
SMN Deficiency Destabilizes ABCA1 Expression in Human Fibroblasts: Novel Insights in Pathophysiology of Spinal Muscular Atrophy


 ,
,  , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
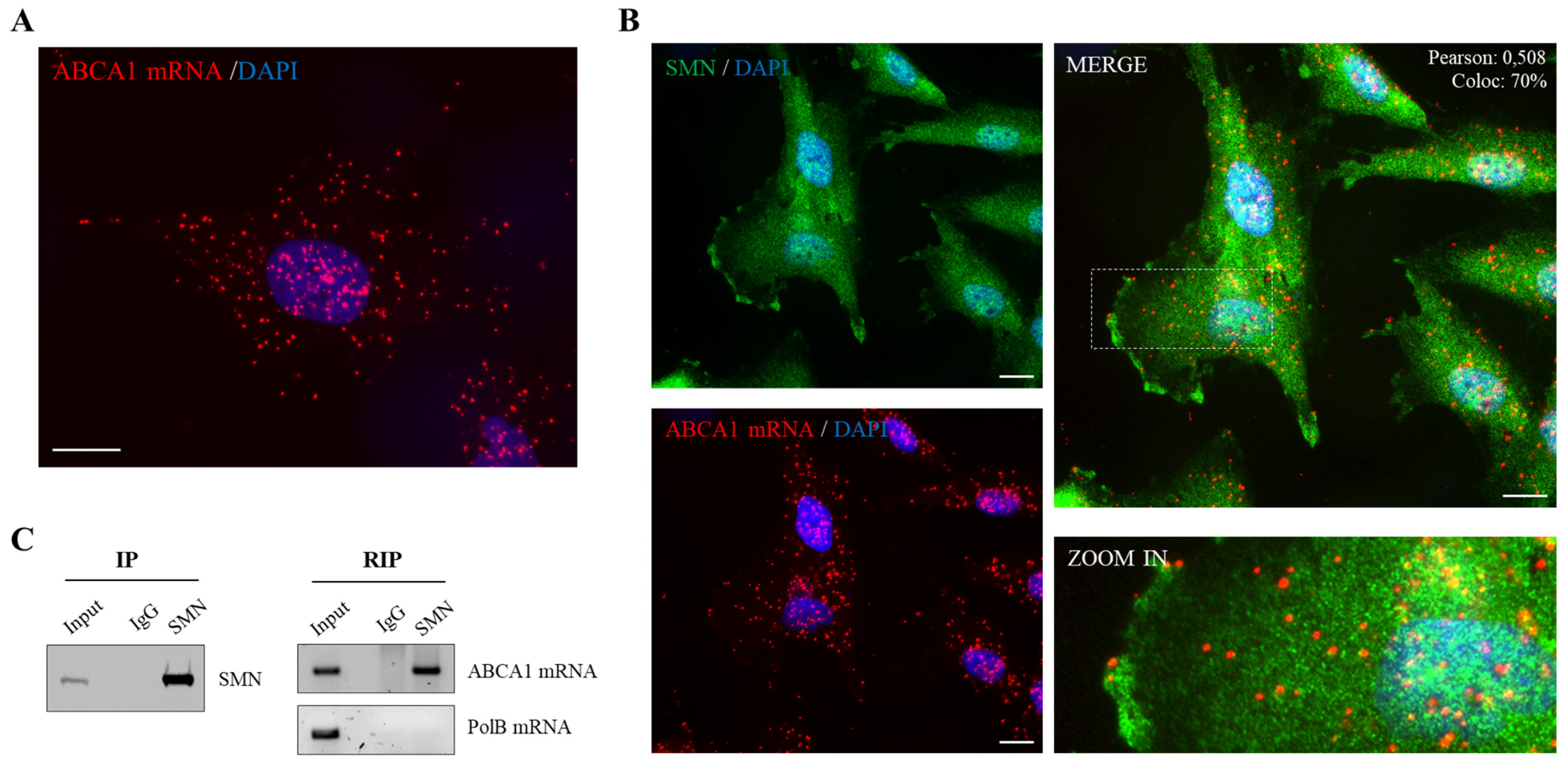
2.1. SMN Associates to ABCA1 mRNA
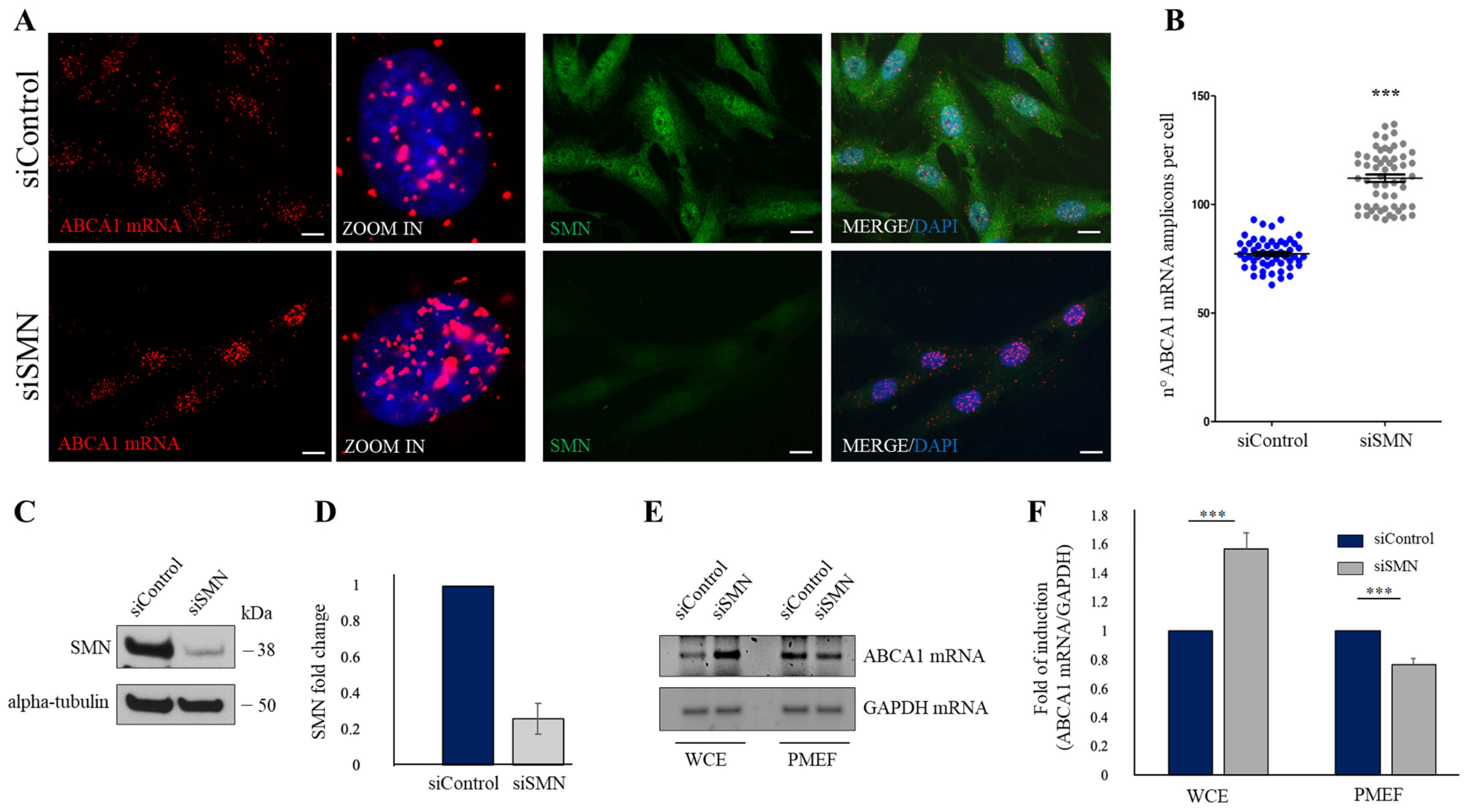
2.2. SMN Knockdown Affects the Subcellular Distribution of ABCA1 mRNA
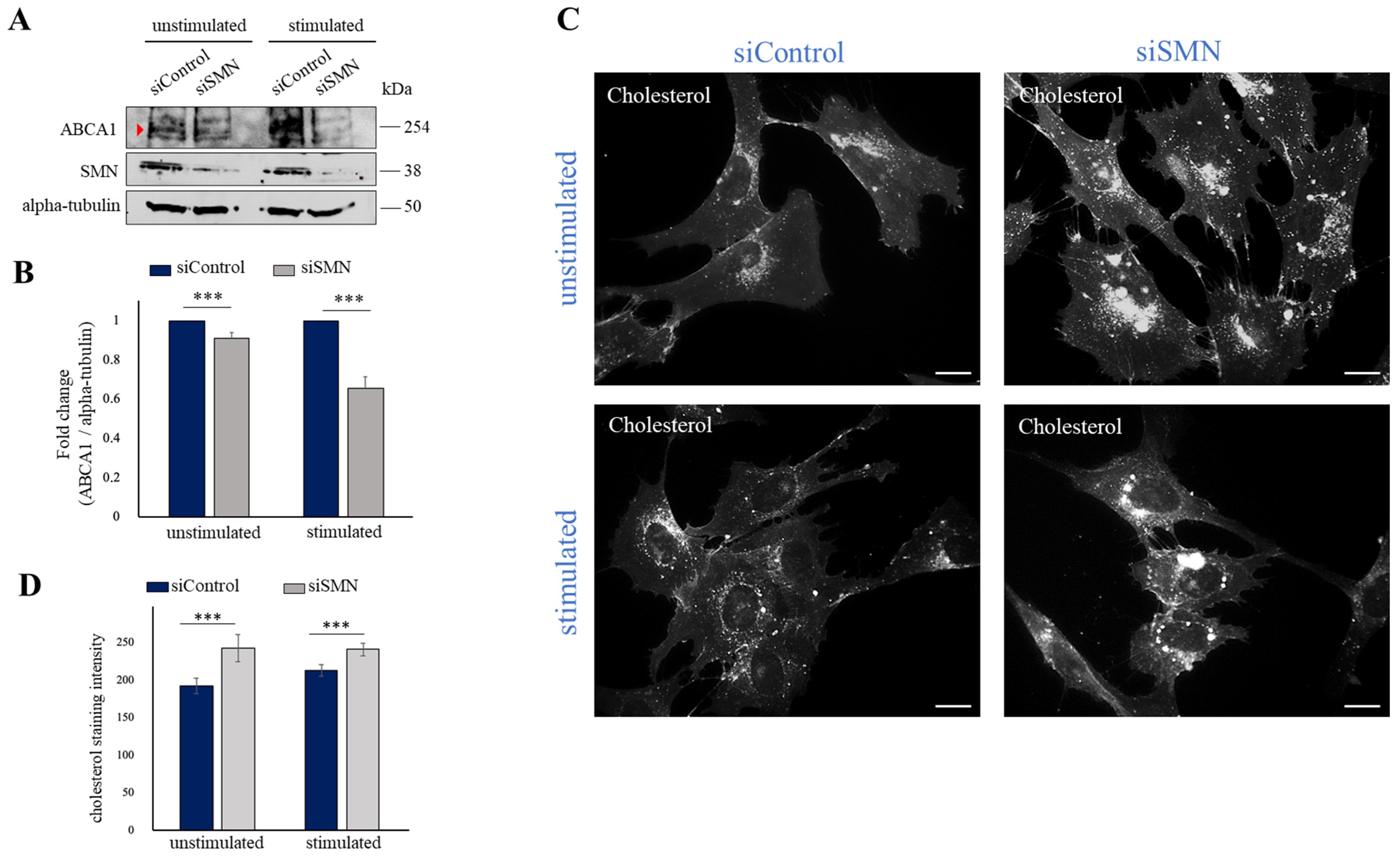
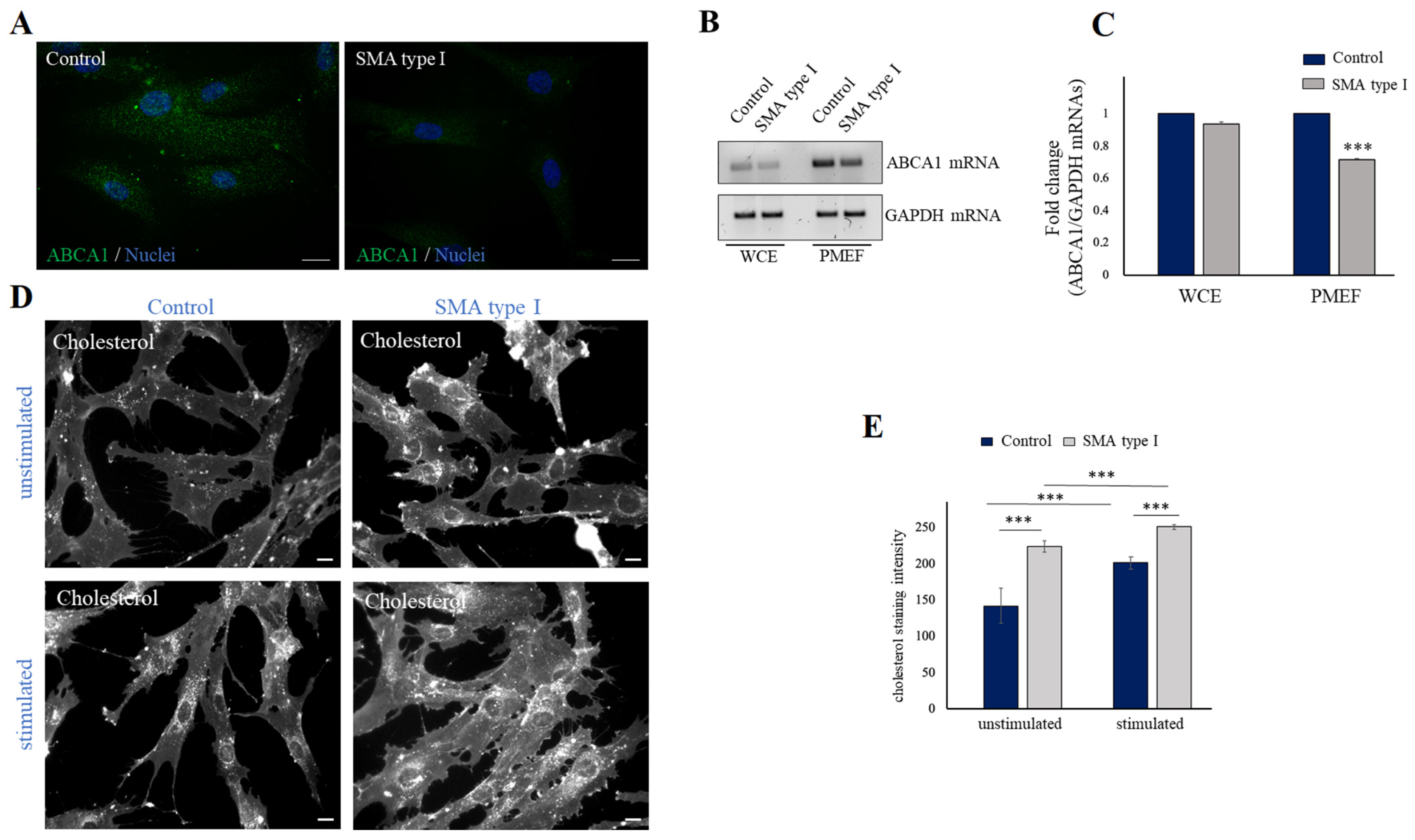
2.3. SMN Deficiency Down-Regulates ABCA1 Protein Levels and Causes Intracellular Accumulation of Cholesterol
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Cultures and Transfection
4.3. ATP Depletion and Recovery Assay
4.4. Preparation of Plasma-Membrane-Enriched Fractions
4.5. Immunofluorescence and Filipin Staining
4.6. RNA Immunoprecipitation (RIP) Assay
4.7. Western Blot Analysis
4.8. RNA Extraction, Retrotranscription and Semiquantitative Real-Time PCR (RT-PCR)
4.9. Padlock Assay
4.10. Quantification and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef]
- Chaytow, H.; Huang, Y.T.; Gillingwater, T.H.; Faller, K.M.E. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Pera, M.C.; Scoto, M.; Finkel, R.; Muntoni, F. Spinal muscular atrophy—Insights and challenges in the treatment era. Nat. Rev. Neurol. 2020, 16, 706–715. [Google Scholar] [CrossRef]
- Pearn, J.H. Fetal movements and Werdnig-Hoffmann disease. J. Neurol. Sci. 1973, 18, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Shababi, M.; Lorson, C.L.; Rudnik-Schöneborn, S.S. Spinal muscular atrophy: A motor neuron disorder or a multi-organ disease? J. Anat. 2014, 224, 15–28. [Google Scholar] [CrossRef]
