-cell RNA-seq data. Brief. Bioinform. 2021, 22, bbaa306. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Srivatsan, S.R.; Regier, M.C.; Barkan, E.; Franks, J.M.; Packer, J.S.; Grosjean, P.; Duran, M.; Saxton, S.; Ladd, J.J.; Spielmann, M.; et al. Embryo-scale, single-cell spatial transcriptomics. Science 2021, 373, 111–117. [Google Scholar] [CrossRef]
- Liao, J.; Lu, X.; Shao, X.; Zhu, L.; Fan, X. Uncovering an Organ’s Molecular Architecture at Single-Cell Resolution by Spatially Resolved Transcriptomics. Trends Biotechnol. 2021, 39, 43–58. [Google Scholar] [CrossRef]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Peng, M.; Tang, L.; Ouyang, J.; Xiong, F.; Guo, C.; Tang, Y.; Zhou, Y.; Liao, Q.; et al. Single-cell RNA sequencing in cancer research. J. Exp. Clin. Cancer Res. CR 2021, 40, 81. [Google Scholar] [CrossRef]
- Van Galen, P.; Hovestadt, V.; Wadsworth Ii, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef] [PubMed]
- La, H.; Yoo, H.; Lee, E.; Thang, N.; Choi, H.; Oh, J.; Park, J.; Hong, K. Insights from the Applications of Single-Cell Transcriptomic Analysis in Germ Cell Development and Reproductive Medicine. Int. J. Mol. Sci. 2021, 22, 823. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.E.; Klein, A.M. Lineage tracing meets single-cell omics: Opportunities and challenges. Nat. Rev. Genet. 2020, 21, 410–427. [Google Scholar] [CrossRef] [PubMed]
- Tam, P.P.L.; Ho, J.W.K. Cellular diversity and lineage trajectory: Insights from mouse single cell transcriptomes. Development 2020, 147, dev179788. [Google Scholar] [CrossRef]
- Liang, P.; Zheng, L.; Long, C.; Yang, W.; Yang, L.; Zuo, Y. HelPredictor models single-cell transcriptome to predict human embryo lineage allocation. Brief. Bioinform. 2021, 22, bbab196. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, S.; Min, M.; Ni, Y.; Lu, Z.; Sun, X.; Wu, J.; Liu, B.; Ying, X.; Liu, Y. Dissecting transcriptional heterogeneity in primary gastric adenocarcinoma by single cell RNA sequencing. Gut 2021, 70, 464–475. [Google Scholar] [CrossRef]
- Ilsley, G.R.; Suyama, R.; Noda, T.; Satoh, N.; Luscombe, N.M. Finding cell-specific expression patterns in the early Ciona embryo with single-cell RNA-seq. Sci. Rep. 2020, 10, 4961. [Google Scholar] [CrossRef]
- Malkowska, A.; Penfold, C.; Bergmann, S.; Boroviak, T.E. A hexa-species transcriptome atlas of mammalian embryogenesis delineates metabolic regulation across three different implantation modes. Nat. Commun. 2022, 13, 3407. [Google Scholar] [CrossRef]
- Devis-Jauregui, L.; Eritja, N.; Davis, M.L.; Matias-Guiu, X.; Llobet-Navàs, D. Autophagy in the physiological endometrium and cancer. Autophagy 2021, 17, 1077–1095. [Google Scholar] [CrossRef]
- Jalouli, M.; Mofti, A.; Elnakady, Y.A.; Nahdi, S.; Feriani, A.; Alrezaki, A.; Sebei, K.; Bizzarri, M.; Alwasel, S.; Harrath, A.H. Allethrin Promotes Apoptosis and Autophagy Associated with the Oxidative Stress-Related PI3K/AKT/mTOR Signaling Pathway in Developing Rat Ovaries. Int. J. Mol. Sci. 2022, 23, 6397. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, M.; Zhao, Y.; Wang, W. Taiji: System-level identification of key transcription factors reveals transcriptional waves in mouse embryonic development. Sci. Adv. 2019, 5, eaav3262. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, G. Regulation of energy metabolism in human pluripotent stem cells. Cell. Mol. Life Sci. 2021, 78, 8097–8108. [Google Scholar] [CrossRef] [PubMed]
- Jarajapu, Y.P.R. Targeting Angiotensin-Converting Enzyme-2/Angiotensin-(1-7)/Mas Receptor Axis in the Vascular Progenitor Cells for Cardiovascular Diseases. Mol. Pharmacol. 2021, 99, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Hadland, B.; Varnum-Finney, B.; Dozono, S.; Dignum, T.; Nourigat-McKay, C.; Heck, A.M.; Ishida, T.; Jackson, D.L.; Itkin, T.; Butler, J.M.; et al. Engineering a niche supporting hematopoietic stem cell development using integrated single-cell transcriptomics. Nat. Commun. 2022, 13, 1584. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, T.-Y.; Wan, S.; Yang, J.Y.H.; Wong, W.H.; Wang, Y.X.R. scJoint integrates atlas-scale single-cell RNA-seq and ATAC-seq data with transfer learning. Nat. Biotechnol. 2022, 40, 703–710. [Google Scholar] [CrossRef]
- Wu, H.; Villalobos, R.G.; Yao, X.; Reilly, D.; Chen, T.; Rankin, M.; Myshkin, E.; Breyer, M.D.; Humphreys, B.D. Mapping the single-cell transcriptomic response of murine diabetic kidney disease to therapies. Cell Metab. 2022, 34, 1064–1078.e6. [Google Scholar] [CrossRef]
- Yu, Q.; Kilik, U.; Holloway, E.M.; Tsai, Y.-H.; Harmel, C.; Wu, A.; Wu, J.H.; Czerwinski, M.; Childs, C.J.; He, Z.; et al. Charting human development using a multi-endodermal organ atlas and organoid models. Cell 2021, 184, 3281–3298.e22. [Google Scholar] [CrossRef]
- Mu, Q.; Chen, Y.; Wang, J. Deciphering Brain Complexity Using Single-Cell Sequencing. Genom. Proteom. Bioinform. 2019, 17, 344–366. [Google Scholar] [CrossRef]
