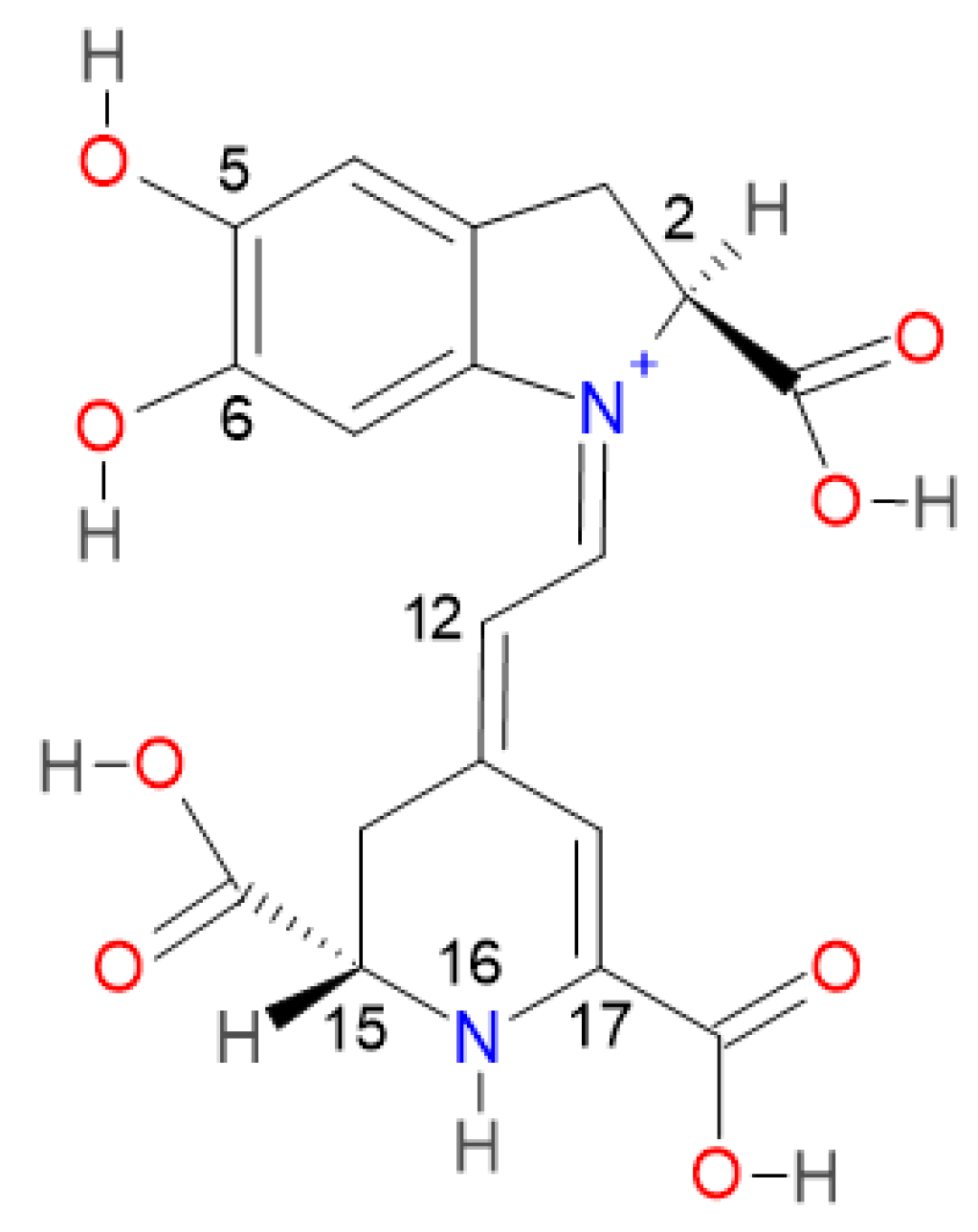
2.2. Prediction Model for Clusterization and Computation of pKa
Consider the first round of deprotonation for the Bd. According to [
12,
20,
42] (see also our simulations), pK
a can be split in two groups: the inferior cluster of pK
a involves the carboxyl groups C15-COOH and C17-COOH (placed in the dihydropyridine ring nitrogen) and the carboxyl group C2-COOH (placed in the pyrrole ring). The superior cluster of pK
a considers the hydroxyl groups C5-OH and C6-OH (placed in the catechol ring), and the group HN16. The carboxyl groups are more acidic [
12,
20,
42], and this can be one chemical justification for this clusterization.
Denote the inferior and superior clusters of the round as and , respectively; where the subindex , denotes the deprotonation route of round , and .
Based on the chemical results given in [
12,
20,
42]:
for all
.
Namely, the
pKa of any deprotonation route of the inferior cluster in the first round is strictly less than any deprotonation route of the superior cluster in the same round. Therefore, the clusters of the first round are given by:
Under the same regularity and chemical conditions of the of the direct approach, the predicted clusterizations for the second round are listed by the left multiplication of
and
with all the non-repeated elements of the those sets, namely:
The same applies for the inferior cluster of the third route of deprotonation, where each element of
and
multiplies
by the left element (without repeated elements). The superior cluster is also given by the left multiplication with
:
Law for clusterization prediction: In general, the deprotonation routeis located at the inferior cluster; for arbitrary previous deprotonations, i.e., every route finishing inis placed in the inferior cluster of pKa. In the opposite way, the deprotonation routegets higher pKa, and it is located at the superior cluster.
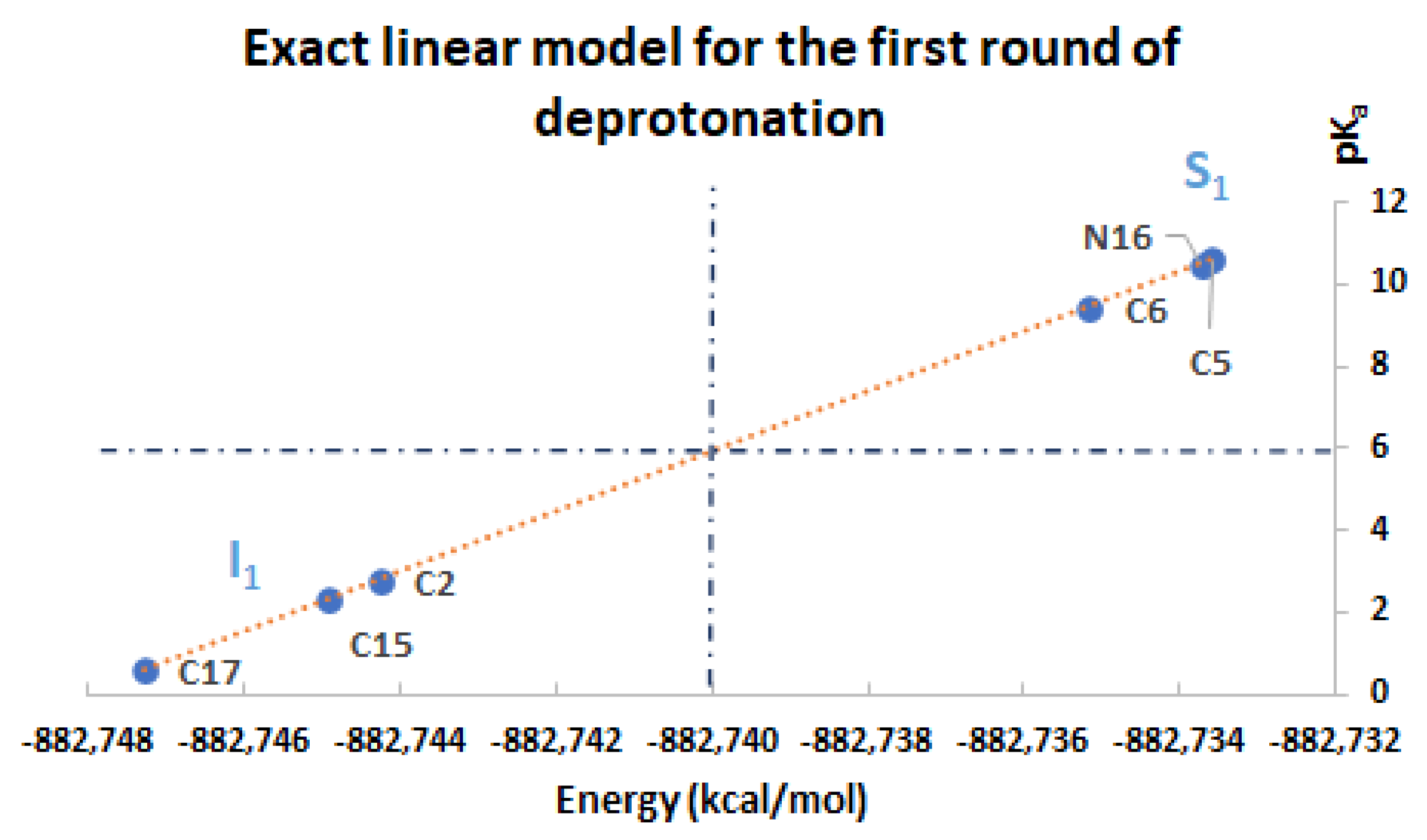
Moreover, assume that
(see the simulations); in this case, the prediction of the high
pKa values for
can be obtained directly from the energies of
by using the linear model:
If the
pKa values for
are available, then we can predict the values for
by using the opposite model:
In the same manner, we can predict the superior (inferior) clusters of pKa by using the available energies under certain permutations, which are supported by the simulations.
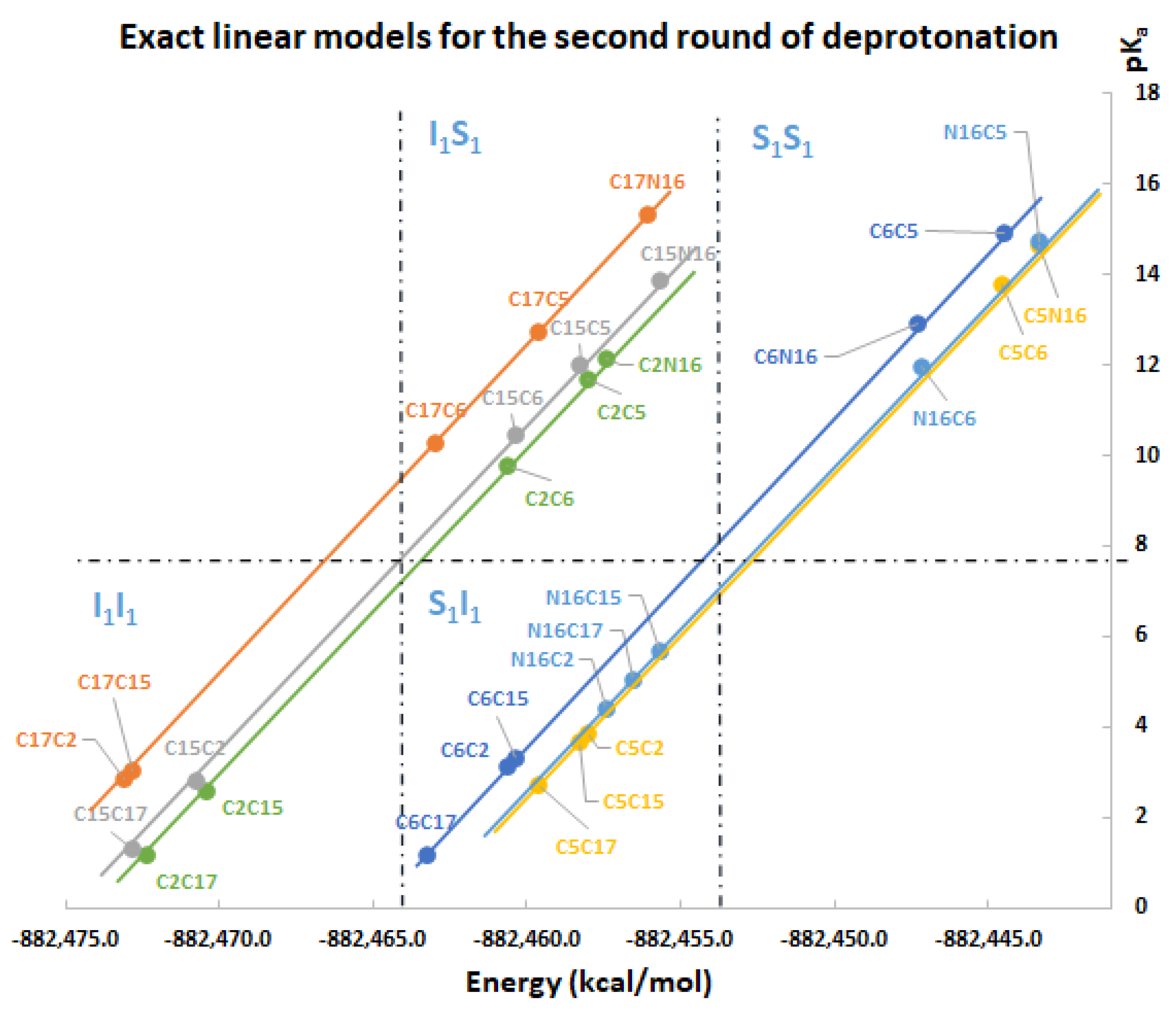
Table 3 shows the equations for prediction of all the deprotonation routes based on the parallel linear models with positive constant slope m. As before, each pair of successive Equations ((10)–(11),(12)–(13),(14)–(15),(16)–(17)) is based on certain energy assumption supported by the data of the complete exploration of the deprotonation routes.
Proof. The proof of this result just follows from the linear model for pKa in terms of energies and the inequality for the corresponding energies:
for all
. See the
Supporting Information. □
Remark 1. In practical situations, we do not require the application of all the pairs of models; rather, we just need to compute the energy in the expert software Gaussian for the lowest pKa. We prefer the energy computation of the inferior clusters (more stable), which will provide the best deprotonation route. Explicitly, the method recommends the computation in Gaussian of the following energies by applying the definition:
And the approximations for lowest energies verified in the second round:
and provide 18 inferior deprotonations.
and provide 18 inferior deprotonations.
and via Equation (10) provide 9 deprotonations.
Finally, if we include the 6 computations of , then only 51 of the 120 possible deprotonations can be performed if the conditions of the linear model are accomplished.