- Gabanella, F.; Pisani, C.; Borreca, A.; Farioli-Vecchioli, S.; Ciotti, M.T.; Ingegnere, T.; Onori, A.; Canu, N.; Monaco, L.; Passananti, C.; et al. SMN affects membrane remodelling and anchoring of the protein synthesis machinery. J. Cell Sci. 2016, 129, 804–816. [Google Scholar]
- Gabanella, F.; Onori, A.; Ralli, M.; Greco, A.; Passananti, C.; Di Certo, M.G. SMN protein promotes membrane compartmentalization of ribosomal protein S6 transcript in human fibroblasts. Sci. Rep. 2020, 10, 19000. [Google Scholar] [CrossRef]
- Koppers, M.; Holt, C.E. Receptor-Ribosome Coupling: A Link Between Extrinsic Signals and mRNA Translation in Neuronal Compartments. Annu. Rev. Neurosci. 2022, 45, 41–61. [Google Scholar] [CrossRef]
- Deguise, M.O.; Baranello, G.; Mastella, C.; Beauvais, A.; Michaud, J.; Leone, A.; De Amicis, R.; Battezzati, A.; Dunham, C.; Selby, K.; et al. Abnormal fatty acid metabolism is a core component of spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 1519–1532. [Google Scholar] [CrossRef] [PubMed]
- Tein, I.; Sloane, A.E.; Donner, E.J.; Lehotay, D.C.; Millington, D.S.; Kelley, R.I. Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy:primary or secondary defect(s)? Pediatr. Neurol. 1995, 12, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O.; Sladky, J.T.; Hurko, O.; Besner-Johnston, A.; Kelley, R.I. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann. Neurol. 1999, 45, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Zolkipli, Z.; Sherlock, M.; Biggar, W.D.; Taylor, G.; Hutchison, J.S.; Peliowski, A.; Alman, B.A.; Ling, S.C.; Tein, I. Abnormal fatty acid metabolism in spinal muscular atrophy may predispose to perioperative risks. Eur. J. Paediatr. Neurol. 2012, 16, 549–553. [Google Scholar] [CrossRef]