- Paik, D.T.; Tian, L.; Williams, I.M.; Rhee, S.; Zhang, H.; Liu, C.; Mishra, R.; Wu, S.M.; Red-Horse, K.; Wu, J.C. Single-Cell RNA Sequencing Unveils Unique Transcriptomic Signatures of Organ-Specific Endothelial Cells. Circulation 2020, 142, 1848–1862. [Google Scholar] [CrossRef]
- Zelco, A.; Börjesson, V.; de Kanter, J.K.; Lebrero-Fernandez, C.; Lauschke, V.M.; Rocha-Ferreira, E.; Nilsson, G.; Nair, S.; Svedin, P.; Bemark, M.; et al. Single-cell atlas reveals meningeal leukocyte heterogeneity in the developing mouse brain. Genes Dev. 2021, 35, 1190–1207. [Google Scholar] [CrossRef]
- Cuevas-Diaz Duran, R.; Wei, H.; Wu, J.Q. Single-cell RNA-sequencing of the brain. Clin. Transl. Med. 2017, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Armand, E.J.; Li, J.; Xie, F.; Luo, C.; Mukamel, E.A. Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron 2021, 109, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e19. [Google Scholar] [CrossRef]
- Luo, T.; Zheng, F.; Wang, K.; Xu, Y.; Xu, H.; Shen, W.; Zhu, C.; Zhang, X.; Sui, W.; Tang, D.; et al. A single-cell map for the transcriptomic signatures of peripheral blood mononuclear cells in end-stage renal disease. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2021, 36, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, L.; Wang, Y.-C.; Xu, Z.-R.; Feng, Y.; Zhang, J.; Wang, Y.; Xu, C.-R. Comparative analysis of cell lineage differentiation during hepatogenesis in humans and mice at the single-cell transcriptome level. Cell Res. 2020, 30, 1109–1126. [Google Scholar] [CrossRef]
- Huang, S.; Huang, F.; Zhang, H.; Yang, Y.; Lu, J.; Chen, J.; Shen, L.; Pei, G. In vivo development and single-cell transcriptome profiling of human brain organoids. Cell Prolif. 2022, 55, e13201. [Google Scholar] [CrossRef]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950.e5. [Google Scholar] [CrossRef]
- Lindström, N.O.; Sealfon, R.; Chen, X.; Parvez, R.K.; Ransick, A.; Brandine, G.D.S.; Guo, J.; Hill, B.; Tran, T.; Kim, A.D.; et al. Spatial transcriptional mapping of the human nephrogenic program. Dev. Cell 2021, 56, 2381–2398.e6. [Google Scholar] [CrossRef]
- Magaletta, M.E.; Lobo, M.; Kernfeld, E.M.; Aliee, H.; Huey, J.D.; Parsons, T.J.; Theis, F.J.; Maehr, R. Integration of single-cell transcriptomes and chromatin landscapes reveals regulatory programs driving pharyngeal organ development. Nat. Commun. 2022, 13, 457. [Google Scholar] [CrossRef]
- Liu, Y.; E Kossack, M.; E McFaul, M.; Christensen, L.N.; Siebert, S.; Wyatt, S.R.; Kamei, C.N.; Horst, S.; Arroyo, N.; A Drummond, I.; et al. Single-cell transcriptome reveals insights into the development and function of the zebrafish ovary. Elife 2022, 11, e76014. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Kocarnik, J.M.; Compton, K.; Dean, F.E.; Fu, W.; Gaw, B.L.; Harvey, J.D.; Henrikson, H.J.; Lu, D.; Pennini, A.; Xu, R.; et al. Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life Years for 29 Cancer Groups from 2010 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 2022, 8, 420–444. [Google Scholar] [PubMed]
- Li, X.-Y.; Shen, Y.; Zhang, L.; Guo, X.; Wu, J. Understanding initiation and progression of hepatocellular carcinoma through single cell sequencing. Biochim. Et Biophys. Acta BBA Rev. Cancer 2022, 1877, 188720. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. Ebiomedicine 2021, 66, 103315. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, D.L.; Blackburn, H.L.; Shriver, C.D.; Rabizadeh, S.; Soon-Shiong, P.; Ellsworth, R.E. Single-cell sequencing and tumorigenesis: Improved understanding of tumor evolution and metastasis. Clin. Transl. Med. 2017, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R. Deciphering Tumor Heterogeneity in Hepatocellular Carcinoma (HCC)-Multi-Omic and Singulomic Approaches. Semin. Liver Dis. 2021, 41, 9–18. [Google Scholar] [CrossRef]
- Chen, P.; Wang, Y.; Li, J.; Bo, X.; Wang, J.; Nan, L.; Wang, C.; Ba, Q.; Liu, H.; Wang, H. Diversity and intratumoral heterogeneity in human gallbladder cancer progression revealed by single-cell RNA sequencing. Clin. Transl. Med. 2021, 11, e462. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-Cell Transcriptome Analysis Reveals Disease-Defining T-Cell Subsets in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef]
- Bischoff, P.; Trinks, A.; Obermayer, B.; Pett, J.P.; Wiederspahn, J.; Uhlitz, F.; Liang, X.; Lehmann, A.; Jurmeister, P.; Elsner, A.; et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene 2021, 40, 6748–6758. [Google Scholar] [CrossRef]
- Lin, W.; Noel, P.; Borazanci, E.H.; Lee, J.; Amini, A.; Han, I.W.; Heo, J.S.; Jameson, G.S.; Fraser, C.; Steinbach, M.; et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 2020, 12, 80. [Google Scholar] [CrossRef]