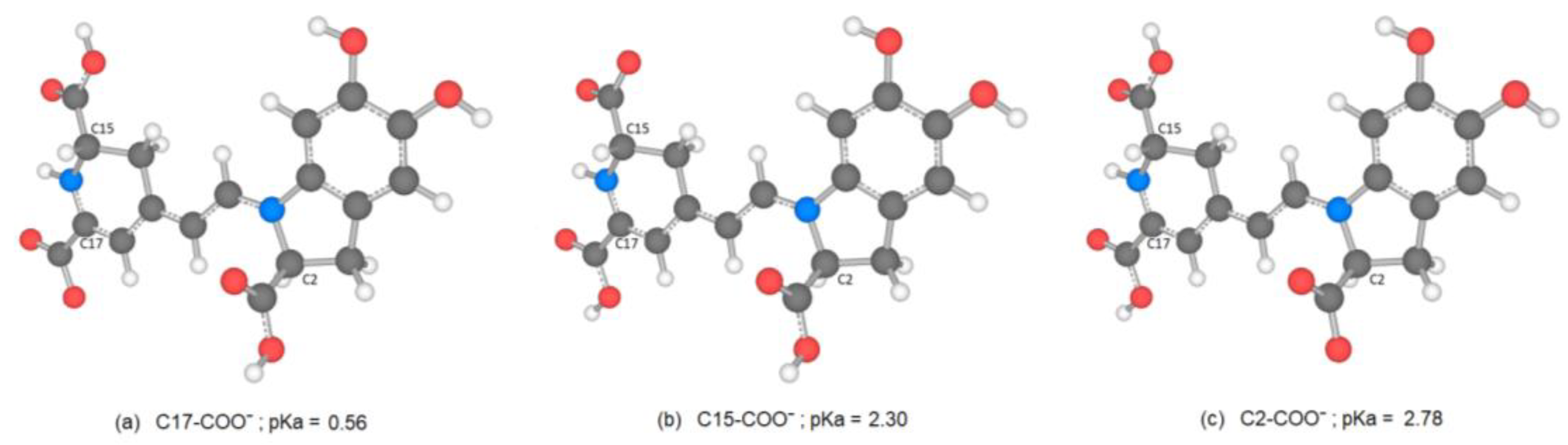
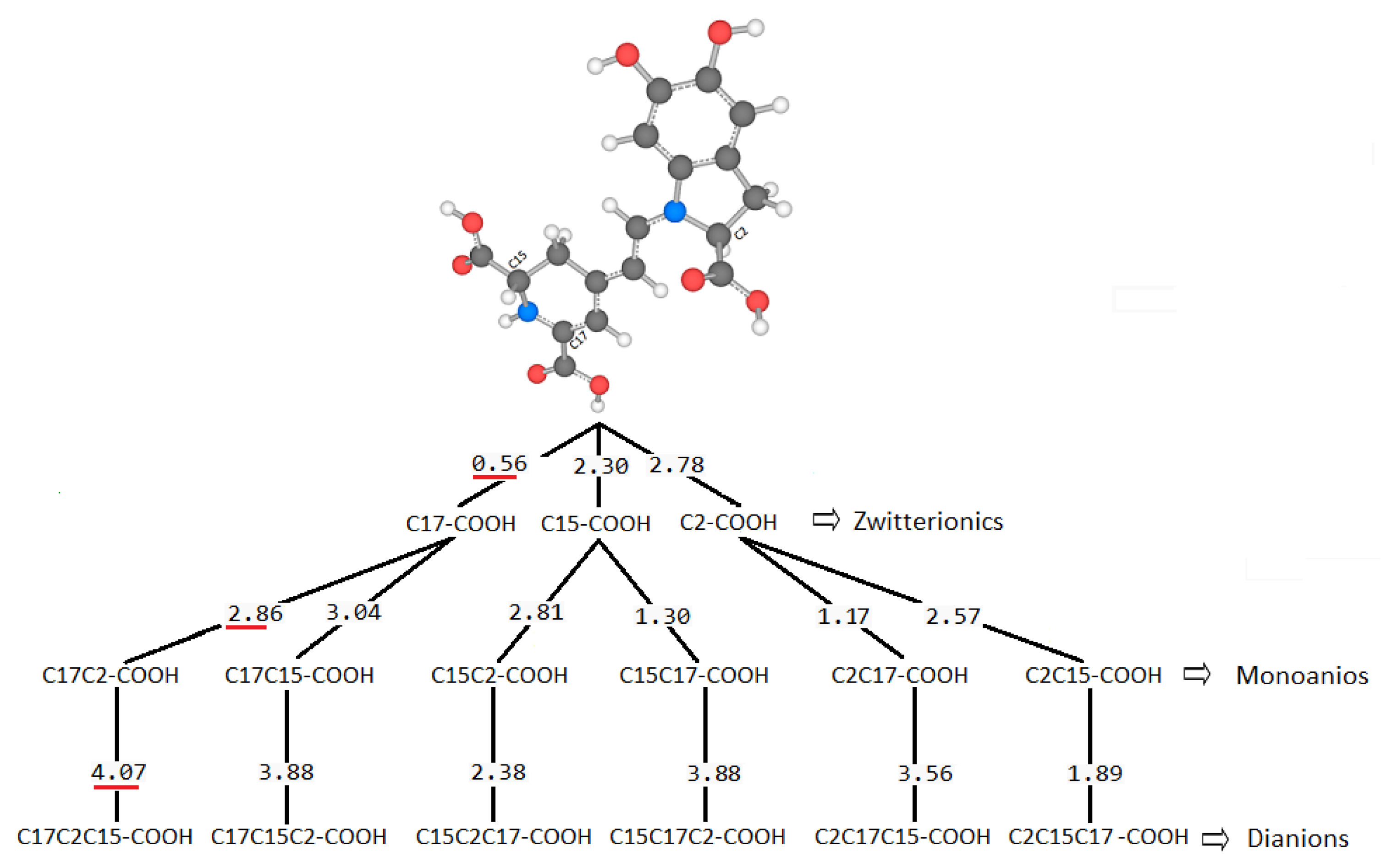
There are several discrepancies in the best route of deprotonation. Reference [
20] suggests different best deprotonation routes that are reached in the carboxyl group, i.e., C17C2C15 (see
Figure 6). However, according to [
10,
20] the pathway starting in H-N16 is as possible as the referenced carboxyl routes.
It is easy to trace the source of this important assertion about H-N16 and C2-COOH in a number of papers. With the addressed discrepancies related to C17-COOH, H-N16 and C2-COOH, in general, there is not a consensus about the best deprotonation route.
We highlight, for example, the work [
41] that supports the N16-H pathway by experiments. Just as other areas propose other options without moving away from the chemical context, they open the possibility of discovering different routes that are not classically possible. The references are open to all the options of deprotonation routes from a theoretical and/or experimental point of view.
However, suppose we extend our observation to other possible routes generated by the permutations of the order of the deprotonations. In that case, we can notice two aspects, one related to the property of pKa commutativity and the other with the inheritance of the local minimum of energy between the different extraction levels. Both properties will not fulfill all the deprotonations.
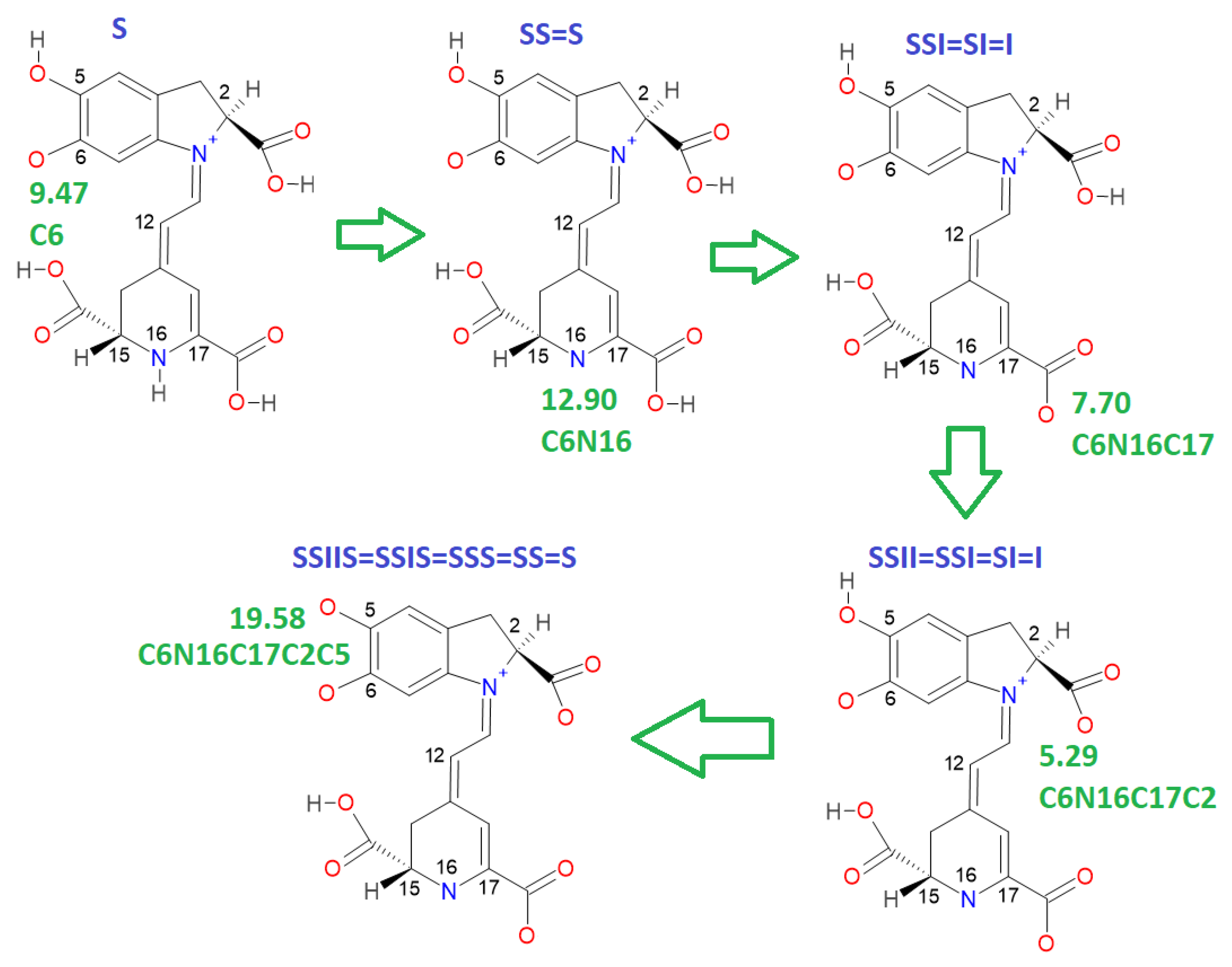
This paper also advances the prediction of non-complete explored rounds. In particular, the model proved in
Section 2.2 provides the location of higher deprotonation routes by using the clusterization of the first round. As can be seen in the statement after Equation (7), if we optimize an arbitrary deprotonation for 4, 5, and 6 rounds, we can predict the corresponding cluster. Given that the pK
a is just a simple linear model of the energy, then any associated stable and reasonable value can be promoted to a valid pK
a. We submitted the addressed statement to a test of an interesting route, starting with a high pK
a.
Figure 7 and
Figure 8 show the complete procedure of deprotonations, moving between superior and inferior clusters. The evolution of the pK
a is also described, and the final clusterization is obtained as we have expected; see also
Table 4. This simple law is useful for avoiding unnecessary computations; we just need to select a priori deprotonation routes ending with a carboxyl group, no matter the initial location of the first or middle deprotonations.
For simplicity in the notation, and the left operation in the third column, we have removed the sub-indexes; see the left multiplication operation after Equation (7).
2.3. Alternative Clusterization of the Deprotonation Route via Riemann–Mulliken Distance
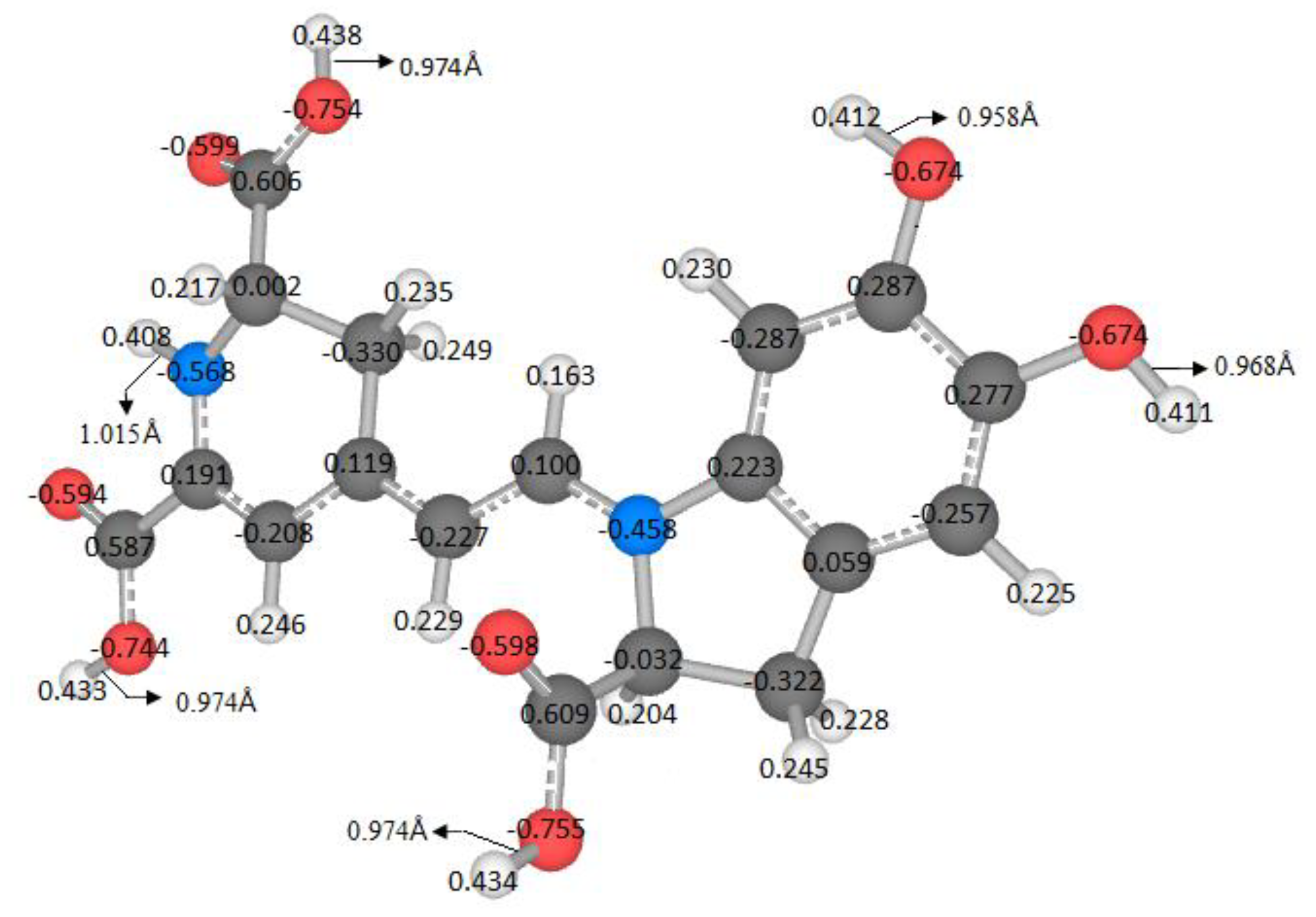
The literature does not explore all possible routes of deprotonation. It is assumed that the local minima of the first deprotonation are successively inherited. The energy or pKa is related to the locus of the molecules. A variable can measure similarity or dissimilarity by using the geometry, the atomic mass, and the Mulliken charges. In this case we defined the Riemann–Mulliken distance (RMD) to compare the parent and the deprotonated configurations. It provides a descriptor using the recent theory of Riemannian geometry of the shape.