- Krivoi, I.I.; Petrov, A.M. Cholesterol and the Safety Factor for Neuromuscular Transmission. Int. J. Mol. Sci. 2019, 20, 1046. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. How cells handle cholesterol. Science 2000, 290, 1721–1726. [Google Scholar] [CrossRef]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Liu, W.; Chakraborty, B.; Safi, R.; Kazmin, D.; Chang, C.-Y.; McDonnell, D.P. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat. Commun. 2021, 12, 5103. [Google Scholar] [CrossRef]
- Pfrieger, F.W. Neurodegenerative Diseases and Cholesterol: Seeing the Field Through the Players. Front. Aging Neurosci. 2021, 13, 766587. [Google Scholar] [CrossRef]
- Juhl, A.D.; Wüstner, D. Pathways and Mechanisms of Cellular Cholesterol Efflux-Insight From Imaging. Front. Cell Dev. Biol. 2022, 10, 834408. [Google Scholar] [CrossRef]
- Broccardo, C.; Luciani, M.; Chimini, G. The ABCA subclass of mammalian transporters. Biochim. Biophys Acta 1999, 1461, 395–404. [Google Scholar] [CrossRef]
- Oram, J.F.; Vaughan, A.M. ABCA1-mediated transport of cellular cholesterol and phosopholipids to HDL apolipoproteins. Curr. Opin. Lipidol. 2000, 11, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ferrans, V.J.; Fredrickson, D.S. The pathology of Tangier disease. A light and electron microscopic study. Am. J. Pathol. 1975, 78, 101–158. [Google Scholar] [PubMed]
