
Dates and Rates of Tick-Borne Encephalitis Virus—The Slowest Changing Tick-Borne Flavivirus




Abstract
1. Introduction
2. Results
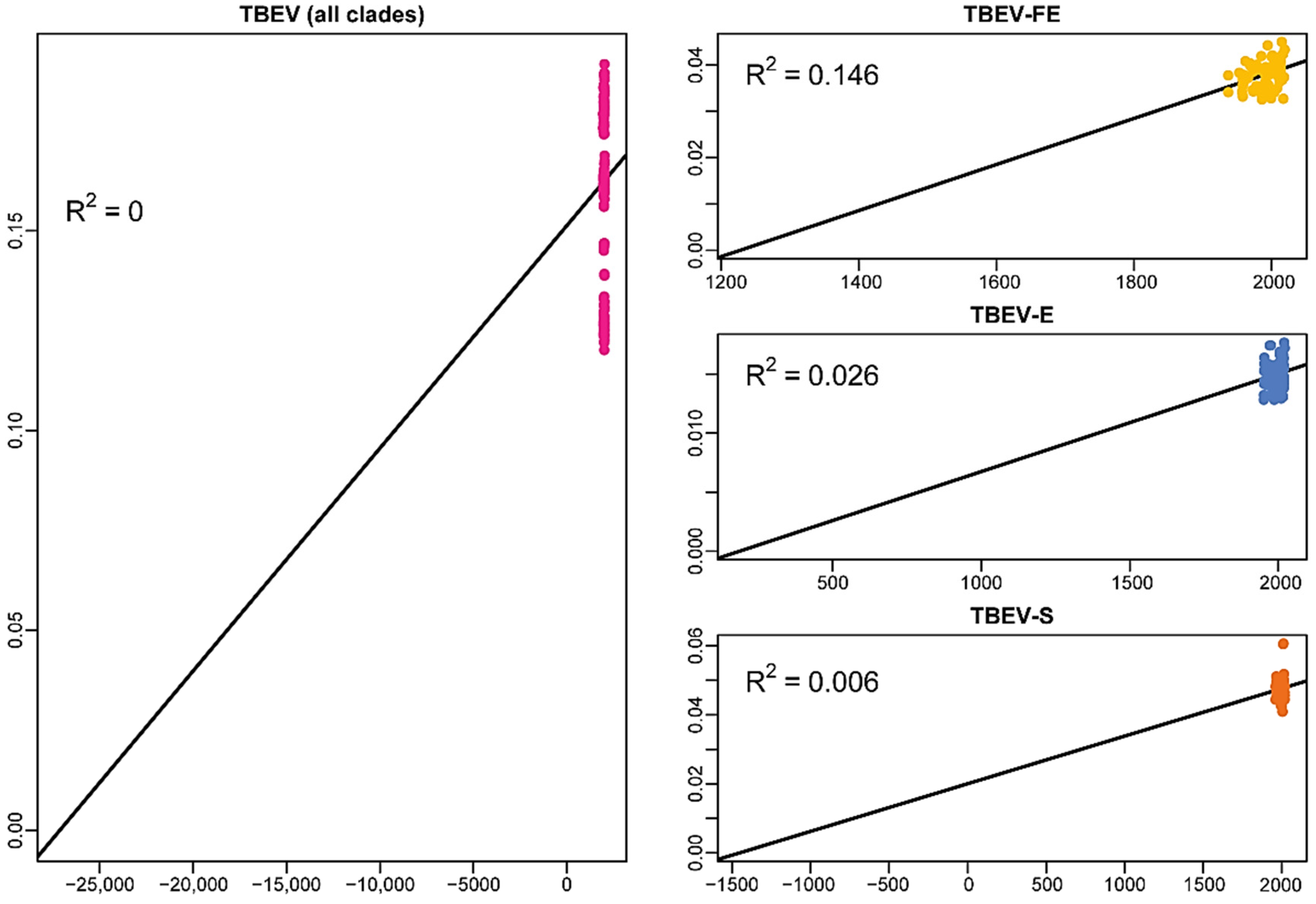
2.1. Temporal Signal in the Data Sets of the Common Clade and Different Subtypes of TBEV
2.2. Nucleotide Saturation Test
2.3. Substitution Rates
2.4. Divergence Times
2.5. Relative Genetic Diversity
3. Discussion
- Hidden population structure within TBEV-E and -FE (so-called confounding effect of population structure [42]);
- Large-scale forest treatment by acaricides including dichlorodiphenyltrichloroethane (DDT) to prevent human infection.