- Cui, Y.; Li, C.; Jiang, Z.; Zhang, S.; Li, Q.; Liu, X.; Zhou, Y.; Li, R.; Wei, L.; Li, L.; et al. Single-cell transcriptome and genome analyses of pituitary neuroendocrine tumors. Neuro Oncol. 2021, 23, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, J.; Zhao, Z.; Yang, M.; Chen, M.; Liu, C.; Ji, J.; Zhu, D. Single-cell transcriptome analysis reveals tumor immune microenvironment heterogenicity and granulocytes enrichment in colorectal cancer liver metastases. Cancer Lett. 2020, 470, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, G.; Yang, Y.; Wang, F.; Xiao, Y.-T.; Zhang, N.; Bian, X.; Zhu, Y.; Yu, Y.; Liu, F.; et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nature 2021, 23, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Olbrecht, S.; Busschaert, P.; Qian, J.; Vanderstichele, A.; Loverix, L.; Van Gorp, T.; Van Nieuwenhuysen, E.; Han, S.; Van den Broeck, A.; Coosemans, A.; et al. High-grade serous tubo-ovarian cancer refined with single-cell RNA sequencing: Specific cell subtypes influence survival and determine molecular subtype classification. Genome Med. 2021, 13, 111. [Google Scholar] [CrossRef]
- Yang, L.; He, Y.-T.; Dong, S.; Wei, X.-W.; Chen, Z.-H.; Zhang, B.; Chen, W.-D.; Yang, X.-R.; Wang, F.; Shang, X.-M.; et al. Single-cell transcriptome analysis revealed a suppressive tumor immune microenvironment in EGFR mutant lung adenocarcinoma. J. Immunother. Cancer 2022, 10, e003534. [Google Scholar] [CrossRef]
- Chen, J.; Tan, Y.; Sun, F.; Hou, L.; Zhang, C.; Ge, T.; Yu, H.; Wu, C.; Zhu, Y.; Duan, L.; et al. Single-cell transcriptome and antigen-immunoglobin analysis reveals the diversity of B cells in non-small cell lung cancer. Genome Biol. 2020, 21, 152. [Google Scholar] [CrossRef]
- Pritchett, J.C.; Yang, Z.-Z.; Kim, H.J.; Villasboas, J.C.; Tang, X.; Jalali, S.; Cerhan, J.R.; Feldman, A.L.; Ansell, S.M. High-dimensional and single-cell transcriptome analysis of the tumor microenvironment in angioimmunoblastic T cell lymphoma (AITL). Leukemia 2022, 36, 165–176. [Google Scholar] [CrossRef]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef]
- Yu, T.J.; Ma, D.; Liu, Y.Y.; Xiao, Y.; Gong, Y.; Jiang, Y.Z.; Shao, Z.M.; Hu, X.; Di, G.H. Bulk and single-cell transcriptome profiling reveal the metabolic heterogeneity in human breast cancers. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 2350–2365. [Google Scholar] [CrossRef]
- Davis, R.T.; Blake, K.; Ma, D.; Gabra, M.B.I.; Hernandez, G.A.; Phung, A.T.; Yang, Y.; Maurer, D.; Lefebvre, A.E.Y.T.; Alshetaiwi, H.; et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nature 2020, 22, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Wang, Z.; Sun, Z.; Wei, J.; Zhang, L.; Ju, H.; Wang, T.; Zhang, C.; Guan, M.; Pan, S. Single-cell RNA sequencing reveals the characteristics of cerebrospinal fluid tumour environment in breast cancer and lung cancer leptomeningeal metastases. Clin. Transl. Med. 2022, 12, e885. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Z.; Zhu, Y.; Fu, J.; Zhao, X.; Zhang, Y.; Wang, S.; Wu, J.; Wang, K.; Wu, R.; et al. Single-Cell Transcriptome Analysis Uncovers Intratumoral Heterogeneity and Underlying Mechanisms for Drug Resistance in Hepatobiliary Tumor Organoids. Adv. Sci. 2021, 8, e2003897. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Liu, S.; Zhang, S.; Min, L.; Zhu, S. Cellular and Extracellular Components in Tumor Microenvironment and Their Application in Early Diagnosis of Cancers. Anal. Cell. Pathol. 2020, 2020, 6283796. [Google Scholar] [CrossRef]
- Zhang, M.; Zhai, W.; Miao, J.; Cheng, X.; Luo, W.; Song, W.; Wang, J.; Gao, W. Single cell analysis reveals intra-tumour heterogeneity, microenvironment and potential diagnosis markers for clear cell renal cell carcinoma. Clin. Transl. Med. 2022, 12, e713. [Google Scholar] [CrossRef]
- Zhang, J.; Song, C.; Tian, Y.; Yang, X. Single-Cell RNA Sequencing in Lung Cancer: Revealing Phenotype Shaping of Stromal Cells in the Microenvironment. Front. Immunol. 2022, 12, 802080. [Google Scholar] [CrossRef]
- Kaymak, I.; Williams, K.S.; Cantor, J.R.; Jones, R.G. Immunometabolic Interplay in the Tumor Microenvironment. Cancer Cell 2021, 39, 28–37. [Google Scholar] [CrossRef]
- Motwani, K.; Peters, L.D.; Vliegen, W.H.; El-Sayed, A.G.; Seay, H.R.; Lopez, M.C.; Baker, H.V.; Posgai, A.L.; Brusko, M.A.; Perry, D.J.; et al. Human Regulatory T Cells from Umbilical Cord Blood Display Increased Repertoire Diversity and Lineage Stability Relative to Adult Peripheral Blood. Front. Immunol. 2020, 11, 611. [Google Scholar] [CrossRef]
- Hoover, A.R.; Liu, K.; DeVette, C.I.; Krawic, J.R.; Medcalf, A.D.; West, C.L.; Hode, T.; Lam, S.S.; Welm, A.L.; Sun, X.; et al. Single-cell RNA sequencing reveals localized tumour ablation and intratumoural immunostimulant delivery potentiate T cell mediated tumour killing. Clin. Transl. Med. 2022, 12, e937. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, F.; Liu, W.; Ma, M.; Gao, J.; Lu, Y.; Huang, L.; Li, X.; Shi, Y.; Wang, X.; et al. Analysis of single-cell RNAseq identifies transitional states of T cells associated with hepatocellular carcinoma. Clin. Transl. Med. 2020, 10, e133. [Google Scholar] [CrossRef]