This matter is wholly contextualized in the optimization problem because a small change in the coordinates, mass, and charges of the molecules, concerning a reference, implies a low RMD. From the chemical point of view, these few changes can be a slight variation in the pKa. Therefore, we expect a relationship between the pKa and the RMD. However, we do not enter into the details of this function; we will just provide some figures. We will research this topic in a future study.
The variable pK
a strongly depends on the energy; therefore, it is necessary to find an independent variable to study this affinity. Furthermore, an easy-to-measure predictor related to geometry, atomic mass, and Mulliken charges is also required. Due to this situation and the previous analyses, the RMD satisfies the requirements because it can detect minor differences in the geometric arrangement of a molecule and changes in the mass and charge. These variations translate into changes in physicochemical properties. Moreover, if we explore the entire energy surface, we can provide a non-statistical but algebraic-geometric total population space, which allows general conclusions about the deprotonation process. We start with the first deprotonation, and
Figure 9 shows the claimed relation.
However, we have obtained two clusters for pK
a, which are explained by a chemical argument [
12,
20,
42]. The RMD is a good splitter for worst and better affinity. We notice that this separation is an expected behavior in the deprotonation process. We can apply several methods for clusterization, but according to the above-expected relation between pK
a and RMD of the parent (See
Table S1 in Supporting Information) and optimized molecules, we must demand an additional constraint for the two clusters; they must be parallel models. This restriction, which we will not consider in the classical methods for clusterization, motivates us to consider a new technique based on shape theory.
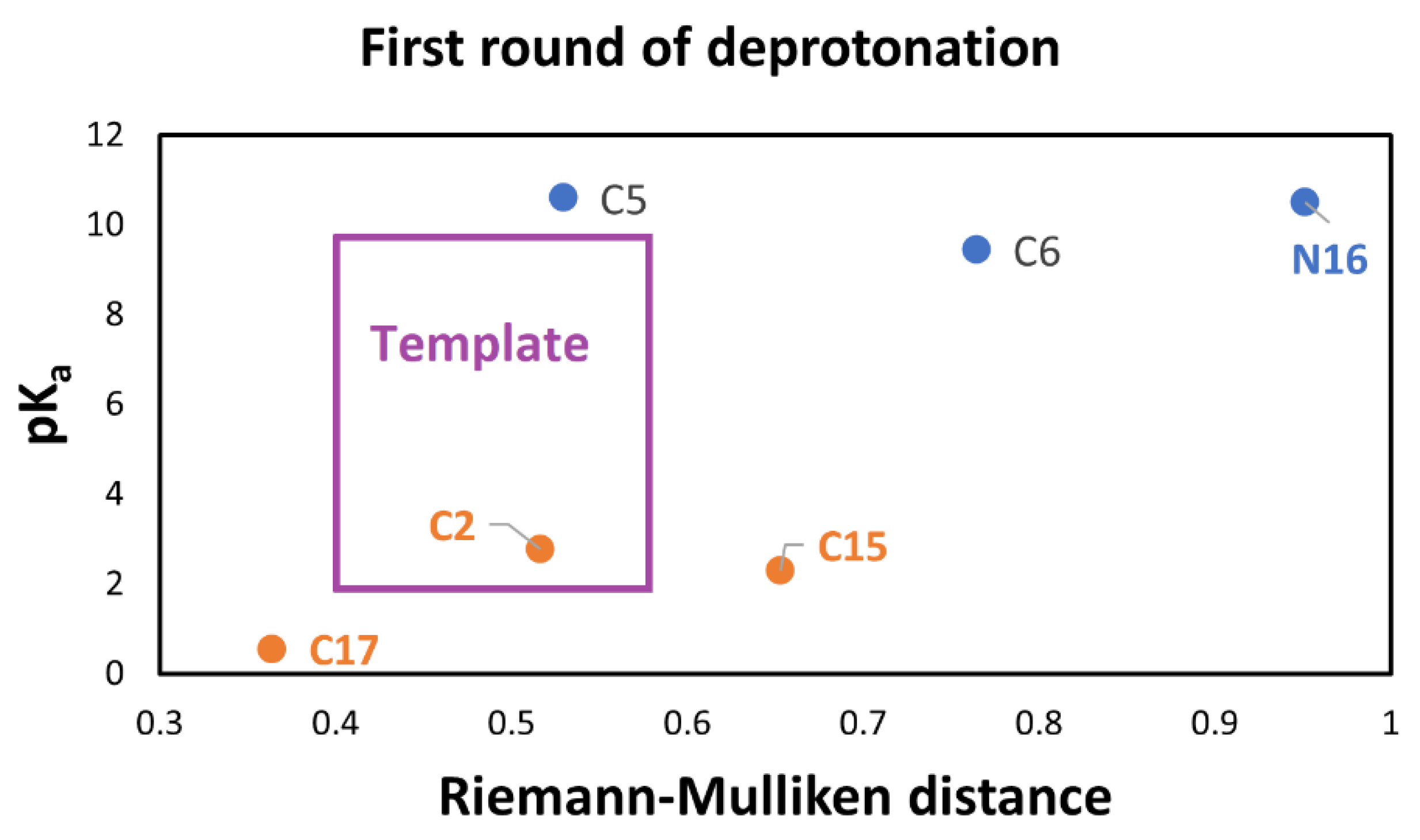
To get to this end, we used algebraic statistics of the six points mixed with the Riemannian geometry information of the population. Given that we expect two parallel linear models, we find all the possible subsets of four points and consider a simple rectangle as the template. The low Riemannian distance (RD) between the template and the four polygons will separate the clusters. The Riemannian distance is not invariant under the permutation of the landmarks. Then an additional step must involve orientable quadrilaterals.
Figure 8 shows the orientable quadrilateral template. We have 15 possible orientable quadrilaterals compared with the template via Riemannian distance. Note that we have used the RD for the clusterization from a geometric point of view based on a large rectangle. The result is the same as the pK
a clusterization. However, we also use the enriched RMD for a geometric-chemical descriptor of the pK
a. In this case, the clusterization is reached by grouping the extremal RMD shown in
Table S18.
The automatic detection by the RD gives two clusters: 1 (low): corresponding to the groups C2-COO-, C17-COO-, and C15-COO-; and 2 (high): C5-COO-, C6-COO-, and N16- as expected.
According to
Figure 9, C17-COO- is the best first deprotonation and also obtains the minimum RMD. However, we do not consider any preferable way of getting the next best deprotonation. To get to this end, we consider the second deprotonation. In this case, we have 30 possible routes.
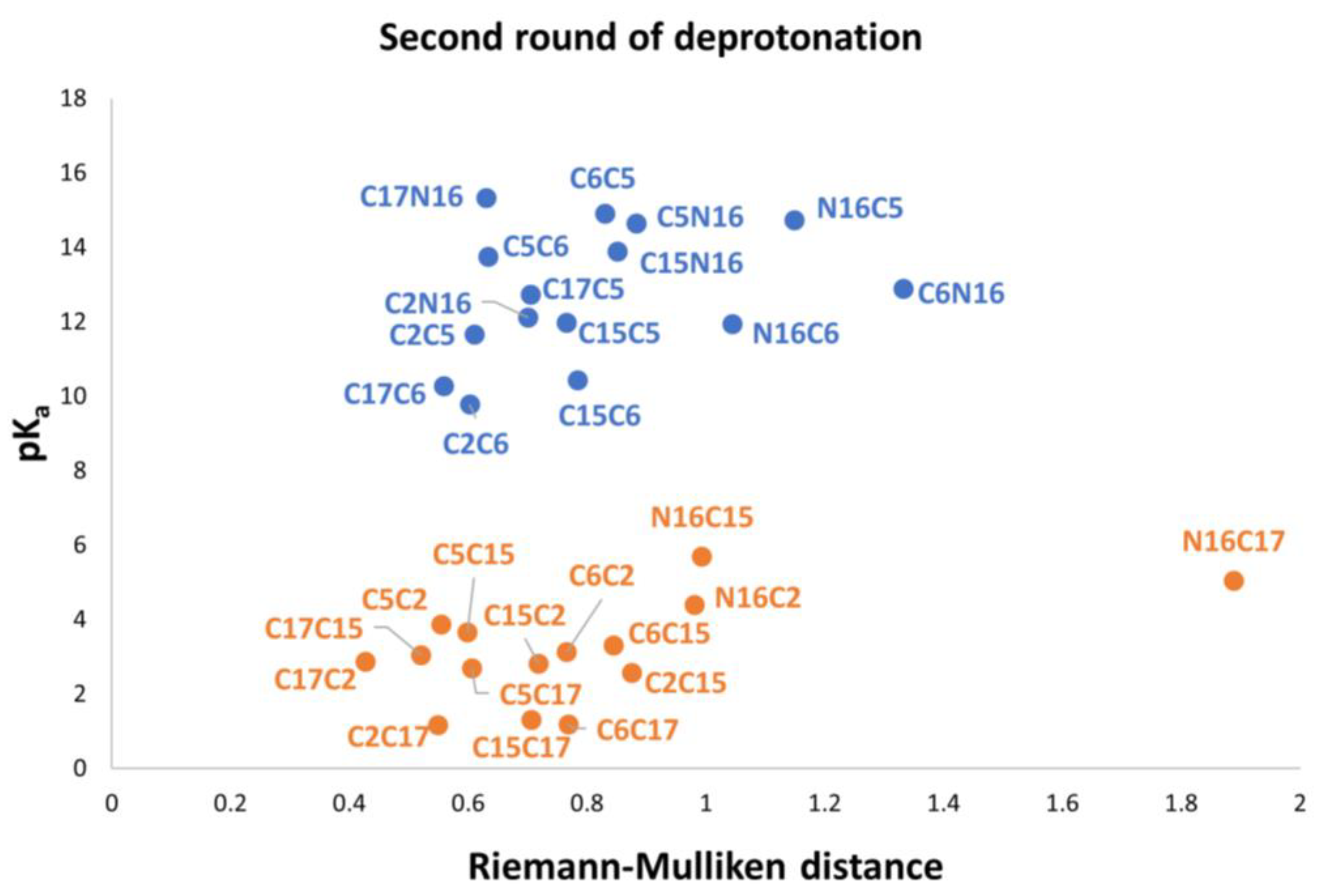
Figure 10 shows the relationship between pK
a and the RMD between the parent and the deprotonated molecules.
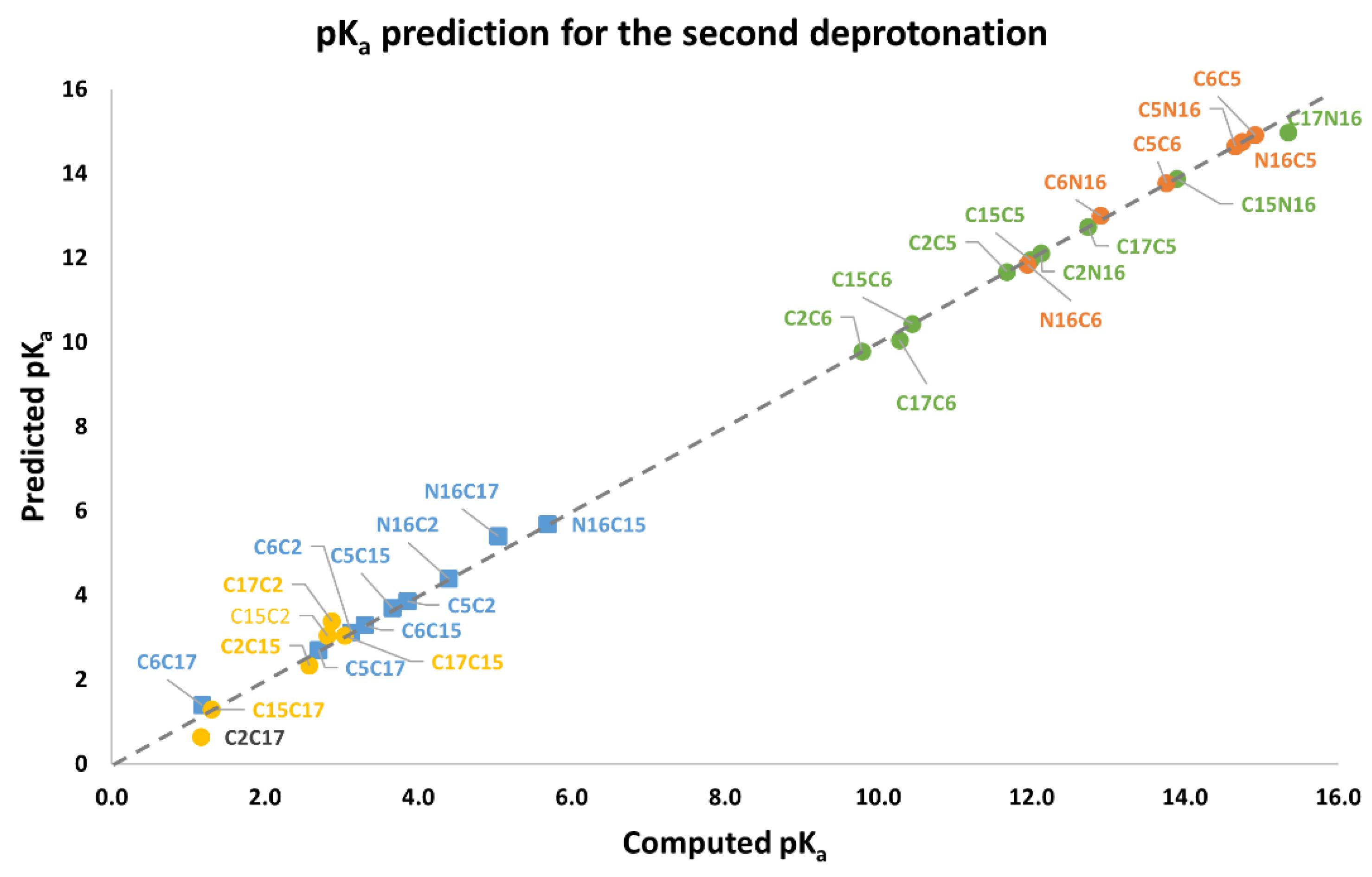
Again, note that the clusters can be easily listed: Each permutation finishing in C2, C15, and C17 belongs to the inferior cluster; the remaining permutations belong to the superior clusters and end with C5, C6, and N16.
We can obtain the same clusterization by using the RD applied to the rectangular template of
Figure 9. We compare the 27,405 oriented quadrilaterals with the same template of the first round of deprotonation.
As in the first deprotonation, we found two clusters. Therefore, we apply the clusterization method via a perfect template and the RD of all possible quadrilaterals. See classification in
Table S19.
Note that the pKa is a linear function of the RMD. It is geometrically distinct on each cluster of the second deprotonation.
Also observed is that the first and second deprotonation numbers have been uniformly distributed in the two clusters. There is no preferable route of minimum energy, and the results will not transferfrom the given first deprotonation.
The best second deprotonation route includes C2-COO- and C17-COO-; however, C6-O-, C17-COO-, and C15-COO-, C17-COO- are also very near in pK
a and RMD. The table shows that the commutativity does not hold. For example, C17-COO- and C6-O- reach one of the highest pK
a; several other issues can be inferred from both clusters (see Equations (8) and (9)). Finally, we considered the third deprotonation.