- Brooks-Wilson, A.; Marcil, M.; Clee, S.M.; Zhang, L.; Roomp, K.; van Dam, M.; Yu, L.; Brewer, C.; Collins, J.A.; Molhuizen, H.O.F.; et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 1999, 22, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.; Bissonnette, R.; Haidar, B.; Krimbou, L.; Bouvier, M.; Genest, J. Expression, regulation, and activity of ABCA1 in human cell lines. Mol. Genet. Metab. 2003, 78, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Jacobo-Albavera, L.; Domínguez-Pérez, M.; Medina-Leyte, D.J.; González-Garrido, A.; Villarreal-Molina, T. The Role of the ATP-Binding Cassette A1 (ABCA1) in Human Disease. Int. J. Mol. Sci. 2021, 22, 1593. [Google Scholar] [CrossRef] [PubMed]
- Wellington, C.L.; Walker, E.K.Y.; Suarez, A.; Kwok, A.; Bissada, N.; Singaraja, R.; Yang, Y.-Z.; Zhang, L.-H.; James, E.; Wilson, J.E.; et al. ABCA1 mRNA and Protein Distribution Patterns Predict Multiple Different Roles and Levels of Regulation. Lab. Invest. 2002, 82, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Svitkina, T.M.; Neyfakh, A.A., Jr.; Bershadsky, A.D. Actin cytoskeleton of spread fibroblasts appears to assemble at the cell edges. J. Cell Sci. 1986, 82, 235–248. [Google Scholar] [CrossRef]
- Bear, J.E.; Svitkina, T.M.; Krause, M.; Schafer, D.A.; Loureiro, J.J.; Strasser, G.A.; Maly, I.V.; Chaga, O.Y.; Cooper, J.A.; Borisy, G.G.; et al. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell 2002, 109, 509–521. [Google Scholar] [CrossRef]
- Lauria, F.; Bernabò, P.; Tebaldi, T.; Groen, E.J.N.; Perenthaler, E.; Maniscalco, F.; Rossi, A.; Donzel, D.; Clamer, M.; Marchioretto, M.; et al. SMN-primed ribosomes modulate the translation of transcripts related to spinal muscular atrophy. Nat. Cell