- Trzupek, D.; Dunstan, M.; Cutler, A.J.; Lee, M.; Godfrey, L.; Jarvis, L.; Rainbow, D.B.; Aschenbrenner, D.; Jones, J.L.; Uhlig, H.H.; et al. Discovery of CD80 and CD86 as recent activation markers on regulatory T cells by protein-RNA single-cell analysis. Genome Med. 2020, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Cho, E.J.; Lee, H.; Kim, W.K.; Oh, J.H.; Kim, S.H.; Lee, D.; Sung, C.O. Integrated single-cell RNA sequencing analyses suggest developmental paths of cancer-associated fibroblasts with gene expression dynamics. Clin. Transl. Med. 2021, 11, e487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Y.; Chen, T.; Miao, B.; Tang, Z.; Hu, X.; Luo, Y.; Zheng, T.; Na, N. Single-cell transcriptomics provides new insights into the role of fibroblasts during peritoneal fibrosis. Clin. Transl. Med. 2021, 11, e321. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sun, L.; Zhang, R.; Hu, Y.; Wu, Y.; Dong, X.; Dong, D.; Chen, C.; Geng, Z.; Li, E.; et al. Thrombospondin 4/integrin α2/HSF1 axis promotes proliferation and cancer stem-like traits of gallbladder cancer by enhancing reciprocal crosstalk between cancer-associated fibroblasts and tumor cells. J. Exp. Clin. Cancer Res. CR 2021, 40, 14. [Google Scholar] [CrossRef]
- Derks, S.; de Klerk, L.K.; Xu, X.; Fleitas, T.; Liu, K.X.; Liu, Y.; Dietlein, F.; Margolis, C.; Chiaravalli, A.M.; Da Silva, A.C.; et al. Characterizing diversity in the tumor-immune microenvironment of distinct subclasses of gastroesophageal adenocarcinomas. Ann. Oncol. 2020, 31, 1011–1020. [Google Scholar] [CrossRef]
- Fridman, W.H.; Meylan, M.; Petitprez, F.; Sun, C.-M.; Italiano, A.; Sautès-Fridman, C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat. Rev. Clin. Oncol. 2022, 19, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Xu, P.; Wang, N.; Yi, H.; Dong, L.; Fu, D.; Huang, J.; Huang, H.; Janin, A.; Cheng, S.; et al. Influence of oncogenic mutations and tumor microenvironment alterations on extranodal invasion in diffuse large B-cell lymphoma. Clin. Transl. Med. 2020, 10, e221. [Google Scholar] [CrossRef]
- Tong, Y.; Gao, W.-Q.; Liu, Y. Metabolic heterogeneity in cancer: An overview and therapeutic implications. Biochim. Et Biophys. Acta BBA Rev. Cancer 2020, 1874, 188421. [Google Scholar] [CrossRef]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019, 10, 3763. [Google Scholar] [CrossRef]
- Jiang, H.; Yu, D.; Yang, P.; Guo, R.; Kong, M.; Gao, Y.; Yu, X.; Lu, X.; Fan, X. Revealing the transcriptional heterogeneity of organ-specific metastasis in human gastric cancer using single-cell RNA Sequencing. Clin. Transl. Med. 2022, 12, e730. [Google Scholar] [CrossRef]
- Di Conza, G.; Tsai, C.-H.; Gallart-Ayala, H.; Yu, Y.-R.; Franco, F.; Zaffalon, L.; Xie, X.; Li, X.; Xiao, Z.; Raines, L.N.; et al. Tumor-induced reshuffling of lipid composition on the endoplasmic reticulum membrane sustains macrophage survival and pro-tumorigenic activity. Nat. Immunol. 2021, 22, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Powell, C.A.; Wang, X. Forward single-cell sequencing into clinical application: Understanding of cancer microenvironment at single-cell solution. Clin. Transl. Med. 2022, 12, e782. [Google Scholar] [PubMed]
- Chen, D.; Zhang, X.; Li, Z.; Zhu, B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics 2021, 11, 1016–1030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, M.; Wang, Q.; Zhang, J.; Zhou, T.; Sun, X. Single-cell transcriptome-based multilayer network biomarker for predicting prognosis and therapeutic response of gliomas. Brief. Bioinform. 2020, 21, 1080–1097. [Google Scholar] [CrossRef]
- Akar-Ghibril, N. Defects of the Innate Immune System and Related Immune Deficiencies. Clin. Rev. Allergy Immunol. 2022, 63, 36–54. [Google Scholar] [CrossRef]
- See, P.; Lum, J.; Chen, J.; Ginhoux, F. A Single-Cell Sequencing Guide for Immunologists. Front. Immunol. 2018, 9, 2425. [Google Scholar] [CrossRef]
- Pincha, N.; Marangoni, P.; Haque, A.; Klein, O.D. Parallels in signaling between development and regeneration in ectodermal organs. Curr. Top. Dev. Biol. 2022, 149, 373–419. [Google Scholar] [CrossRef]
- Xu, G.; Liu, Y.; Li, H.; Liu, L.; Zhang, S.; Zhang, Z. Dissecting the human immune system with single cell RNA sequencing technology. J. Leukoc. Biol. 2020, 107, 613–623. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, J.; Hu, Y.; Li, X.; Sun, L.; Peng, Y.; Sun, Y.; Liu, B.; Bian, Z.; Rong, Z. Single-cell transcriptome analysis reveals the dynamics of human immune cells during early fetal skin development. Cell Rep. 2021, 36, 109524. [Google Scholar] [CrossRef]
- Bourges, C.; Groff, A.F.; Burren, O.; Gerhardinger, C.; Mattioli, K.; Hutchinson, A.; Hu, T.; Anand, T.; Epping, M.W.; Wallace, C.; et al. Resolving mechanisms of immune-mediated disease in primary CD4 T cells. EMBO Mol. Med. 2020, 12, e12112. [Google Scholar] [CrossRef]