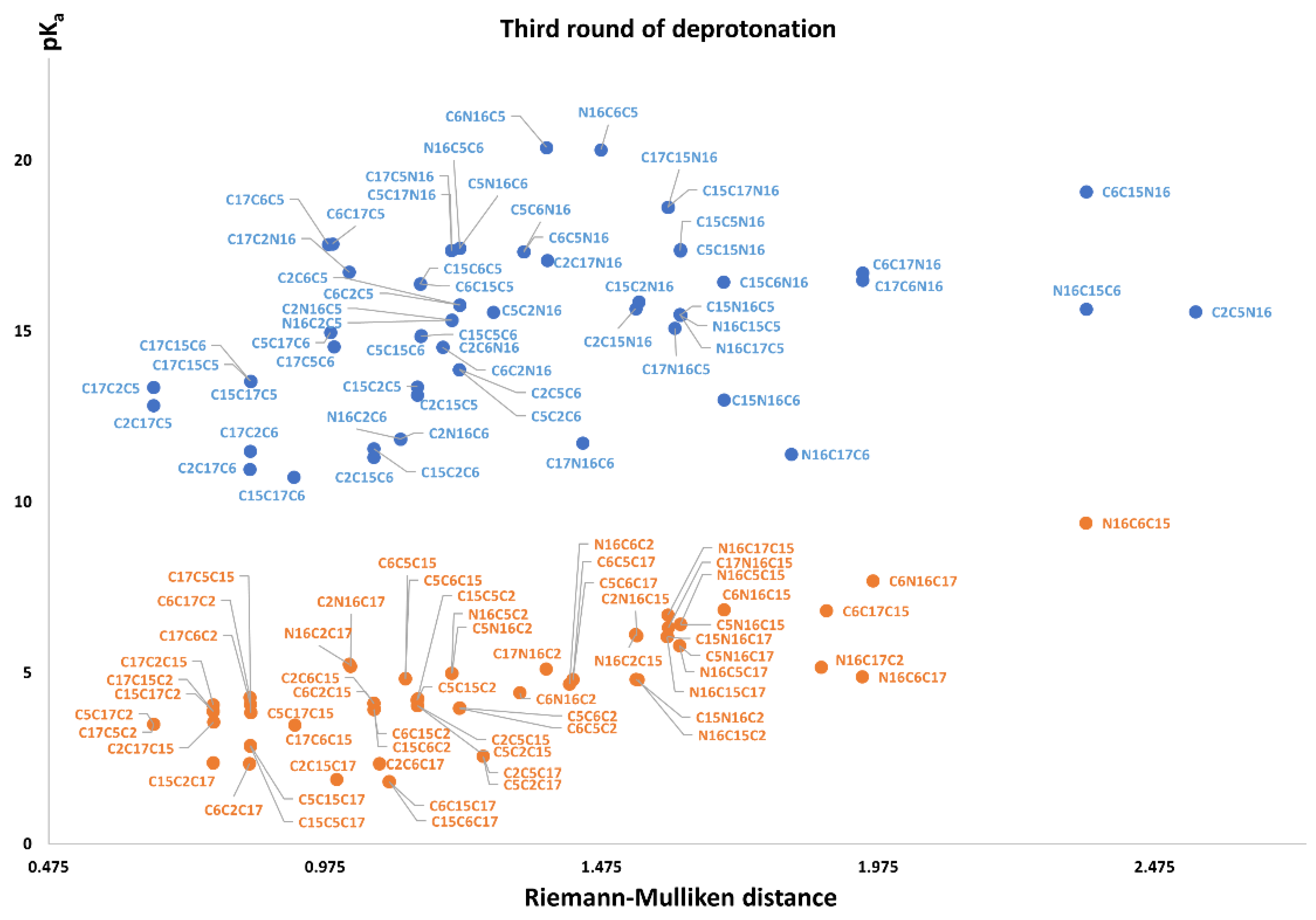
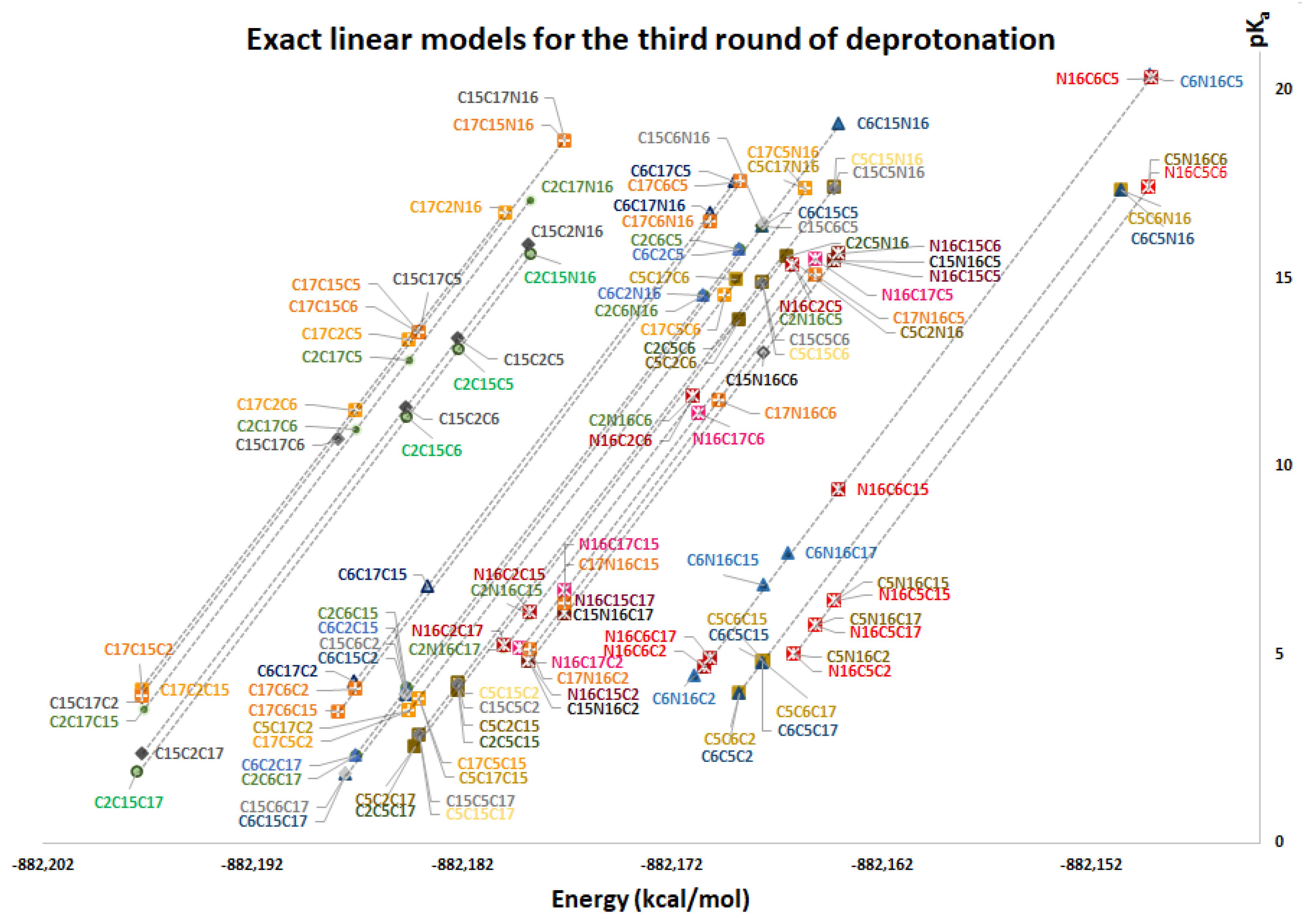
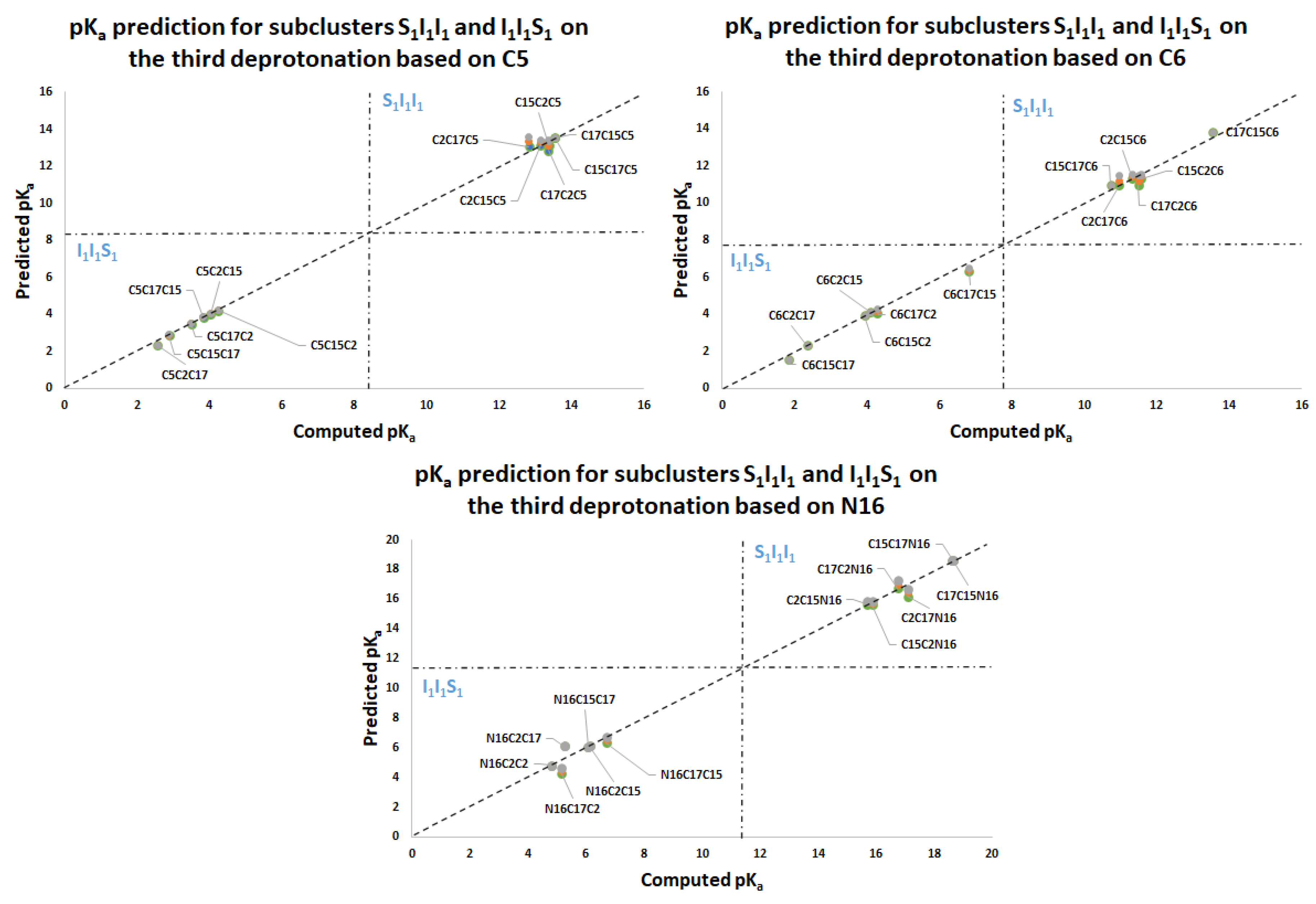
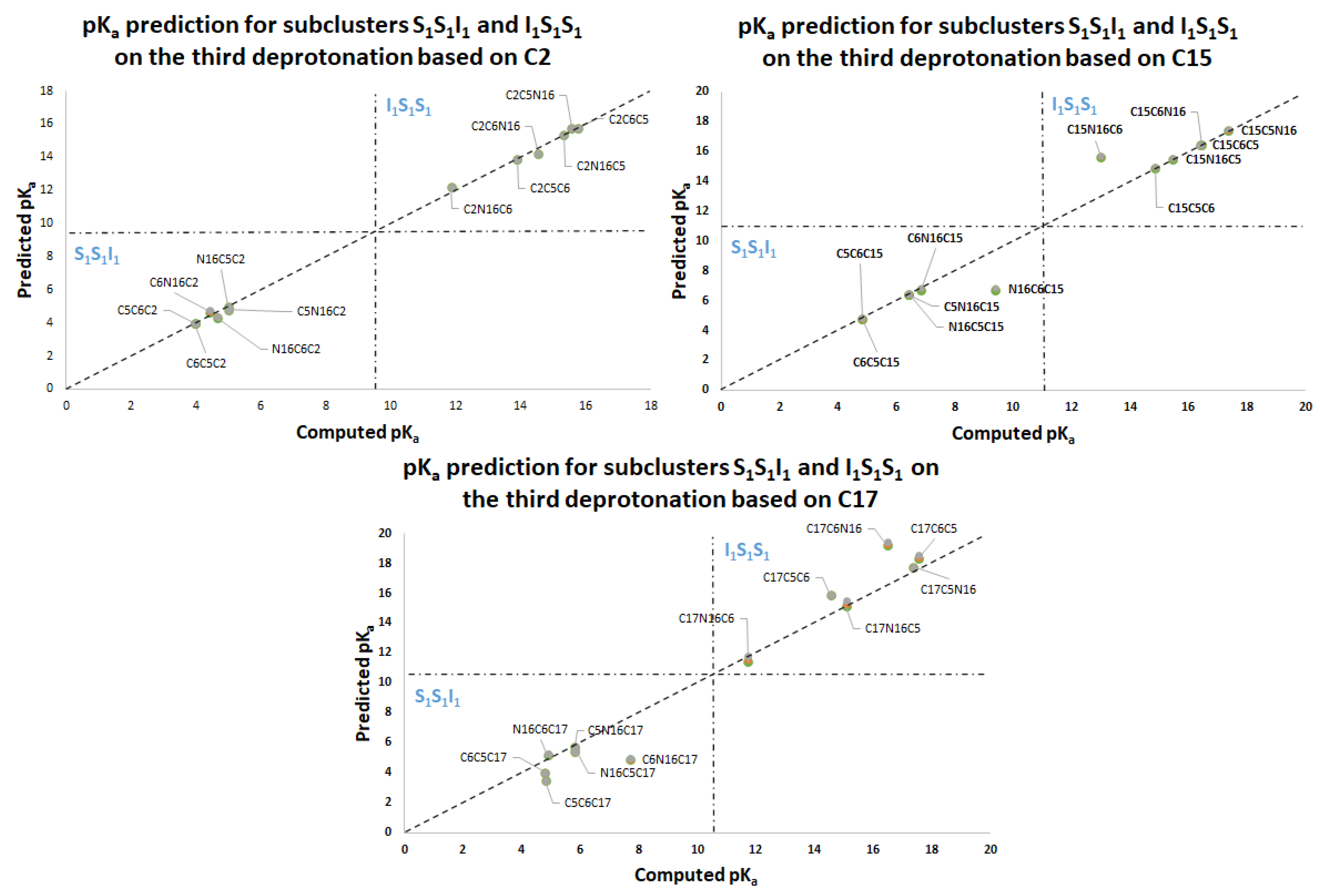
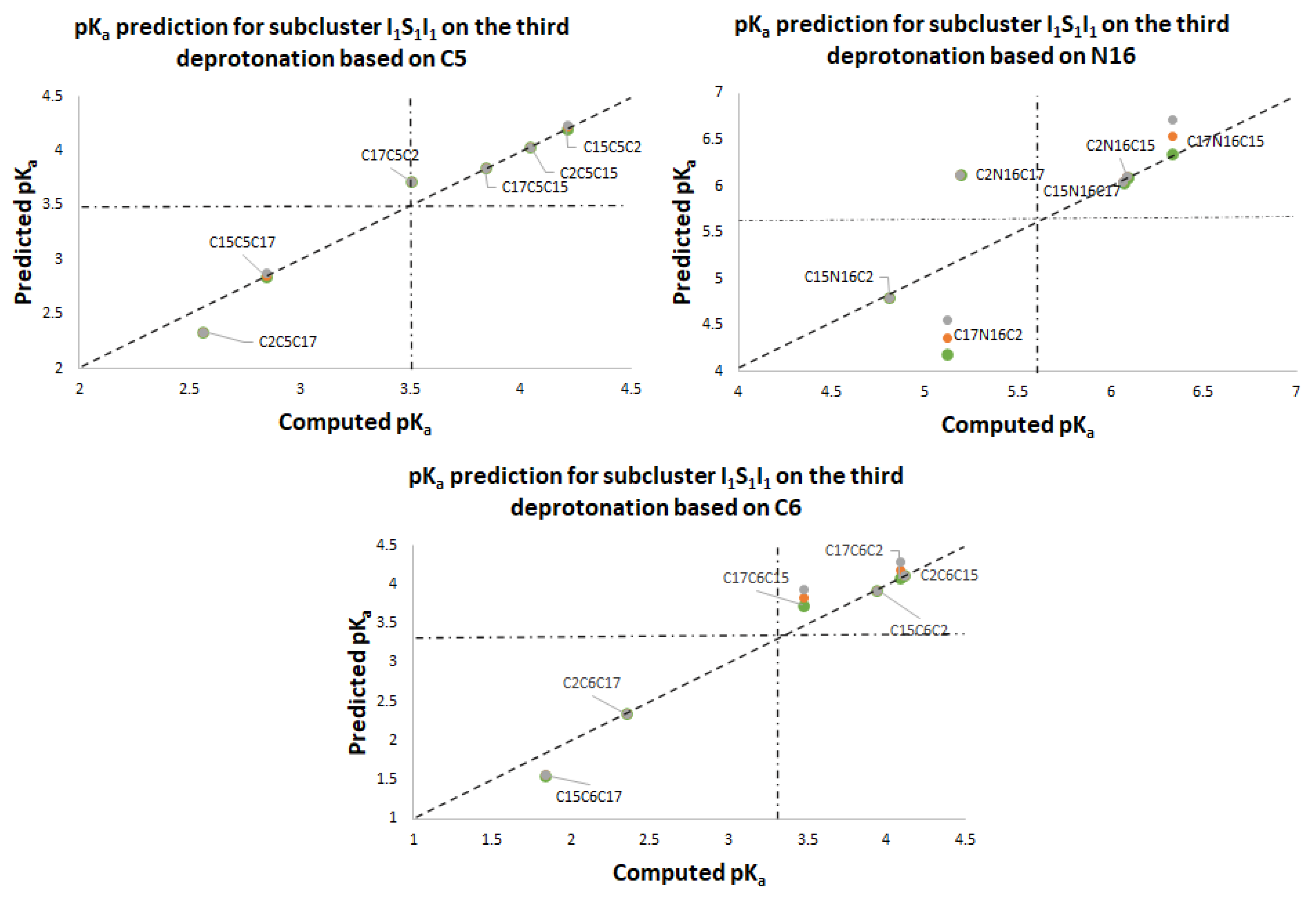
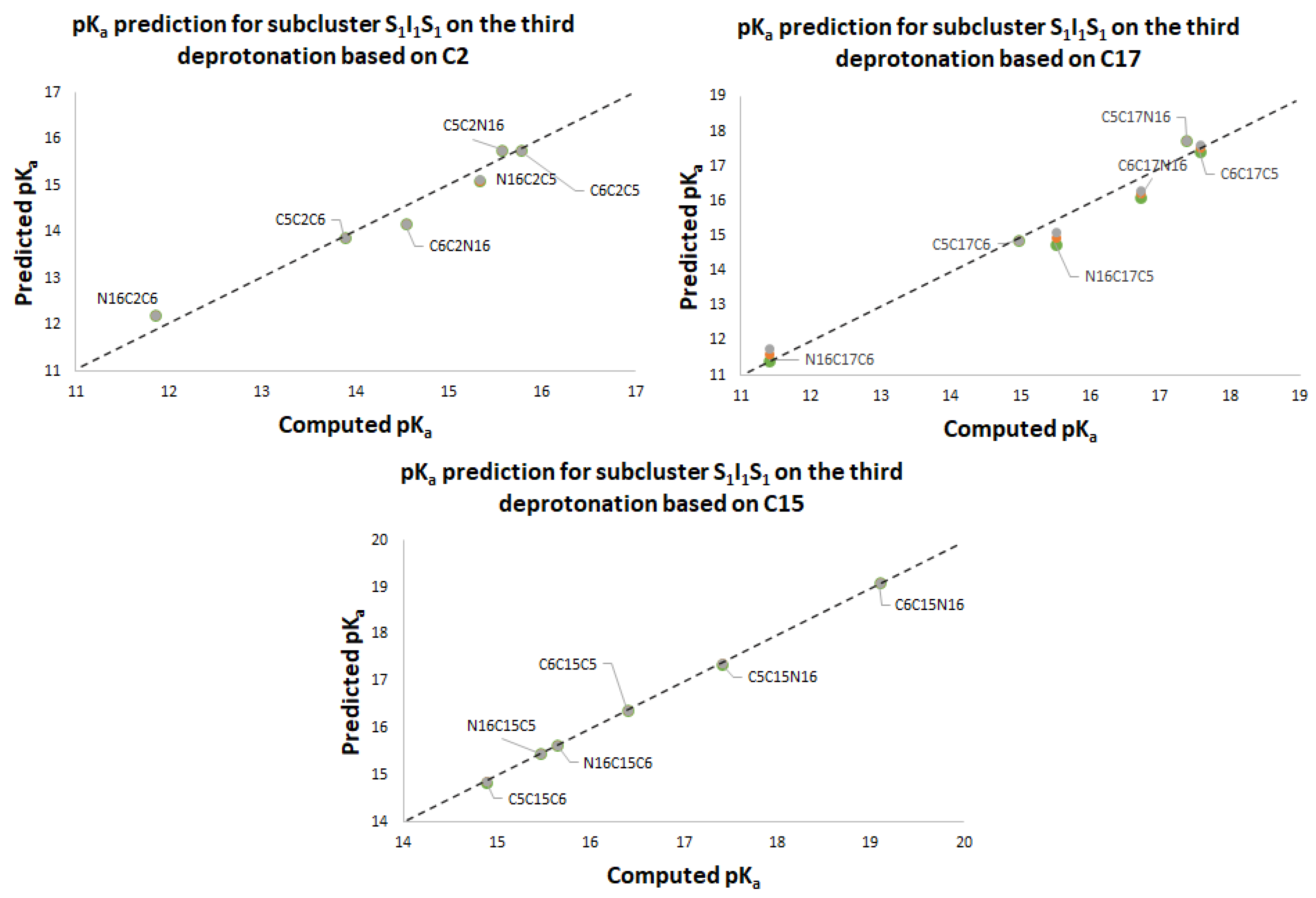
Figure 11 again shows two differentiated clusters.
The inferior cluster (orange) is strictly constituted by all the permutations of the low cluster in the second round (dihydropyridine ring nitrogen: C2, C15, C17) and the superior and inferior cluster of the first round. The complementary set constitutes the superior cluster (blue). Note that the third deprotonation follows a rule that can be used for optimization of the computations. The superior cluster does not require computations, because we know that the pK
a will be high. According to the prediction model, we need to compute 51 of the 60 deprotonation routes of the inferior clusters (see
Section 2.2).
The RD of 8,214,570 orientable quadrilaterals with the template gives the two clusters recognized in
Table S20. Again, the permutations of each triple provide a uniform distribution around the two clusters. Non-expert behavior can be traced by knowing only performance in the previous two deprotonations. Commutativity is also problematic. The best third deprotonation route corresponds to C6-COO-, C15-COO-, and C17-COO-. The prediction model of