Biol. 2020, 22, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Gabanella, F.; Barbato, C.; Fiore, M.; Petrella, C.; de Vincentiis, M.; Greco, A.; Minni, A.; Corbi, N.; Passananti, C.; Di Certo, M.G. Fine-Tuning of mTOR mRNA and Nucleolin Complexes by SMN. Cells 2021, 10, 3015. [Google Scholar] [CrossRef] [PubMed]
- Gondran, P.; Amiot, F.; Weil, D.; Dautry, F. Accumulation of mature mRNA in the nuclear fraction of mammalian cells. FEBS Lett. 1999, 458, 324–328. [Google Scholar] [CrossRef]
- Didiot, M.C.; Ferguson, C.M.; Ly, S.; Coles, A.H.; Smith, A.O.; Bicknell, A.A.; Hall, L.M.; Sapp, E.; Echeverria, D.; Pai, A.A.; et al. Nuclear Localization of Huntingtin mRNA Is Specific to Cells of Neuronal Origin. Cell Rep. 2018, 24, 2553–2560.e5. [Google Scholar] [CrossRef]
- Frambach, S.J.C.M.; de Haas, R.; Smeitink, J.A.M.; Rongen, G.A.; Russel, F.G.M.; Schirris, T.J.J. Brothers in Arms: ABCA1- and ABCG1-Mediated Cholesterol Efflux as Promising Targets in Cardiovascular Disease Treatment. Pharmacol. Rev. 2020, 72, 152–190. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, S.; Guo, Z.; Xing, D.; Chen, W. The crosstalk of ABCA1 and ANXA1: A potential mechanism for protection against atherosclerosis. Mol. Med. 2020, 26, 84. [Google Scholar] [CrossRef]
- Li, L.; Xu, L.; Chen, W.; Li, X.; Xia, Q.; Zheng, L.; Duan, Q.; Zhang, H.; Zhao, Y. Reduced Annexin A1 Secretion by ABCA1 Causes Retinal Inflammation and Ganglion Cell Apoptosis in a Murine Glaucoma Model. Front. Cell Neurosci. 2018, 12, 347. [Google Scholar] [CrossRef]