- Soskic, B.; Cano-Gamez, E.; Smyth, D.J.; Ambridge, K.; Ke, Z.; Matte, J.C.; Bossini-Castillo, L.; Kaplanis, J.; Ramirez-Navarro, L.; Lorenc, A.; et al. Immune disease risk variants regulate gene expression dynamics during CD4+ T cell activation. Nat. Genet. 2022, 54, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, C.; Wang, F.; Wang, X.; Zhu, J.; Luo, L.; Ding, X.; Zhang, Y.; Ding, P.; Wang, H.; et al. Molecular mechanisms governing circulating immune cell heterogeneity across different species revealed by single-cell sequencing. Clin. Transl. Med. 2022, 12, e689. [Google Scholar] [CrossRef] [PubMed]
- Suo, C.; Dann, E.; Goh, I.; Jardine, L.; Kleshchevnikov, V.; Park, J.-E.; Botting, R.A.; Stephenson, E.; Engelbert, J.; Tuong, Z.K.; et al. Mapping the developing human immune system across organs. Science 2022, 376, eabo0510. [Google Scholar] [CrossRef]
- Zhao, M.; Jiang, J.; Zhao, M.; Chang, C.; Wu, H.; Lu, Q. The Application of Single-Cell RNA Sequencing in Studies of Autoimmune Diseases: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2021, 60, 68–86. [Google Scholar] [CrossRef]
- Stubbington, M.J.T.; Rozenblatt-Rosen, O.; Regev, A.; Teichmann, S.A. Single-cell transcriptomics to explore the immune system in health and disease. Science 2017, 358, 58–63. [Google Scholar] [CrossRef]
- Perez, R.K.; Gordon, M.G.; Subramaniam, M.; Kim, M.C.; Hartoularos, G.C.; Targ, S.; Sun, Y.; Ogorodnikov, A.; Bueno, R.; Lu, A.; et al. Single-cell RNA-seq reveals cell type—Specific molecular and genetic associations to lupus. Science 2022, 376, eabf1970. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zheng, Y.; Li, D.; Hong, Q.; Zhang, M.; Li, Q.; Fu, B.; Wu, L.; Wang, X.; Shen, W.; et al. Expression characteristics of interferon-stimulated genes and possible regulatory mechanisms in lupus patients using transcriptomics analyses. EBioMedicine 2021, 70, 103477. [Google Scholar] [CrossRef]
- Zhao, M.; Wu, J.; Wu, H.; Sawalha, A.H.; Lu, Q. Clinical Treatment Options in Scleroderma: Recommendations and Comprehensive Review. Clin. Rev. Allergy Immunol. 2022, 62, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Siegert, E.; Uruha, A.; Goebel, H.H.; Preuße, C.; Casteleyn, V.; Kleefeld, F.; Alten, R.; Burmester, G.R.; Schneider, U.; Höppner, J.; et al. Systemic sclerosis-associated myositis features minimal inflammation and characteristic capillary pathology. Acta Neuropathol. 2021, 141, 917–927. [Google Scholar] [CrossRef]
- Gaydosik, A.M.; Tabib, T.; Domsic, R.; Khanna, D.; Lafyatis, R.; Fuschiotti, P. Single-cell transcriptome analysis identifies skin-specific T-cell responses in systemic sclerosis. Ann. Rheum. Dis. 2021, 80, 1453–1460. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, X.; Lei, X.; Xiao, X.; Jiao, T.; Ma, R.; Dong, X.; Jiang, Q.; Wang, W.; Shi, Y.; et al. Sensing of cytoplasmic chromatin by cGAS activates innate immune response in SARS-CoV-2 infection. Signal Transduct. Target. Ther. 2021, 6, 382. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhuang, M.W.; Deng, J.; Zheng, Y.; Zhang, J.; Nan, M.L.; Zhang, X.J.; Gao, C.; Wang, P.H. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J. Med. Virol. 2021, 93, 5376–5389. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Shi, Y.; Gong, B.; Jiang, L.; Zhang, Z.; Liu, X.; Yang, J.; He, Y.; Jiang, Z.; Zhong, L.; et al. Dynamic blood single-cell immune responses in patients with COVID-19. Signal Transduct. Target. Ther. 2021, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-N.; You, Y.; Cui, X.-M.; Gao, H.-X.; Wang, G.-L.; Zhang, S.-B.; Yao, L.; Duan, L.-J.; Zhu, K.-L.; Wang, Y.-L.; et al. Single-cell immune profiling reveals distinct immune response in asymptomatic COVID-19 patients. Signal Transduct. Target. Ther. 2021, 6, 342. [Google Scholar] [CrossRef]
- Li, J.; Ma, X.; Chakravarti, D.; Shalapour, S.; DePinho, R.A. Genetic and biological hallmarks of colorectal cancer. Genes Dev. 2021, 35, 787–820. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, C.; Zhu, M.; Fu, Z.; Shi, Y.; Li, M.; Wang, W.; Zhu, S.; Bin Jiang, B.; Luo, Y.; et al. Traditional Chinese medicine syndromes classification associates with tumor cell and microenvironment heterogeneity in colorectal cancer: A single cell RNA sequencing analysis. Chin. Med. 2021, 16, 133. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, K.; Chen, J.; Xu, Y.; Liu, Y.; Zheng, R. Traditional Chinese medicine classification of knee osteoarthritis with proteomics analysis. Ann. Palliat. Med. 2020, 9, 3750–3756. [Google Scholar] [CrossRef]
- Zhao, X.; Ma, L.; Guo, H.; Wang, J.; Zhang, S.; Yang, X.; Yang, L.; Jin, Q. Osteoclasts secrete leukemia inhibitory factor to promote abnormal bone remodeling of subchondral bone in osteoarthritis. BMC Musculoskelet. Disord. 2022, 23, 87. [Google Scholar] [CrossRef]