4. Materials and Methods
4.1. Data Set Design
4.2. Codon Saturation Measuring
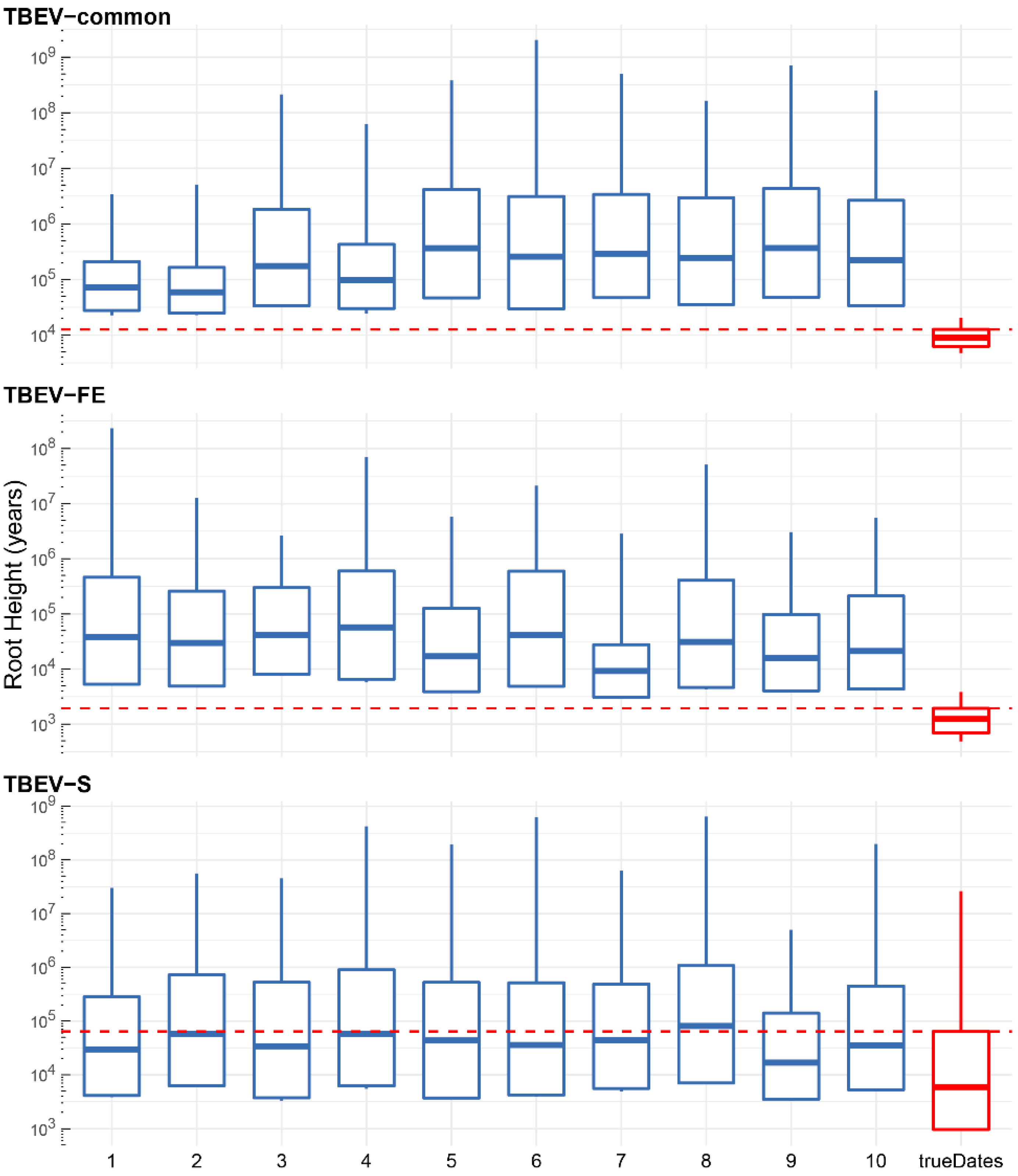
4.3. Temporal Signal Assessment
4.4. Evolution Model Selection
4.5. Phylogenetic Analysis in BEAST
4.6. Bayesian Skyline Reconstruction
5. Conclusions
- TBEV has the lowest substitution rate among other tick-borne flavivirus species;
- The actual data set of TBEV-S ORF sequences has no temporal structure;
- TBEV common clade is characterized by the lowest substitution rate (1.87 × 10−5 s/s/y or 1 nucleotide substitution per ORF per 4.9 years; 95% HPD, 1.3–2.4 × 10−5 s/s/y) in comparison with the other tick-borne flaviviruses;
- TBEV-E has a lower evolutionary rate (1.3 × 10−5 s/s/y or 1 nucleotide substitution per ORF per 7.5 years; 95% HPD, 1.0–1.8 × 10−5 s/s/y) compared with TBEV-common (1.87 × 10−5 s/s/y or 1 nucleotide substitution per ORF per 4.9 years; 95% HPD, 1.3–2.4 × 10−5 s/s/y) and TBEV-FE (3.0 × 10−5 s/s/y or 1 nucleotide substitution per ORF per 3.2 years; 95% HPD, 1.6–4.5 × 10−5 s/s/y);
- TBEV-common representing the species tick-borne encephalitis virus according to median values diverged 9623 years ago with a 95% HPD interval of 6373–13,208 years;
- TBEV-B is the youngest TBEV subtype occurred about 489 years ago (95% HPD, 291–697 years) that is significantly different from TBEV-E (848 years; 95% HPD, 596–1112 years), LIV (2424 years; 95% HPD, 1572–3400 years), TBEV-FE (1936 years, 95% HPD, 1344–2598 years), and the joint clade of TBEV-S (2505 years, 95% HPD, 1700–3421 years);
- The population dynamics of TBEV-common as a metapopulation with a strong population structure has a “chimeric” pattern comprising features of dynamics of highly isolated TBEV subtypes (TBEV-S, -E, and -FE) so Ne × τ dynamics inferred from TBEV-common cannot be interpreted as changes in the effective number of infections.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dobler, G.; Erber, W.; Bröker, M.; Schmitt, H.J. TBE-epidemiology by country—An overview. In The TBE Book, 5th ed.; Global Health Press: Singapore, 2022. [Google Scholar]
- Dobler, G.; Erber, W.; Bröker, M.; Schmitt, H.J. Virology. In The TBE Book, 5th ed.; Global Health Press: Singapore, 2022. [Google Scholar]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, I.V.; Demina, T.V.; Tkachev, S.E.; Doroshchenko, E.K.; Lisak, O.V.; Verkhozina, M.M.; Karan, L.S.; Dzhioev, Y.P.; Paramonov, A.I.; Suntsova, O.V.; et al. Characteristics of the Baikal Subtype of Tick-Borne Encephalitis Virus Circulating in Eastern Siberia. Acta Biomed. Sci. East Sib. Biomed. J. 2018, 3, 53–60. [Google Scholar] [CrossRef]