Section 2.2 guarantees this route because the last deprotonation corresponds to a carboxyl group. The reader can verify that all the routes of the inferior cluster finish in C2, C15, or C17.
Note also that the pKa is linearly related to the RMD. We will study this interesting relation in future work.
Furthermore, the pK
a commutativity of deprotonation is not maintained. For example, if we switch the deprotonation of mono-anion C6-O-, C15-COO- in the second round, the behavior of the mono-anion C15-COO-, C6-COO- is opposite since it ranks in the high cluster. The respective pK
a values are 3.30 and 10.44.
Tables S21–S28 show the energy differences under permutations. Only some equalities appear, supporting the hypothesis of the prediction models given in
Section 2.2. Of course, this is a simple consequence of the prediction model in
Section 2.2.
2.5. Molecular Docking
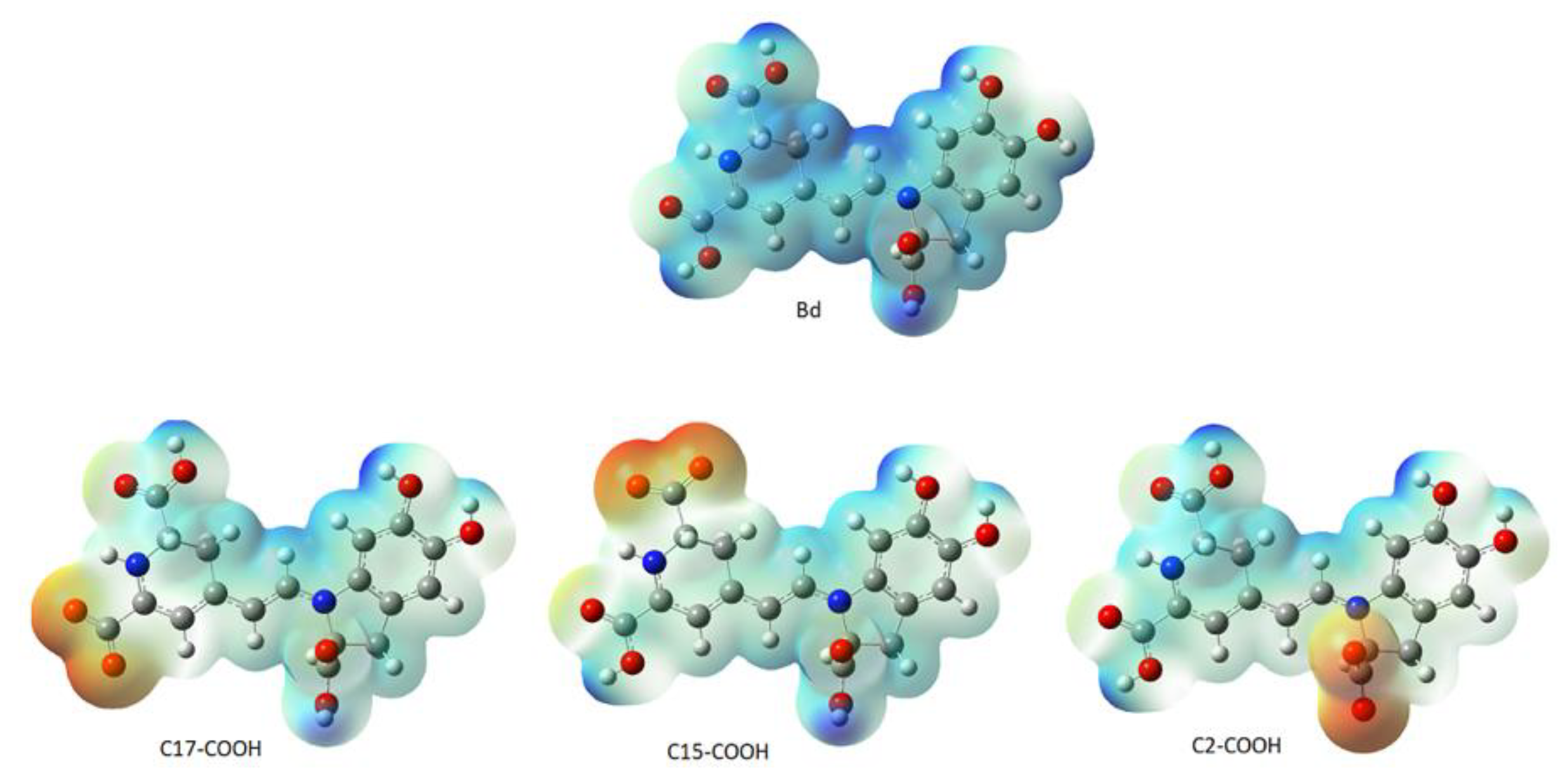
We carried out in silico molecular docking studies to identify binding residues in the active site of the enzyme aldose reductase (AR) in a complex with Bd. Applying this process, the binding affinity of the first ligand pose was −9.6 kcal/mol. The results show that the studied compound interacts favorably with the target through various non-bonded interactions, such as hydrophobic and hydrogen-bonding interactions.