- Ji, Q.; Zheng, Y.; Zhang, G.; Hu, Y.; Fan, X.; Hou, Y.; Wen, L.; Li, L.; Xu, Y.; Wang, Y.; et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann. Rheum. Dis. 2019, 78, 100–110. [Google Scholar] [CrossRef]
- Liu, Z.; Xiang, H.; Xiang, D.; Xiao, S.; Xiang, H.; Xiao, J.; Ren, H.; Hu, P.; Liu, H.; Peng, M. Revealing potential anti-fibrotic mechanism of Ganxianfang formula based on RNA sequence. Chin. Med. 2022, 17, 23. [Google Scholar] [CrossRef]
- Qiu, Z.C.; Tang, X.Y.; Wu, Q.C.; Tang, Z.L.; Wong, M.S.; Chen, J.X.; Yao, X.S.; Dai, Y. A new strategy for discovering effective substances and mechanisms of traditional Chinese medicine based on standardized drug containing plasma and the absorbed ingredients composition, a case study of Xian-Ling-Gu-Bao capsules. J. Ethnopharmacol. 2021, 279, 114396. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Liang, P.; Ma, Y.; Sun, Q.; Pu, Q.; Dong, L.; Luo, G.; Mazhar, M.; Liu, J.; Wang, R.; et al. Research progress of traditional Chinese medicine against COVID-19. Biomed. Pharmacother. 2021, 137, 111310. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T.; Durantel, D.; Pasmant, E.; Lau, G.; Schinazi, R.F. COVID-19: Discovery, diagnostics and drug development. J. Hepatol. 2021, 74, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Zhang, P.; Zhang, Z.; Youn, J.Y.; Wang, C.; Zhang, H.; Cai, H. Traditional Chinese medicine (TCM) in the treatment of COVID-19 and other viral infections: Efficacies and mechanisms. Pharmacol. Ther. 2021, 225, 107843. [Google Scholar] [CrossRef]
- Qiao, L.; Huang, W.; Zhang, X.; Guo, H.; Wang, D.; Feng, Q.; Jin, R.; Xie, L.; Li, W.; Cheng, J. Evaluation of the immunomodulatory effects of anti-COVID-19 TCM formulae by multiple virus-related pathways. Signal Transduct. Target. Ther. 2021, 6, 50. [Google Scholar] [CrossRef]
- Wu, H.; Gong, K.; Qin, Y.; Yuan, Z.; Xia, S.; Zhang, S.; Yang, J.; Yang, P.; Li, L.; Xie, M. In silico analysis of the potential mechanism of a preventive Chinese medicine formula on coronavirus disease 2019. J. Ethnopharmacol. 2021, 275, 114098. [Google Scholar] [CrossRef]
- Friedman, S.L.; Pinzani, M. Hepatic fibrosis 2022: Unmet needs and a blueprint for the future. Hepatology 2022, 75, 473–488. [Google Scholar] [CrossRef]
- Cho, H.; Kim, H.; Lee, K.; Lasli, S.; Ung, A.; Hoffman, T.; Nasiri, R.; Bandaru, P.; Ahadian, S.; Dokmeci, M.R.; et al. Bioengineered Multicellular Liver Microtissues for Modeling Advanced Hepatic Fibrosis Driven Through Non-Alcoholic Fatty Liver Disease. Small 2021, 17, e2007425. [Google Scholar] [CrossRef]
- Sam, S.; Edelstein, S.L.; Arslanian, S.A.; Barengolts, E.; Buchanan, T.A.; Caprio, S.; Ehrmann, D.A.; Hannon, T.S.; Tjaden, A.H.; Kahn, S.E.; et al. Baseline Predictors of Glycemic Worsening in Youth and Adults with Impaired Glucose Tolerance or Recently Diagnosed Type 2 Diabetes in the Restoring Insulin Secretion (RISE) Study. Diabetes Care 2021, 44, 1938–1947. [Google Scholar] [CrossRef]
- Potter, K.J.; Reynaud, Q.; Boudreau, V.; Racine, F.; Tremblay, F.; Lavoie, A.; Carricart, M.; Mailhot, G.; Durieu, I.; A Senior, P.; et al. Combined Indeterminate and Impaired Glucose Tolerance Is a Novel Group at High Risk of Cystic Fibrosis-Related Diabetes. J. Clin. Endocrinol. Metab. 2021, 106, e3901–e3910. [Google Scholar] [CrossRef]
- Liang, Q.; Qu, Z.; Liang, Y.; Feng, Q.; Niu, X.; Bai, T.; Wang, Y.; Song, Q.; Adelson, D.L. Zuo Gui Wan Alters Expression of Energy Metabolism Genes and Prevents Cell Death in High-Glucose Loaded Mouse Embryos. Evid. Based Complement. Altern. Med. eCAM 2018, 2018, 2409471. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Li, P.; Wang, Z.; Cheng, Y.; Wu, H.; Yang, B.; Du, S.; Lu, Y. Brain distribution pharmacokinetics and integrated pharmacokinetics of Panax Notoginsenoside R1, Ginsenosides Rg1, Rb1, Re and Rd in rats after intranasal administration of Panax Notoginseng Saponins assessed by UPLC/MS/MS. J. Chromatogr. B 2014, 969, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhu, D.; Li, H.; Zhang, H.; Feng, C.; Zhang, W. Analyses of mRNA Profiling through RNA Sequencing on a SAMP8 Mouse Model in Response to Ginsenoside Rg1 and Rb1 Treatment. Front. Pharmacol. 2017, 8, 88. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Wang, Y.; Zhao, W.; Li, H.; Zhang, L.; Li, X.; Zhang, T.; Zhang, H.; Huang, H.; et al. Application of immune checkpoint targets in the anti-tumor novel drugs and traditional Chinese medicine development. Acta Pharm. Sin. B 2021, 11, 2957–2972. [Google Scholar] [CrossRef]
- Xie, Y.; Valdimarsdóttir, U.A.; Wang, C.; Zhong, X.; Gou, Q.; Zheng, H.; Deng, L.; He, P.; Hu, K.; Fall, K.; et al. Public health insurance and cancer-specific mortality risk among patients with breast cancer: A prospective cohort study in China. Int. J. Cancer 2021, 148, 28–37. [Google Scholar] [CrossRef]