- Rihan, K.; Antoine, E.; Maurin, T.; Bardoni, B.; Bordonné, R.; Soret, J.; Rage, F. A new cis-acting motif is required for the axonal SMN-dependent Anxa2 mRNA localization. RNA 2017, 23, 899–909. [Google Scholar] [CrossRef]
- Kye, M.J.; Niederst, E.D.; Wertz, M.H.; Gonçalves Ido, C.; Akten, B.; Dover, K.Z.; Peters, M.; Riessland, M.; Neveu, P.; Wirth, B.; et al. SMN regulates axonal local translation via miR-183/mTOR pathway. Hum. Mol. Genet. 2014, 23, 6318–6331. [Google Scholar] [CrossRef]
- Bi, D.P.; Yin, C.H.; Zhang, X.Y.; Yang, N.N.; Xu, J.Y. MiR-183 functions as an oncogene by targeting ABCA1 in colon cancer. Oncol. Rep. 2016, 35, 2873–2879. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.E.; Li, H.; Chen, L.Y.; Xia, X.D.; Zhao, Z.W.; Zheng, X.L.; Zhao, G.J.; Tang, C.K. IL-8 negatively regulates ABCA1 expression and cholesterol efflux via upregulating miR-183 in THP-1 macrophage-derived foam cells. Cytokine 2019, 122, 154385. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, H.; Ho, W.Y.; Chang, J.C.; Ling, S.C. Cholesterol dyshomeostasis in amyotrophic lateral sclerosis: Cause, consequence, or epiphenomenon? FEBS J. 2022, 289, 7688–7709. [Google Scholar] [CrossRef] [PubMed]
- Willmann, R.; Pun, S.; Stallmach, L.; Sadasivam, G.; Santos, A.F.; Caroni, P.; Fuhrer, C. Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 2006, 25, 4050–4060. [Google Scholar] [CrossRef]
- Boido, M.; Vercelli, A. Neuromuscular Junctions as Key Contributors and Therapeutic Targets in Spinal Muscular Atrophy. Front. Neuroanat. 2016, 10, 6. [Google Scholar] [CrossRef]