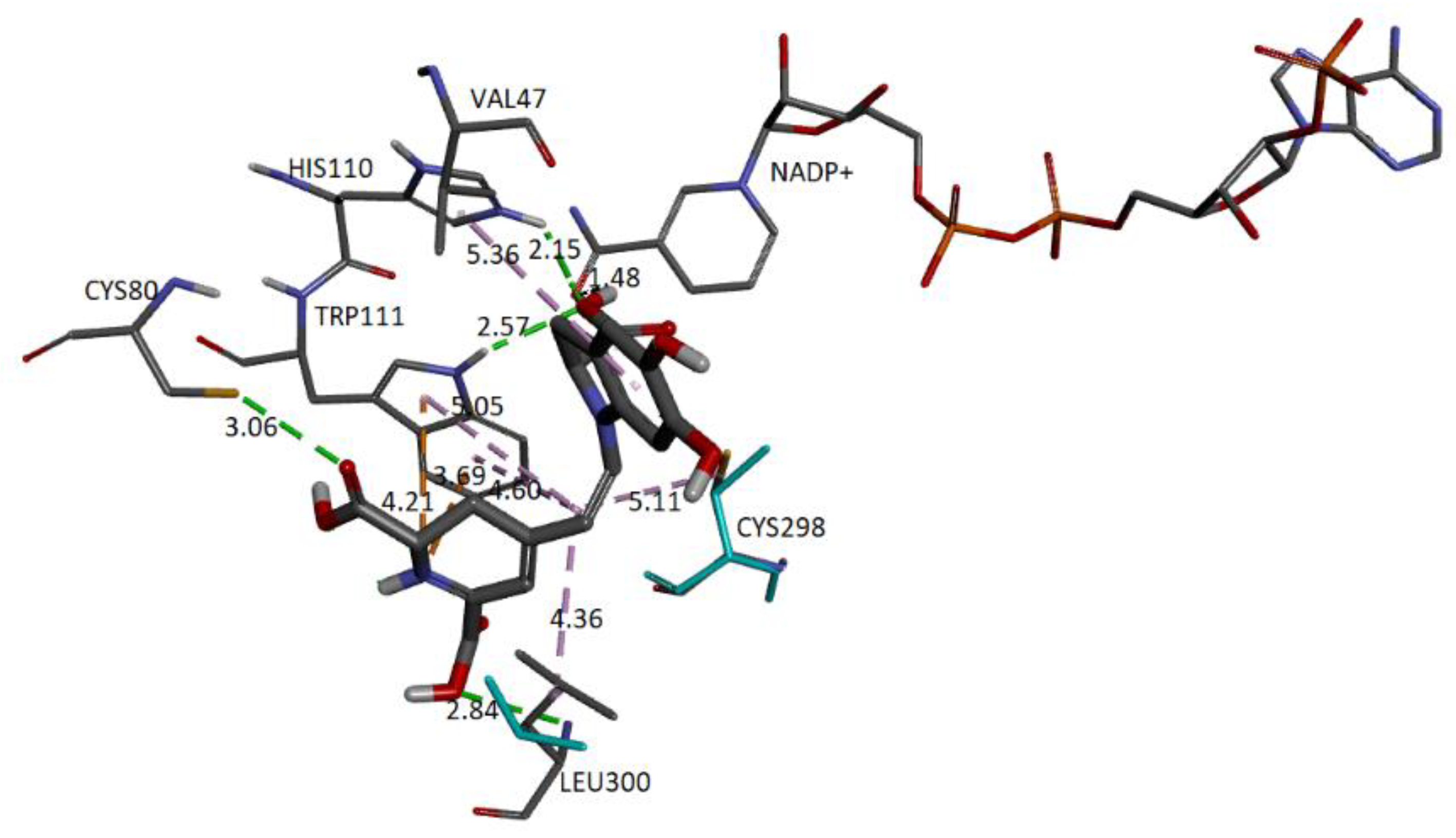
Figure 20 represents the close-up view of the AR active site bound with Bd in the binding pocket.
We can observe that four donor/acceptor hydrogen-bonding interactions are formed between the receptor residues Cys80, Leu300, His110, and Trp111 and carboxyl groups C17-COOH, C15-COOH, and C3-COOH, respectively. In addition, electrostatic pi–cation interactions occur between the dihydropyridine ring nitrogen and Trp111. Other hydrophobic or van der Waals contacts are formed by the spacer C12 of Bd and amino acids Trp111, Cys298, and Leu300. Finally, the catechol group presents a π-Alkyl with Val47. The carboxylate bound to C2 interacts with charged nicotinamide through hydrogen. In
Table 5, we record information about the observed interactions.
Interactions of the complex AR enzyme with other ligands have been reported, consistent with our results. Therefore, with the residue, His110 appears in lidorestat [
43], sulindac [
44], citronellel [
45], dehydrobenzoxazinone [
29], and betanidin. The residue Leu300 appears in lidorestat, sulindac, and betanidin. The residue Trp111 corresponds to lidorestat [
43] and betanidin. The residue Cys298 appears in sulindac [
44], dihydrobenzoxazinone [
29], and betanidin.
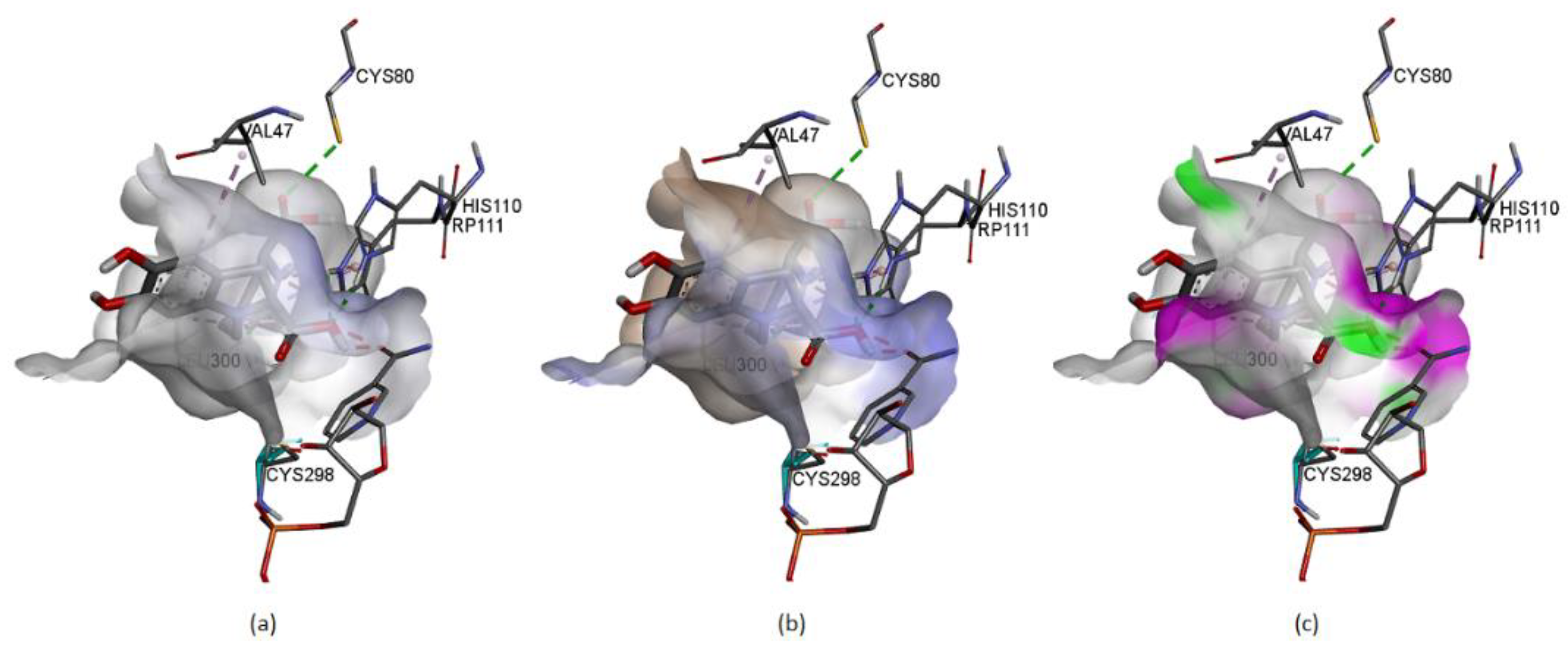
Figure 21 represents some physicochemical properties of binding through surfaces covering the pocket area of the protein and the ligand. In
Figure 21a, the faint blue represents the active site’s basicity. Amber surfaces are regions with hydrophobic binding properties; the molecule occupies the receptor’s active site in the partly polar binding pocket primarily composed of Val47 andLeu300 (
Figure 21b). Hot pink and lime green shades represent hydrogen bond donor and acceptor regions, respectively (
Figure 21c).
To better understand the binding details and point out the relevance of medium acidity in an aqueous solution, we considered the charge alterations of Bd on pH changes proposed in the literature [
42]. It was suggested that at 3.5 < pH < 7.0, a dianion with deprotonated C17-COOH, C15-COOH, and C2-COOH groups appears. Additionally, all carboxyl groups are dissociated in an environment of implicit water at physiological pH, and stabilized conjugated structures favor the type of scavenging reactions [
12]. The molecular docking schema to analyze the interactions between the deprotonated Bd and the active site of the receptor can be seen in
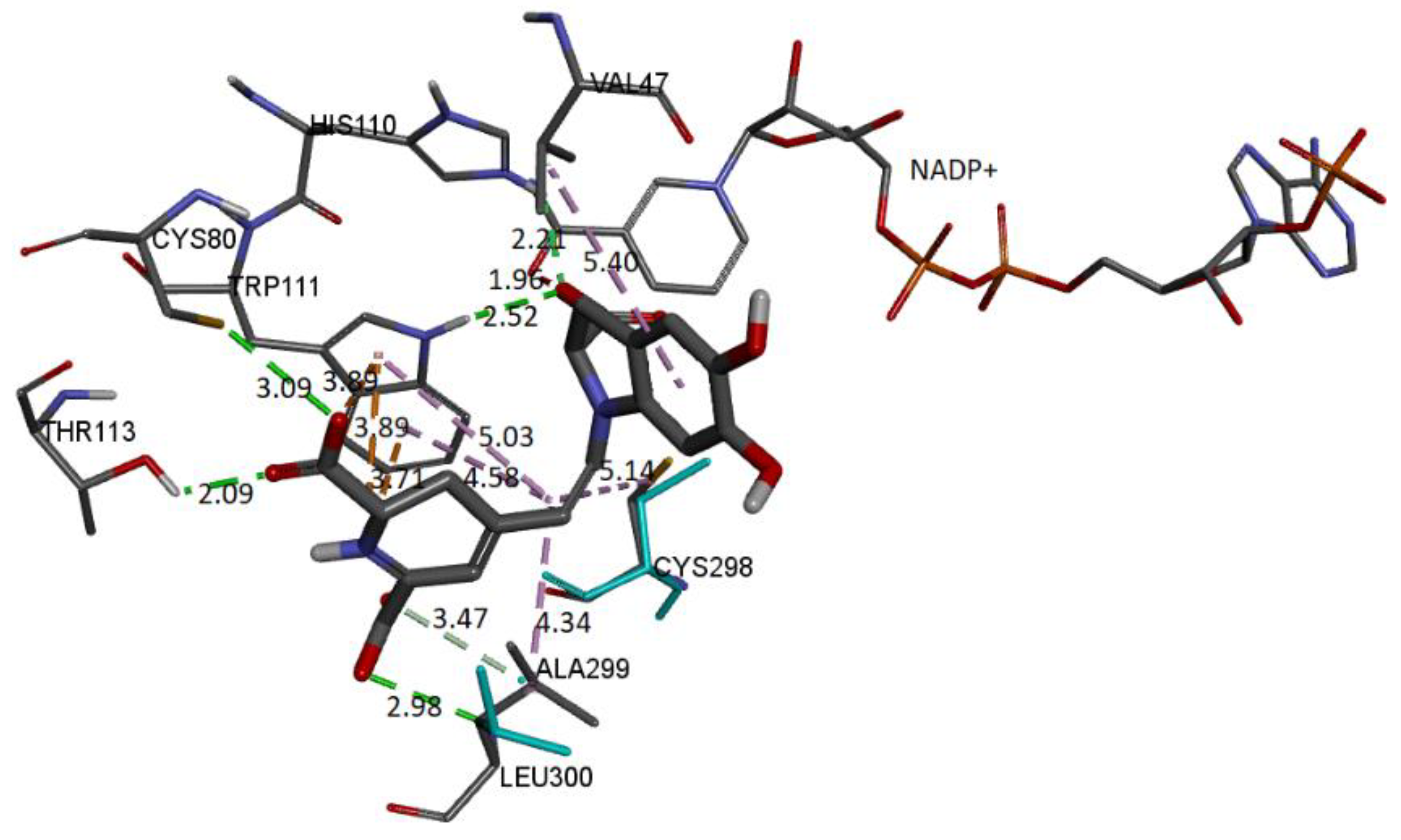
Figure 22.
The molecular docking schema to analyze the interactions between the deprotonated Bd and the active site of the receptor can be seen in
Figure 22. The deprotonated carboxylate attached to the pyrrole ring interacts with NADP+ and forms an H-bond with Trp111 and His110. The other two deprotonated carboxylates also form H-bonds with Cys80 and Leu300. Residues Ala299 and Thr113 interact via H-bond with the ligand. Trp111 residue is in contact with the dihydropyridine ring’s nitrogen and the carboxylic group’s oxygen via π-cation and π-anion interactions, respectively. Hydrophobic interactions occur between the C12 spacer and the Cys298, Trp111, and Leu300 residues. Another hydrophobic interaction occurs between the catechol group and Val47.
Table 6 includes information about the observed interactions.
Further, the binding affinity of the first ligand pose was −9.3 kcal/mol. The receptor amino acids interacting with the ligand are similar to those analyzed for the protonated molecule. However, both structures insert themselves deep into the pocket through mainly hydrogen bonding, electrostatic, and van der Waals interactions. However, it is not a coincidence that all the amino acids participate in these interactions.
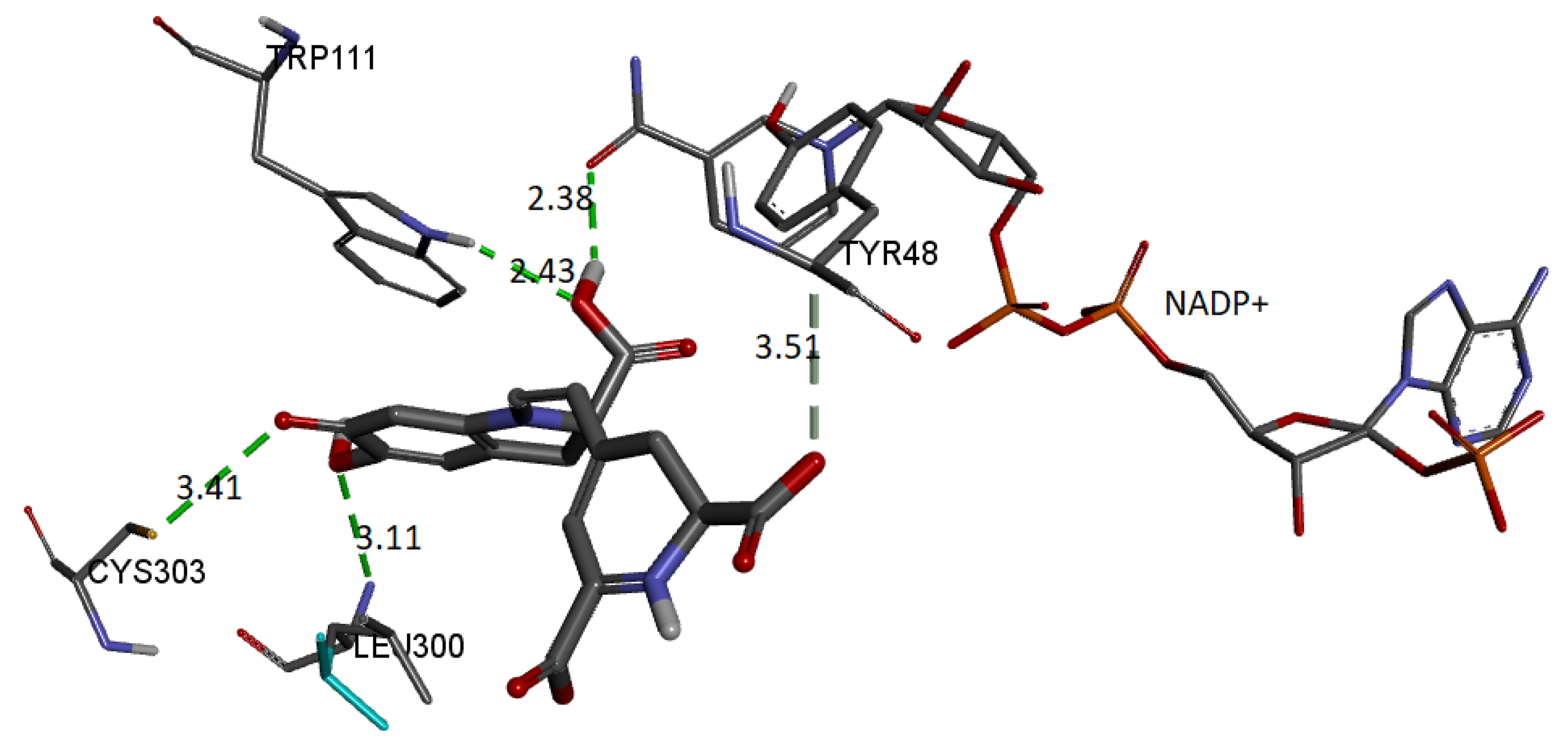
Finally, we show the interaction between the deprotonated molecule (C6C15C17-COOH) and the protein’s active site.