- Kui, L.; Kong, Q.; Yang, X.; Pan, Y.; Xu, Z.; Wang, S.; Chen, J.; Wei, K.; Zhou, X.; Yang, X.; et al. High-Throughput in Vitro Gene Expression Profile to Screen of Natural Herbals for Breast Cancer Treatment. Front. Oncol. 2021, 11, 684351. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, H.; Qiu, L.-Z.; Yue, L.-X.; Zhang, G.-J.; Deng, H.-F.; Ni, Y.-H.; Gao, Y. Cardiac efficacy and toxicity of aconitine: A new frontier for the ancient poison. Med. Res. Rev. 2021, 41, 1798–1811. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.; Liu, Y.-T.; Zeng, X.-C.; Li, C.-P.; Ou-Yang, D.-S. The hepatotoxicity of Polygonum multiflorum: The emerging role of the immune-mediated liver injury. Acta Pharmacol. Sin. 2021, 42, 27–35. [Google Scholar] [CrossRef]
- Chen, M.; Geng, D.; Yang, X.; Liu, X.; Liu, S.; Ding, P.; Pang, Y.; Du, M.; Hu, X.; Wang, R. In Vitro Nephrotoxicity Induced by Herb-Herb Interaction between Radix Glycyrrhizae and Radix Euphorbiae Pekinensis. Oxidative Med. Cell. Longev. 2020, 2020, 6894751. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform Name | Separation Method | Amplification Method | Using UMI | Amplification Range | Advantages | Disadvantages | Release Date | References |
|---|---|---|---|---|---|---|---|---|
| VASA-seq | FANS | PCR | YES | All transcripts | Low cost and accurate dosing | / | 2022 | [10] |
| Smart-seq3 | Microfluidics | PCR | YES | 5′ end | High sensitivity | Time-consuming | 2020 | [11,12] |
| DNBelabC4 | Microfluidics | PCR | YES | All transcripts | Precise quantification | / | 2019 | [13] |
| Seq-Well | Microfluidics | PCR | YES | 3′ end | Low cost and precise quantification | Unsuitable for variable splicing and allelic expression | 2017 | [14] |
| MATQ-seq | FACS | PCR | YES | All transcripts | Precise quantification | Low cell throughput | 2017 | [15] |
| 10× Genomics | Microfluidics | PCR | YES | 3′ end | High cell capture efficiency, fast cycle time, high cell suitability, and reproducibility | Sequencing can be performed only for the 3′ end | 2016 | [16] |
| Cyto-Seq | Microfluidics | PCR | YES | 3′ end | Low cost and high throughput | Cross-contamination between RNAs | 2015 | [17] |
| SC3-seq | Micromanipulation | PCR | YES | 3′ end | Good reproducibility and accurate quantification | Recognize DNA at the 3′ end | 2015 | [18] |
| inDrop-seq | Microfluidics | IVT | YES | 3′ end | Low cost and linear amplification | Long operating time and high initial cell concentration | 2015 | [19] |
| Drop-seq | Microfluidics | PCR | YES | 3′ end | Low cost and high throughput | Low cell capture rate | 2015 | [20] |
| MARS-seq | FACS | IVT | YES | 3′ end | High specificity | Low amplification efficiency | 2014 | [21] |
| STRT-seq | Microfluidics | PCR | NO | All transcripts | Accurate positioning of transcripts at the 5′ end to reduce amplification bias | Low sensitivity, only available for identification of 5′ end DNA | 2014 | [22,23] |
| Quartz-seq | FACS | PCR | YES | 3′ end | High sensitivity, reproducibility, and operational simplicity | Higher noise levels | 2013 | [24] |
| Fluidigm C1 | Microfluidics | PCR | NO | All transcripts | Simple process | High cost and low throughput | 2013 | [25] |
| Smart-seq2 | FACS | PCR | NO | All transcripts | Full-length cDNA detects structural and RNA shear variants | High cost, low throughput, and time-consuming | 2013 | [13,26] |
| Smart-seq | FACS | PCR | NO | All transcripts | High sensitivity to reduce the rates of nucleic acid loss | Low throughput and the existence of transcript length bias | 2012 | [22] |
| CEL-seq | FACS | IVT | YES | 3′ end | Good reproducibility and highly sensitive | Low throughput and amplification efficiency, library biased toward the 3′ end of the gene | 2012 | [27] |
| Tang-2009 | FACS | PCR | NO | 3′ end | Good reproducibility | High cost and low throughput | 2009 | [9] |
| Tissues and Organs | Methods | Stromal Cell Subtypes | Key Difference Genes | Mechanisms | References |
|---|---|---|---|---|---|
| Liver | 10× Genomics | ID3+ hepatocytes NCAM1+ cholangiocytes Sox9+ cholangiocytes | ID3 | Inhibition of the Wnt signaling pathway maintains ID3+ cells in an undifferentiated hepatocyte-like state. | [85] |
| COL1A1 | |||||
| HAND2 | |||||
| Brain | 10× Genomics | Astrocyte Radial glia cell BMP single-related cells | CLIC6 | Pathways associated with metabolic stress, such as glycolysis, ER stress, and hypoxia, are significantly less activated in IVD-like organs. | [86] |
| ZBTB20 | |||||
| STRN3 | |||||
| Heart | STAR | Valvular interstitial cells Cardiac fibroblasts Endothelial cells | HAND1 | Endocardial cells highly express ligands and receptors of the Notch signaling pathway, which regulates neuromodulin (NRG)/ERBB signaling to promote cardiomyocyte differentiation from the trabecular layer. | [87] |
| HEY2 | |||||
| IRX3 | |||||
| Kidney | 10× Genomics | CDH1+/JAG1+ cells JAG1+/Jag1+ cells | SLC39A8 | Notch pathways in JAG1 and HES1-expressing proximal/medial renal vesicles are tightly linked. | [88] |
| LAMP5 | |||||
| HNF4A | |||||