- Hunter, G.; Powis, R.A.; Jones, R.A.; Groen, E.J.; Shorrock, H.K.; Lane, F.M.; Zheng, Y.; Sherman, D.L.; Brophy, P.J.; Gillingwater, T.H. Restoration of SMN in Schwann cells reverses myelination defects and improves neuromuscular function in spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 2853–2861. [Google Scholar] [CrossRef]
- Xia, W.; Zhu, J.; Wang, X.; Tang, Y.; Zhou, P.; Hou, M.; Li, S. ANXA1 directs Schwann cells proliferation and migration to accelerate nerve regeneration through the FPR2/AMPK pathway. FASEB J. 2020, 34, 13993–14005. [Google Scholar] [CrossRef]
- Pollock, M.; Nukada, H.; Frith, R.W.; Simcock, J.P.; Allpress, S. Peripheral neuropathy in Tangier disease. Brain 1983, 106, 911–928. [Google Scholar] [CrossRef]
- Ceccanti, M.; Cambieri, C.; Frasca, V.; Onesti, E.; Biasiotta, A.; Giordano, C.; Bruno, S.M.; Testino, G.; Lucarelli, M.; Arca, M.; et al. A Novel Mutation in ABCA1 Gene Causing Tangier Disease in an Italian Family with Uncommon Neurological Presentation. Front. Neurol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Day, J.W.; Howell, K.; Place, A.; Long, K.; Rossello, J.; Kertesz, N.; Nomikos, G. Advances and limitations for the treatment of spinal muscular atrophy. BMC Pediatr. 2022, 22, 632. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′-3′) |
|---|---|
| RT-PCR GAPDH F | CATGAGAAGTATGACAACAGCCT |
| RT-PCR GAPDH R | AGTCCTTCCACGATACCAAAGT |
| RT-PCR POLB F | GTGAGACAAAGTTCATGGGTGT |
| RT-PCR POLB R | GTGAAACCCTTTTCTAGGGCAT |
| RT-PCR ABCA1 F | TACATCTCCCTTCCCGAGCA |
| RT-PCR ABCA1 R | GGAGCTGGAGCTGTTCACAT |
| Padlock Probe ABCA1 | CATGTCACTCCAGCTTTTTTTTCTCAATTCTGCTACTTTACTACCTCAATTCTGCTACTGTACTACTTTTTTCATCACCTCCTGTCG |
| RCA Primer | AGTACAGTAGCAGAATTGAG |