Figure 23 shows these interactions.
The binding affinity of the ligand was −9.5 kcal/mol. The interactions presented are of the hydrogen bond type between the ligand and the residues Trp111 (2.42 Å), Leu300 (3.10 Å), Cys303 (3.40 Å), and Tyr48 (3.50 Å). In addition, the NADP+ molecule also interacts via H-bond with Bd (2.38 Å).
In all cases, the interactions between the receptor and the ligand are consistent with those previously reported. Therefore, we selected the conformation of the AR-lidorestat complex. As a reference, according to our results and those previously reported, the molecule interacts with residues Cys80 and Trp111 through hydrogen bonds and with Leu300, Trp20, and Cys298 through van de Waals contacts. Additionally, it interacts with amino acids Trp79, Phe122, and His110 via another type of contact [
29,
43]. The results obtained from the AR-Bd complex’s behavior agree with those referred to as the AR-lidorestat complex.
Search for Active Sites in Automatic Molecular Docking via Shape Theory
We use a new method to automatically search protein pockets to study the protein-ligand system’s molecular coupling. This method allows us to determine a reference system with Riemann geometric information to analyze the receptor–ligand complex. It is based on the formalism of shape theory and a Lennard–Jones (L–J) potential 6–12 and 6–10. We reported the details of this method in a previous article (under review), and it was implemented in R, a free software environment for statistical computing and graphics [
46]. We applied the algorithm for the automatic search of cavities in the protein and the classification of the pose of the ligand in the protein pocket according to the potential value. The ligands used correspond to betanidin in dianion form with deprotonated C17C15C2-COOH (Ligand 1) and C6C15C17-COOH (Ligand 2).
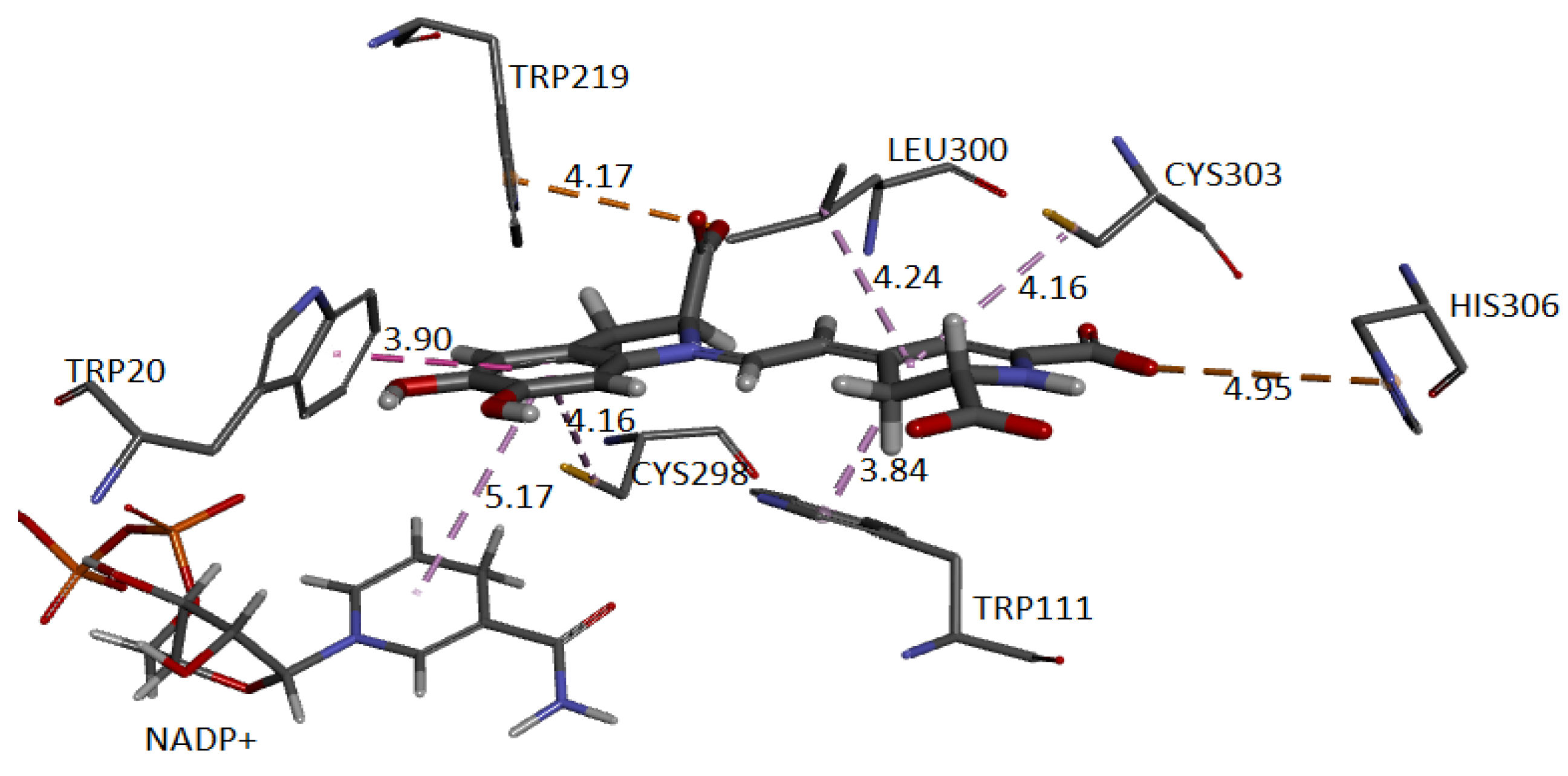
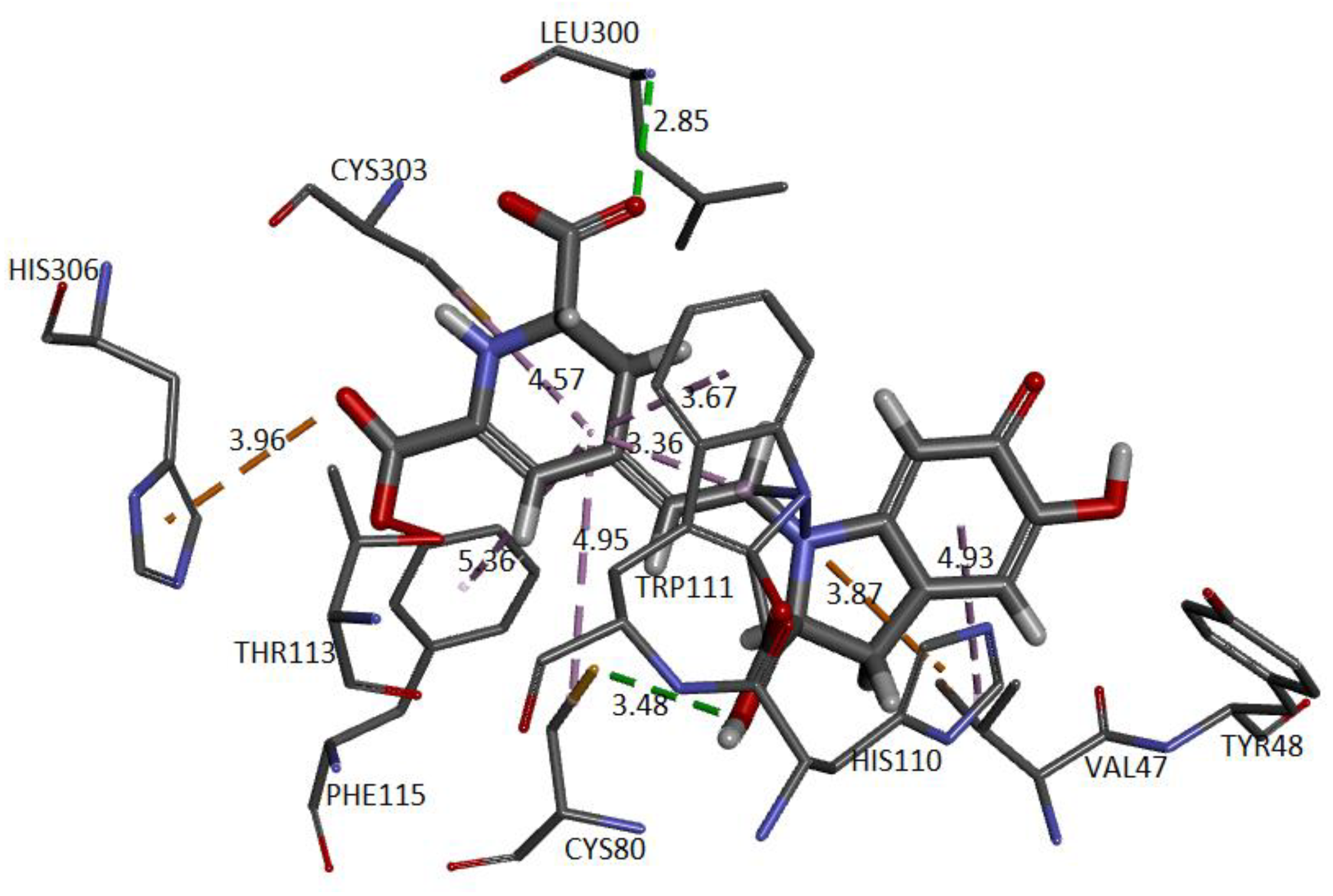
Figure 24 and
Figure 25 show the cavity in which Ligand 1 and Ligand 2 geometrically fit with the corresponding pocket of the same protein and energetically present favorable interactions. Both figures show results consistent with those obtained in conventional software to perform molecular docking. The ligand is surrounded by dots representing the amino acid atoms surrounding the ligand, indicating favorable interactions between the ligand and the receptor’s active site. Although the method is purely geometric, it allows us to visualize the residues that come into contact with Ligand 1: Histidine 306, Tryptophan 219, 20, and 111, Cysteine 298 and 303, and Leucine 300. Ligand 2: Cysteine 80 and 303, Leucine 300, Histidine 110 and 306, Tryptophan 111, Phenylalanine 115, and Valine 47.
The approach has allowed us to identify the cavities and establish the interactions of the pose of each ligand, which we present in
Table 7 and
Table 8.
Our method automatically predicted the AR pocket and the possible interactions between the active site amino acids and the ligands (Ligand 1 and Ligand 2). In general, the interactions are hydrophobic for both cases with amino acids Trp111 ([
29,
43]) and Cys303 [
47]. In Ligand 1, the deprotonated C17 carboxylate oxygen interacts electrostatically with His 306. This same interaction occurs between Trp219 [
44] and the deprotonated C2 carboxyl group oxygen. For Ligand 2, it occurs similarly with the C17 carboxylate. This type of interaction is also generated with Trp219 for Ligand 1 and His110 for Ligand 2. Hydrogen bonds are present in Ligand 2 with Cys80 and Leu300 [
43,
47,
48]. Cys80 and Val 47 [
44] interact hydrophobically with the same ligand. Ligand 1 forms a hydrophobic interaction with NADP
+ [
43] and Trp20 [
44].
Ligand 2, as mentioned before, represents a deprotonation pathway predicted by the geometric approach and located on the receptor and exhibits mainly π-alkyl type interactions at the catalytic site. The carboxylate groups are firmly attached to the active site by hydrogen bonding and electrostatic interaction [
48]. Ligand 1 presents electrostatic interactions but not hydrogen bonds. Not much variation in protein conformation would be expected when interacting with Ligand 1 and Ligand 2, but moderate changes were presented regardless of the computational method used, by applying either conventional molecular docking or the one proposed by us. This situation may be because this protein has presented different binding site conformations for different inhibitors [
43,
44,
49,
50,
51]. To investigate the flexibility of the aldose reductase binding site predicted by our method, we perform molecular dynamics (MD) simulations and report them below.
2.6. Molecular Dynamics
In this section, we present the molecular dynamics analysis applied to the receptor–ligand complex obtained by our method. In the
Supporting Information, we include this information for receptor–ligand models from conventional software for molecular docking calculations.
The molecular dynamics study’s objective is to simulate the local conformational changes in the complex that result from the binding between the protein pocket and the ligand. The procedure gives flexibility to the ligand and the protein residues to arrive at a conformation in a steady state. We chose for molecular dynamics the best conformation obtained from the molecular docking stage via our proposed method. We used the NAMD program, and the force field used was CHARMM36m, which can calculate parameters of bound or unbound molecules. Then we created an environment to put the molecule in to interact with the water. In this case, the protein is in a rectangular system with the correct amount of water molecules needed to solvate the box. We added KCl 0.15 M to the solution, which is necessary to eliminate electrostatic interactions between dissolved molecules.
At the end of the simulation, a file with the extension *.dcd was obtained, containing the residues’ trajectories during molecular dynamics. These trajectories are crucial to calculating the protein’s RMSD during the simulation, which is a widely used measure to quantify the structural deformation of the protein compared to the initial reference point.