| Pharynx | 10× Genomics | Parathyroid mTEC cTEC | Irf628 | Hippo signaling is active in the developing thymus, but absent in the Foxn1 thymus, suggesting that this pathway may function downstream of Foxn1. | [89] |
| Trp63 | |||||
| Vim29 | |||||
| Ovary | 10× Genomics | Vascular smooth muscle cells Ovarian luminal epithelial cells Stromal progenitor cells | Foxl2l | Inhibition of BMP and Wnt signaling pathways can keep prefollicular cells in an undifferentiated stem cell-like state. | [90] |
| Wnt9b | |||||
| Nanos2 |
| Cancer | Method | Stromal Cell Subtypes | Key Differential Genes | Mechanisms | References |
|---|---|---|---|---|---|
| Gallbladder cancer | 10× Genomics | Lymphocytes | CTLA4 TIGIT | Immunoproteins CTLA4 and TIGIT are highly expressed in CD8+ T cells, and bile acid and fatty acid metabolism levels are disturbed. | [98] |
| Macrophages | |||||
| Dendritic cells | |||||
| HL | 10× Genomics | Macrophages | LAG3 FOXP3 | Differential protein LAG3 and FOXP3+ T cells increase, leading to HL. | [99] |
| T cells | |||||
| B cells | |||||
| Lung adenocarcinoma | 10× Genomics | Macrophages | SFTPA2 | High expression of the angiogenic markers VWA1 and HSPG2 through the TGFβ and JAK/STAT signaling pathways lead to an elevated expression of genes, such as EGFR. | [100] |
| NK cells | CXCL9 | ||||
| T cells | EGFR | ||||
| PDAC | 10× Genomics | Endothelial cells Fibroblasts | HIF1A | The expression levels of cell type-specific markers for epithelial–mesenchymal transition (EMT+) cancer cells, activated fibroblasts (CAFs), and endothelial cells are strongly correlated with patient survival. | [101] |
| COL1A1 | |||||
| VEGFA | |||||
| PitNETs | 10× Genomics | Fibro fibroblasts | LHB | The differential gene SOX9 is highly expressed in tumors expressing T-PIT and SF-1 (P11), leading to transcriptional dysregulation in tumors. | [102] |
| Endothelial cells | ZFP36 | ||||
| Immune cells | BTG1 | ||||
| Colorectal cancer | 10× Genomics | Fibroblasts | FABP4 | The Wnt signaling pathway is activated and promotes granulocyte migration, resulting in abnormal ferroptosis. | [103] |
| T cells | SPP1 | ||||
| B Cells | RBP7 | ||||
| Prostate cancer | 10× Genomics | T cells | KRT5 KLK3 TP63 | Elevated KLK3 in T cells inhibits TNF-α, leading to prostate cancer. | [104] |
| B Cells | |||||
| HGSTOC | 10× Genomics | Lymphatic endothelial cells | COMP | Activation of IL6 and JAK/STAT in fibroblast and HGSTOC cancer cell subsets is involved in pathogenesis. | [105] |
| Myofibroblasts | LTBP2 | ||||
| Plasma cells | TGFBI | ||||
| NSCLC | 10× Genomics | CD8+ T cells | SERPINA9 | Increased expression of CD54 and decreased expression of CD62L in CD8+ T cells led to the development of lung cancer. Furthermore, CD20+ B cells produced low levels of SERPINA9 and directly promoted the growth of non-small lung cancer cells. | [106,107] |
| CD4+ T cells | EGFR | ||||
| B cells | CD83 | ||||
| AITL | 10× Genomics | B Cells | XCL2 | Upregulation of the chemokines XCL2 and XCL1 results in deranged metabolic levels of the biomarkers CD73 and CXCR5 in CD8+ T and AITL CD19+ B cell populations. | [108] |
| T cells | XCL1 | ||||
| Plasma cells | CXCR5 | ||||
| Breast cancer | 10× Genomics | Natural killer cells | BDH2 | Upregulation of aerobic glycolysis and mitochondrial oxidative phosphorylation leads to dysregulation of the metabolic level of CD8+ T cells and T cells. | [109,110,111,112] |
| T cells | DECR1 | ||||
| B cells | PHLDA2 | ||||
| Liver and biliary tumors | 10× Genomics | B cells | MALAT1 | The metabolically dominant organoid HCC272 can remodel the tumor microenvironment by accelerating glucose, enhancing hypoxia-induced HIF-1 signaling, and lead to upregulation of NEAT1 in CD44 cells, thereby inducing hyperactivation of Jak-STAT signaling. | [113] |
| CD44 cells | NEAT1 | ||||
| HCC272 cells | SAT1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Sun, S.-T.; Zhang, X.-Y.; Ding, H.-R.; Yuan, Y.; He, J.-J.; Wang, M.-S.; Yang, B.; Li, Y.-B. The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives. Int. J. Mol. Sci. 2023, 24, 2943. https://doi.org/10.3390/ijms24032943
Wang S, Sun S-T, Zhang X-Y, Ding H-R, Yuan Y, He J-J, Wang M-S, Yang B, Li Y-B. The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives. International Journal of Molecular Sciences. 2023; 24(3):2943. https://doi.org/10.3390/ijms24032943
Chicago/Turabian StyleWang, Shuo, Si-Tong Sun, Xin-Yue Zhang, Hao-Ran Ding, Yu Yuan, Jun-Jie He, Man-Shu Wang, Bin Yang, and Yu-Bo Li. 2023. "The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives" International Journal of Molecular Sciences 24, no. 3: 2943. https://doi.org/10.3390/ijms24032943
APA StyleWang, S., Sun, S.-T., Zhang, X.-Y., Ding, H.-R., Yuan, Y., He, J.-J., Wang, M.-S., Yang, B., & Li, Y.-B. (2023). The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives. International Journal of Molecular Sciences, 24(3), 2943. https://doi.org/10.3390/ijms24032943