- Dai, X.; Shang, G.; Lu, S.; Yang, J.; Xu, J. A new subtype of eastern tick-borne encephalitis virus discovered in Qinghai-Tibet Plateau, China. Emerg. Microbes Infect. 2018, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Tkachev, S.E.; Chicherina, G.S.; Golovljova, I.; Belokopytova, P.S.; Tikunov, A.Y.; Zadora, O.V.; Glupov, V.V.; Tikunova, N.V. New genetic lineage within the Siberian subtype of tick-borne encephalitis virus found in Western Siberia, Russia. Infect. Genet. Evol. 2017, 56, 36–43. [Google Scholar] [CrossRef]
- Holding, M.; Dowall, S.D.; Medlock, J.M.; Carter, D.P.; McGinley, L.; Curran-French, M.; Pullan, S.T.; Chamberlain, J.; Hansford, K.M.; Baylis, M.; et al. Detection of new endemic focus of tick-borne encephalitis virus (TBEV), Hampshire/Dorset border, England, September 2019. Eurosurveillance 2019, 24, 1900658. [Google Scholar] [CrossRef]
- Deviatkin, A.A.; Karganova, G.G.; Vakulenko, Y.A.; Lukashev, A.N. TBEV Subtyping in Terms of Genetic Distance. Viruses 2020, 12, 1240. [Google Scholar] [CrossRef]
- Uzcátegui, N.Y.; Sironen, T.; Golovljova, I.; Jääskeläinen, A.E.; Välimaa, H.; Lundkvist, Å.; Plyusnin, A.; Vaheri, A.; Vapalahti, O. Rate of evolution and molecular epidemiology of tick-borne encephalitis virus in Europe, including two isolations from the same focus 44 years apart. J. Gen. Virol. 2012, 93, 786–796. [Google Scholar] [CrossRef]
- Grard, G.; Moureau, G.; Charrel, R.N.; Lemasson, J.J.; Gonzalez, J.P.; Gallian, P.; Gritsun, T.S.; Holmes, E.C.; Gould, E.A.; de Lamballerie, X. Genetic characterization of tick-borne flaviviruses: New insights into evolution, pathogenetic determinants and taxonomy. Virology 2007, 361, 80–92. [Google Scholar] [CrossRef]
- Bondaryuk, A.N.; Andaev, E.I.; Dzhioev, Y.P.; Zlobin, V.I.; Tkachev, S.E.; Kozlova, I.V.; Bukin, Y.S. Delimitation of the tick-borne flaviviruses. Resolving the tick-borne encephalitis virus and louping-ill virus paraphyletic taxa. Mol. Phylogenet. Evol. 2022, 169, 107411. [Google Scholar] [CrossRef]
- Deviatkin, A.A.; Kholodilov, I.S.; Vakulenko, Y.A.; Karganova, G.G.; Lukashev, A.N. Tick-Borne Encephalitis Virus: An Emerging Ancient Zoonosis? Viruses 2020, 12, 247. [Google Scholar] [CrossRef]
- Duchene, S.; Duchene, D.; Holmes, E.C.; Ho, S.Y. The Performance of the Date-Randomization Test in Phylogenetic Analyses of Time-Structured Virus Data. Mol. Biol. Evol. 2015, 32, 1895–1906. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- Duchene, S.; Lemey, P.; Stadler, T.; Ho, S.Y.W.; Duchene, D.A.; Dhanasekaran, V.; Baele, G. Bayesian Evaluation of Temporal Signal in Measurably Evolving Populations. Mol. Biol. Evol. 2020, 37, 3363–3379. [Google Scholar] [CrossRef]
- Clark, J.J.; Gilray, J.; Orton, R.J.; Baird, M.; Wilkie, G.; Filipe, A.D.S.; Johnson, N.; McInnes, C.J.; Kohl, A.; Biek, R. Population genomics of louping ill virus provide new insights into the evolution of tick-borne flaviviruses. PLoS Negl. Trop. Dis. 2020, 14, e0008133. [Google Scholar] [CrossRef]