| AlexaFluor 595-labelled probe | CTCAATTCTGCTACTTTACTAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabanella, F.; Onori, A.; Pisani, C.; Fiore, M.; Ferraguti, G.; Colizza, A.; de Vincentiis, M.; Ceccanti, M.; Inghilleri, M.; Corbi, N.; et al. SMN Deficiency Destabilizes ABCA1 Expression in Human Fibroblasts: Novel Insights in Pathophysiology of Spinal Muscular Atrophy. Int. J. Mol. Sci. 2023, 24, 2916. https://doi.org/10.3390/ijms24032916
Gabanella F, Onori A, Pisani C, Fiore M, Ferraguti G, Colizza A, de Vincentiis M, Ceccanti M, Inghilleri M, Corbi N, et al. SMN Deficiency Destabilizes ABCA1 Expression in Human Fibroblasts: Novel Insights in Pathophysiology of Spinal Muscular Atrophy. International Journal of Molecular Sciences. 2023; 24(3):2916. https://doi.org/10.3390/ijms24032916
Chicago/Turabian StyleGabanella, Francesca, Annalisa Onori, Cinzia Pisani, Marco Fiore, Giampiero Ferraguti, Andrea Colizza, Marco de Vincentiis, Marco Ceccanti, Maurizio Inghilleri, Nicoletta Corbi, and et al. 2023. "SMN Deficiency Destabilizes ABCA1 Expression in Human Fibroblasts: Novel Insights in Pathophysiology of Spinal Muscular Atrophy" International Journal of Molecular Sciences 24, no. 3: 2916. https://doi.org/10.3390/ijms24032916
APA StyleGabanella, F., Onori, A., Pisani, C., Fiore, M., Ferraguti, G., Colizza, A., de Vincentiis, M., Ceccanti, M., Inghilleri, M., Corbi, N., Passananti, C., & Di Certo, M. G. (2023). SMN Deficiency Destabilizes ABCA1 Expression in Human Fibroblasts: Novel Insights in Pathophysiology of Spinal Muscular Atrophy. International Journal of Molecular Sciences, 24(3), 2916. https://doi.org/10.3390/ijms24032916