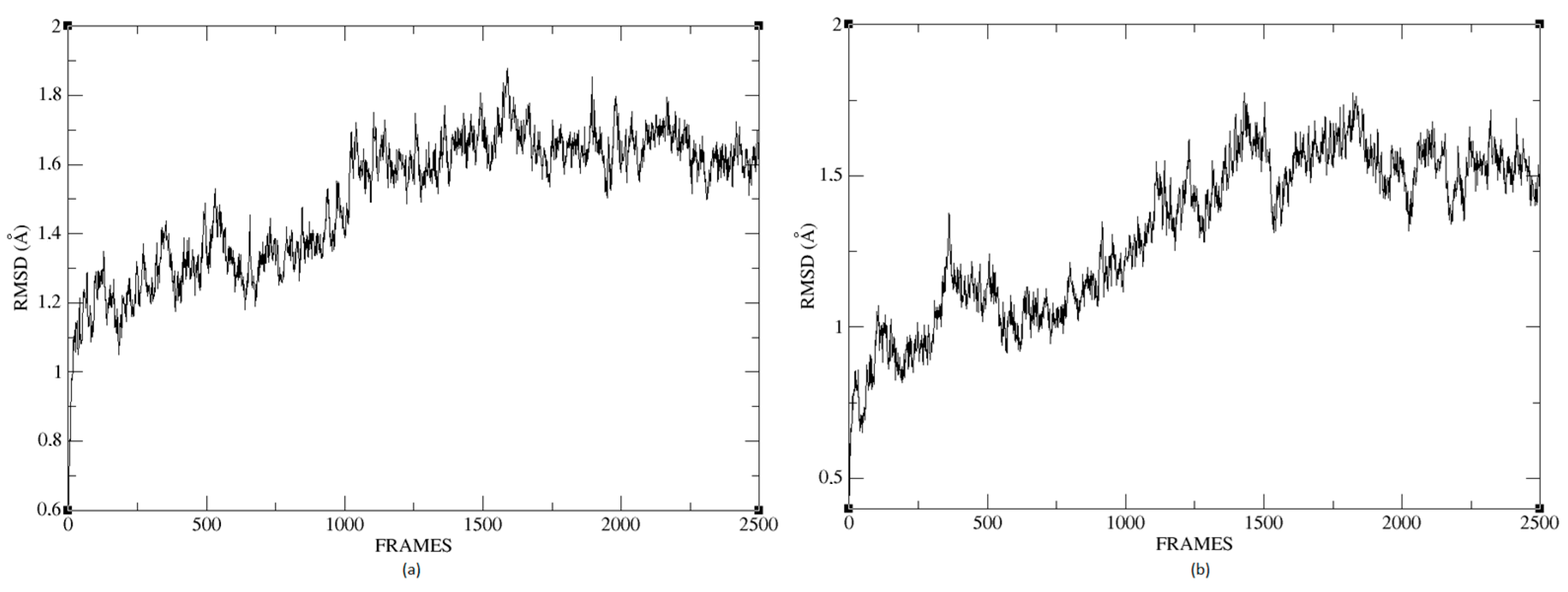
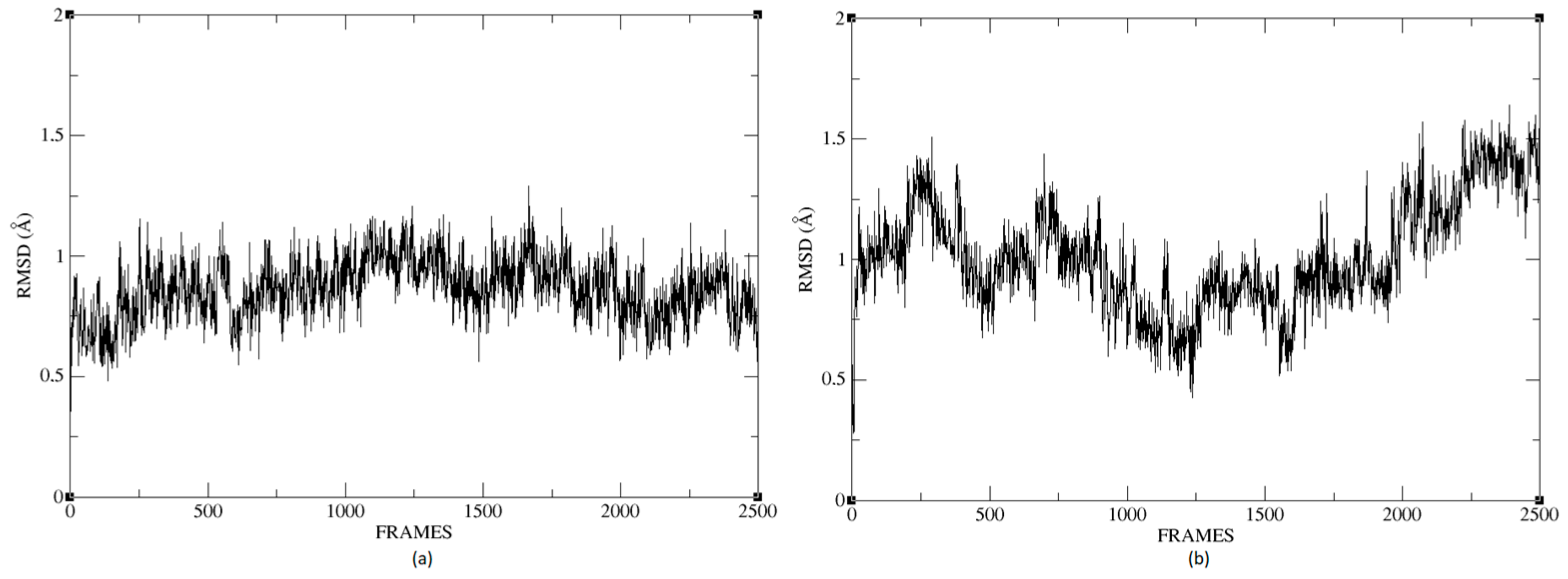
Figure 26a,b shows that the complexes formed with Ligand 1 (C17C15C2-COO-) and Ligand 2 (C6C15C17-COO-) during the molecular dynamics (DM) simulations have high stability during the simulation time. We can see that the structure does not vary significantly; there is an increase in the RMSD until reaching a point where the values fluctuate between 0.50 and 1.49 Å of RMSD (root mean square deviation), in the case of the protein associated with Ligand 1. For the other system, the RMSD value is between 0.44 and 1.77 Å. We can observe in
Figure 26 that the deviations are within normality.
The average RMSD measure is similar for both structures: 1.14 and 1.32, respectively. Therefore, neither of the two complexes suffered a loss of structure during the simulation.
We calculated the deviation for Ligand 1 and Ligand 2 and presented a difference between the maximum and minimum value of RMSD below 5 Å, 0.95 and 1.01 for each molecule.
Figure 27 shows the RMSD of Ligand 1 and Ligand 2. The average of this measure is similar for the two compounds, 0.94 and 1.37, respectively. For a system in equilibrium, the deviations have a maximum difference of 5 Å from RMSD, a situation fulfilled for these simulations, indicating that the compounds are in equilibrium.
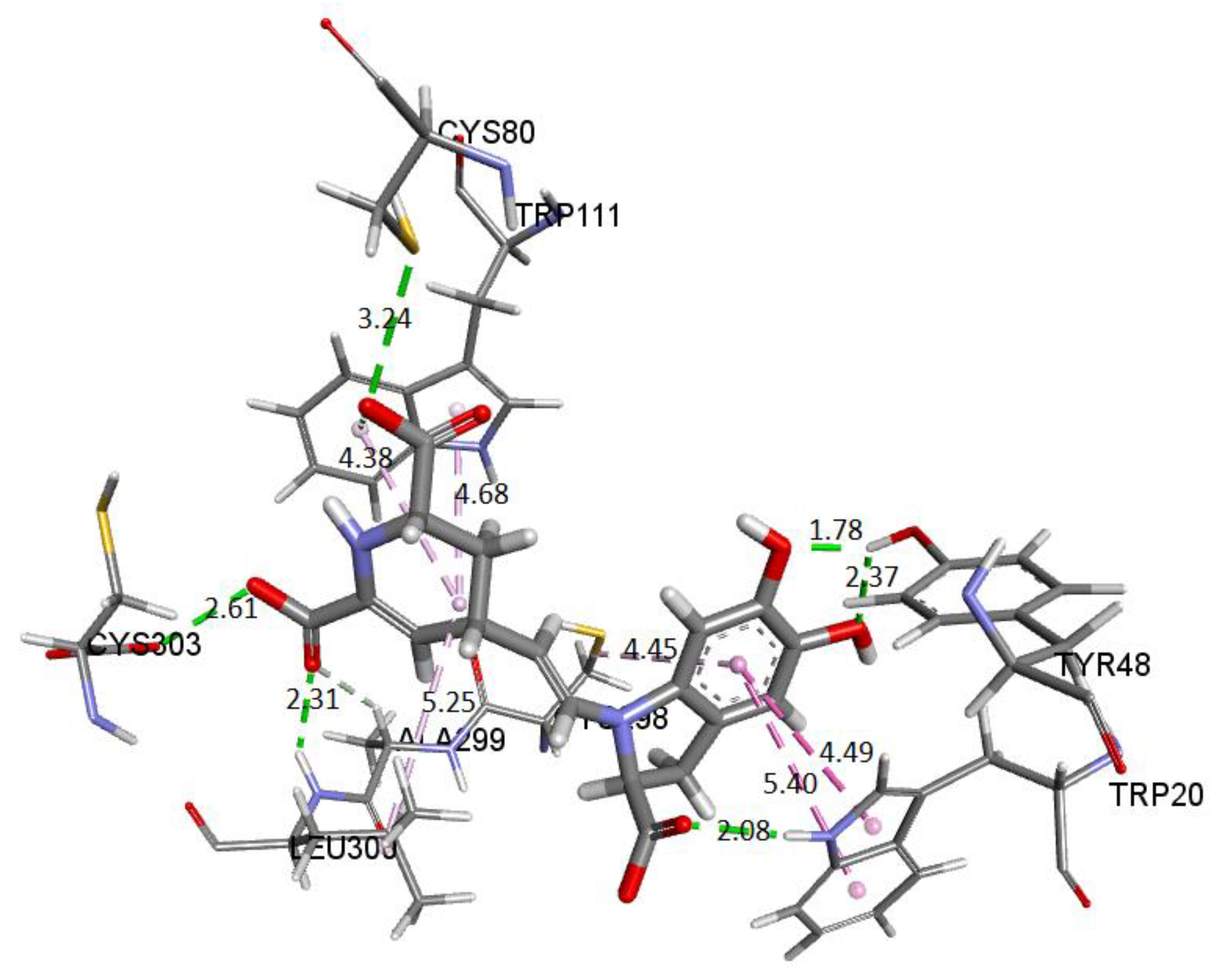
Figure 28 depicts the potential interactions of the AR enzyme with Ligand 1 after molecular dynamics (last frame).
Figure 28 shows the amino acid residues of the active site interacting with the ligand. The green segmented lines show that the hydroxyl groups act as acceptors for forming hydrogen bonds, like the carboxyl groups of the dihydropyridine ring. We observe that Ligand 1 nests well on site after molecular dynamics and exhibits hydrophobic interactions with residues such as Trp20 (4.49 Å), Leu300 (5.24 Å), Trp11 (4.68 Å), and Cys298 (5.09 Å). Hydrogen bonds are with amino acids Leu300 (Å), Cys303 (Å), Cys80 (Å), and Ala299 (Å) [
29,
43].
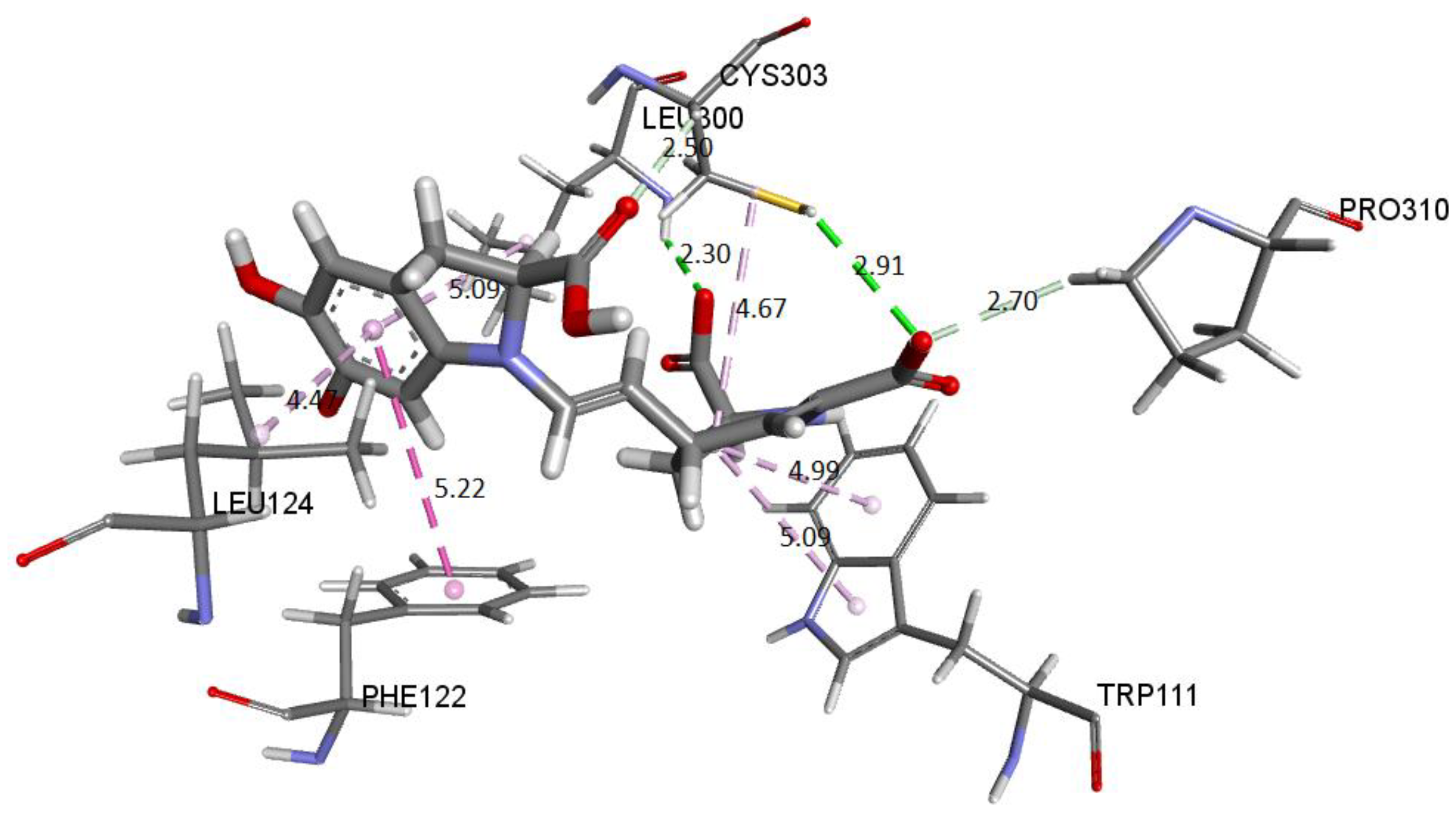
Figure 29 shows the distances and interactions for the residues involved with Ligand 2. The hydrogen bond interactions correspond to residues Leu300 (2.29 Å), Cys303 (2.91 Å–2.5 Å), and Pro310 (2.69 Å). On the other hand, we can visualize a π-alkyl type interaction marked by a segmented pink line with the Trp111 (5.08 Å), Leu124 (Å), Leu300 (5.09 Å), Phe122 (5.22 Å), and Cys302 (4.67 Å) residues, as well as other hydrophobic interactions with Phe122 and Cys303 [
29,
43].
Once we apply molecular dynamics, the stability of each system obtained from our method [
52] is verified, which is an alternative that automatically includes an exhaustive search of possible active sites.
Figures S1 and S2 show that the complexes formed with Ligand 1 (C17C15C2-COO-) and Ligand 2 (C6C15C17-COO-) during the molecular dynamics (DM) simulations have high stability during the simulation time.
Figures S3 and S4 represent the potential interactions of the Ligand 1-AR and Ligand 2-AR complexes.


 ,
, 





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}