- Frost, S.D.W.; Volz, E.M. Viral phylodynamics and the search for an ‘effective number of infections’. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1879–1890. [Google Scholar] [CrossRef]
- Heinze, D.M.; Gould, E.A.; Forrester, N.L. Revisiting the clinal concept of evolution and dispersal for the tick-borne flaviviruses by using phylogenetic and biogeographic analyses. J. Virol. 2012, 86, 8663–8671. [Google Scholar] [CrossRef]
- Adelshin, R.V.; Sidorova, E.A.; Bondaryuk, A.N.; Trukhina, A.; Sherbakov, D.Y.; White, R.A., III; Andaev, E.I.; Balakhonov, S.V. “886-84-like” tick-borne encephalitis virus strains: Intraspecific status elucidated by comparative genomics. Ticks Tick Borne Dis. 2019, 10, 1168–1172. [Google Scholar] [CrossRef]
- Vogels, C.B.F.; Brackney, D.E.; Dupuis, A.P.; Robich, R.M.; Fauver, J.R.; Brito, A.F.; Williams, S.C.; Anderson, J.F.; Lubelczyk, C.B.; Lange, R.E.; et al. Phylogeographic reconstruction of the emergence and spread of Powassan virus in the northeastern United States. bioRxiv 2022. [Google Scholar] [CrossRef]
- Karan, L.S.; Ciccozzi, M.; Yakimenko, V.V.; Lo Presti, A.; Cella, E.; Zehender, G.; Rezza, G.; Platonov, A.E. The deduced evolution history of Omsk hemorrhagic fever virus. J. Med. Virol. 2014, 86, 1181–1187. [Google Scholar] [CrossRef]
- Yadav, P.D.; Patil, S.; Jadhav, S.M.; Nyayanit, D.A.; Kumar, V.; Jain, S.; Sampath, J.; Mourya, D.T.; Cherian, S.S. Phylogeography of Kyasanur Forest Disease virus in India (1957–2017) reveals evolution and spread in the Western Ghats region. Sci. Rep. 2020, 10, 1966. [Google Scholar] [CrossRef]
- Pettersson, J.H.; Bohlin, J.; Dupont-Rouzeyrol, M.; Brynildsrud, O.B.; Alfsnes, K.; Cao-Lormeau, V.M.; Gaunt, M.W.; Falconar, A.K.; de Lamballerie, X.; Eldholm, V.; et al. Re-visiting the evolution, dispersal and epidemiology of Zika virus in Asia. Emerg. Microbes Infect. 2018, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Li, Y. Global evolutionary history and spatio-temporal dynamics of dengue virus type 2. Sci. Rep. 2017, 7, 45505. [Google Scholar] [CrossRef] [PubMed]
- Schuh, A.J.; Ward, M.J.; Brown, A.J.; Barrett, A.D. Phylogeography of Japanese encephalitis virus: Genotype is associated with climate. PLoS Negl. Trop. Dis. 2013, 7, e2411. [Google Scholar] [CrossRef] [PubMed]
- Mir, D.; Delatorre, E.; Bonaldo, M.; Lourenco-de-Oliveira, R.; Vicente, A.C.; Bello, G. Phylodynamics of Yellow Fever Virus in the Americas: New insights into the origin of the 2017 Brazilian outbreak. Sci. Rep. 2017, 7, 7385. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, K.; Yin, Y.; Qin, J.; Zhou, Y.H.; Yang, J.; Li, S.; Poon, L.L.M.; Zhang, C. The Phylodynamics of Seasonal Influenza A/H1N1pdm Virus in China between 2009 and 2019. Front. Microbiol. 2020, 11, 735. [Google Scholar] [CrossRef]
- Worobey, M.; Watts, T.D.; McKay, R.A.; Suchard, M.A.; Granade, T.; Teuwen, D.E.; Koblin, B.A.; Heneine, W.; Lemey, P.; Jaffe, H.W. 1970s and ‘Patient 0’ HIV-1 genomes illuminate early HIV/AIDS history in North America. Nature 2016, 539, 98–101. [Google Scholar] [CrossRef]
- McGuire, K.; Holmes, E.C.; Gao, G.F.; Reid, H.W.; Gould, E.A. Tracing the origins of louping ill virus by molecular phylogenetic analysis. J. Gen. Virol. 1998, 79, 981–988. [Google Scholar] [CrossRef]
- Bondaryuk, A.N.; Peretolchina, T.E.; Romanova, E.V.; Yudinceva, A.V.; Andaev, E.I.; Bukin, Y.S. Phylogeography and Re-Evaluation of Evolutionary Rate of Powassan Virus Using Complete Genome Data. Biology 2021, 10, 1282. [Google Scholar] [CrossRef]
- Ho, S.Y.; Shapiro, B. Skyline-plot methods for estimating demographic history from nucleotide sequences. Mol. Ecol. Resour. 2011, 11, 423–434. [Google Scholar] [CrossRef]
- Büntgen, U.; Hellmann, L. The Little Ice Age in Scientific Perspective: Cold Spells and Caveats. J. Interdiscip. Hist. 2014, 44, 353–368. [Google Scholar] [CrossRef]
- Mann, M.E.; Zhang, Z.; Rutherford, S.; Bradley, R.S.; Hughes, M.K.; Shindell, D.; Ammann, C.; Faluvegi, G.; Ni, F. Global signatures and dynamical origins of the Little Ice Age and Medieval Climate Anomaly. Science 2009, 326, 1256–1260. [Google Scholar] [CrossRef]
- Matthews, J.A.; Briffa, K.R. The ‘little ice age’: Re-evaluation of an evolving concept. Geogr. Ann. Ser. A Phys. Geogr. 2016, 87, 17–36. [Google Scholar] [CrossRef]
- Semken, H.A., Jr.; Falk, C.R. Ecology and Environmental Degradation of Two Little Ice Age Earthlodge Villages in North Dakota: The Micromammal Evidence. Wis. Archeol. 2014, 95, 249–268. [Google Scholar]
- Herman, J.S.; Jóhannesdóttir, F.; Jones, E.P.; McDevitt, A.D.; Michaux, J.R.; White, T.A.; Wójcik, J.M.; Searle, J.B. Post-glacial colonization of Europe by the wood mouse, Apodemus sylvaticus: Evidence of a northern refugium and dispersal with humans. Biol. J. Linn. Soc. 2016, 120, 313–332. [Google Scholar] [CrossRef]
- Michaux, J.R.; Libois, R.; Filippucci, M.G. So close and so different: Comparative phylogeography of two small mammal species, the yellow-necked fieldmouse (Apodemus flavicollis) and the woodmouse (Apodemus sylvaticus) in the Western Palearctic region. Heredity 2005, 94, 52–63. [Google Scholar] [CrossRef]
- Chen, F.; Huang, X.; Zhang, J.; Holmes, J.A.; Chen, J. Humid Little Ice Age in arid central Asia documented by Bosten Lake, Xinjiang, China. Sci. China Ser. D Earth Sci. 2006, 49, 1280–1290. [Google Scholar] [CrossRef]
- Eriksdotter, G. Did the Little Ice Age Affect Indoor Climate and Comfort?: Re-theorizing Climate History and Architecture from the Early Modern Period. J. Early Mod. Cult. Stud. 2013, 13, 24–42. [Google Scholar] [CrossRef]
- Wanner, H.; Pfister, C.; Neukom, R. The variable European Little Ice Age. Quat. Sci. Rev. 2022, 287, 107531. [Google Scholar] [CrossRef]
- Heller, R.; Chikhi, L.; Siegismund, H.R. The confounding effect of population structure on Bayesian skyline plot inferences of demographic history. PLoS ONE 2013, 8, e62992. [Google Scholar] [CrossRef]
- Bondaryuk, A.N.; Adelshin, R.V.; Lopatovskaya, K.V.; Melnikova, O.V.; Sidorova, E.A.; Andaev, E.I. Phylogenetic analysis of European subtype strains of tick-borne encephalitis virus isolated in the territory of Siberia (Russia). Mol. Genet. Microbiol. Virol. 2021, 39, 39–45. [Google Scholar] [CrossRef]
- Xia, X. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Drummond, A.J.; Bouckaert, R.R. Posterior analysis and post-processing. In Bayesian Evolutionary Analysis with BEAST; Cambridge University Press: Cambridge, UK, 2015; p. 249. [Google Scholar]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Dong, X.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus/Subtype 1 | Number of Sequences | Sampling Window 2 | Substitution Model 3 | Molecular Clock 4 |
|---|---|---|---|---|
| LIV | 25 | 84 | – | – |
| TBEV-B | 20 | 26 | – | – |
| TBEV-E | 83 | 70 | SRD06 [43] | SC |
| TBEV-FE | 99 | 81 | GTR + G + I | UCLD |
| TBEV-H | 2 | 0 | – | – |
| TBEV-SB | 7 | 46 | GTR + G + I | UCLD |
| TBEV-SV | 22 | 58 | ||
| TBEV-SZ | 18 | 58 | ||
| TBEV-common | 276 | 90 | GTR + G + I | UCLD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondaryuk, A.N.; Kulakova, N.V.; Belykh, O.I.; Bukin, Y.S. Dates and Rates of Tick-Borne Encephalitis Virus—The Slowest Changing Tick-Borne Flavivirus. Int. J. Mol. Sci. 2023, 24, 2921. https://doi.org/10.3390/ijms24032921
Bondaryuk AN, Kulakova NV, Belykh OI, Bukin YS. Dates and Rates of Tick-Borne Encephalitis Virus—The Slowest Changing Tick-Borne Flavivirus. International Journal of Molecular Sciences. 2023; 24(3):2921. https://doi.org/10.3390/ijms24032921
Chicago/Turabian StyleBondaryuk, Artem N., Nina V. Kulakova, Olga I. Belykh, and Yurij S. Bukin. 2023. "Dates and Rates of Tick-Borne Encephalitis Virus—The Slowest Changing Tick-Borne Flavivirus" International Journal of Molecular Sciences 24, no. 3: 2921. https://doi.org/10.3390/ijms24032921
APA StyleBondaryuk, A. N., Kulakova, N. V., Belykh, O. I., & Bukin, Y. S. (2023). Dates and Rates of Tick-Borne Encephalitis Virus—The Slowest Changing Tick-Borne Flavivirus. International Journal of Molecular Sciences, 24(3), 2921. https://doi.org/10.3390/ijms24032921