
Cellular Senescence as a Brake or Accelerator for Oncogenic Transformation and Role in Lymphatic Metastasis




Abstract
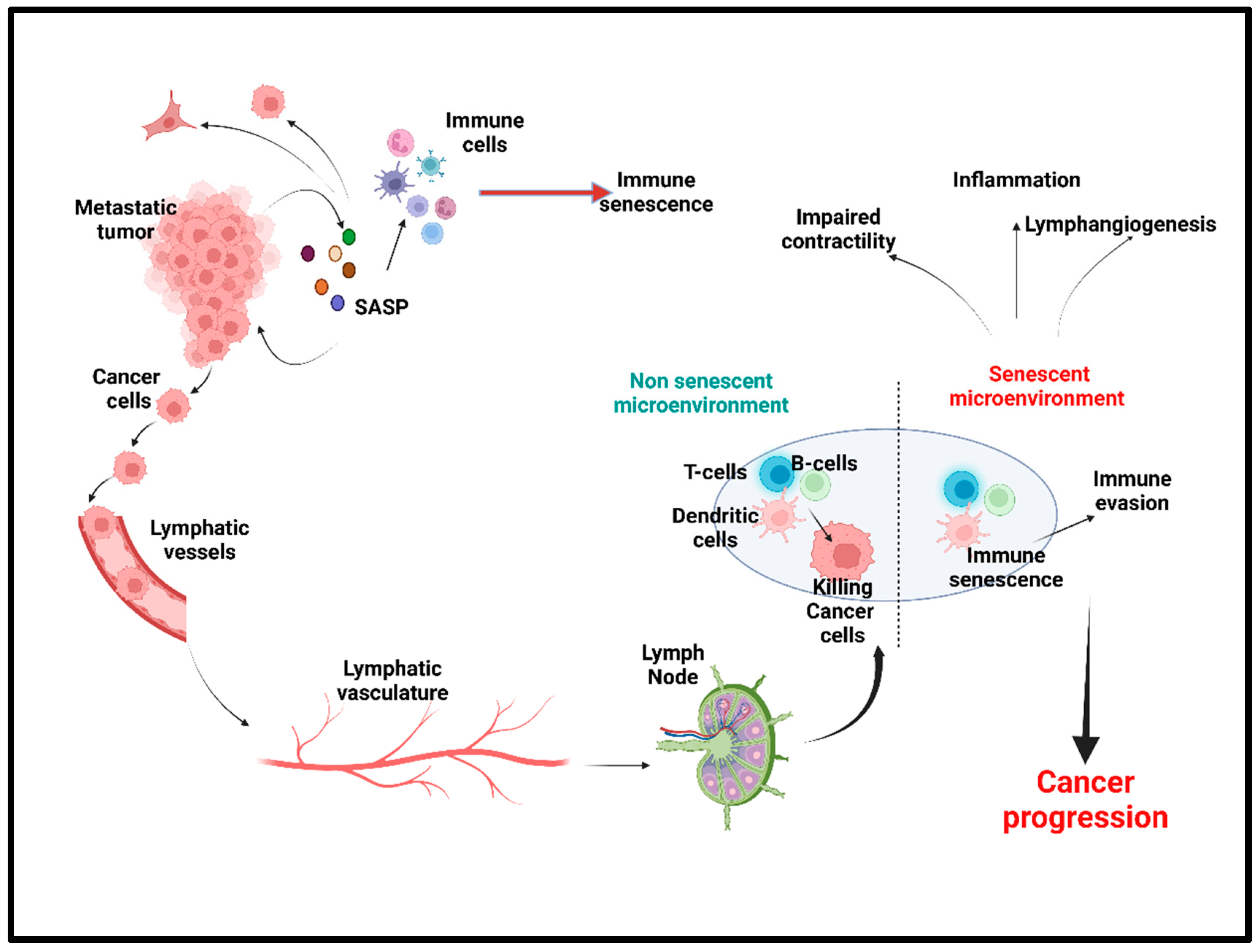
1. Introduction
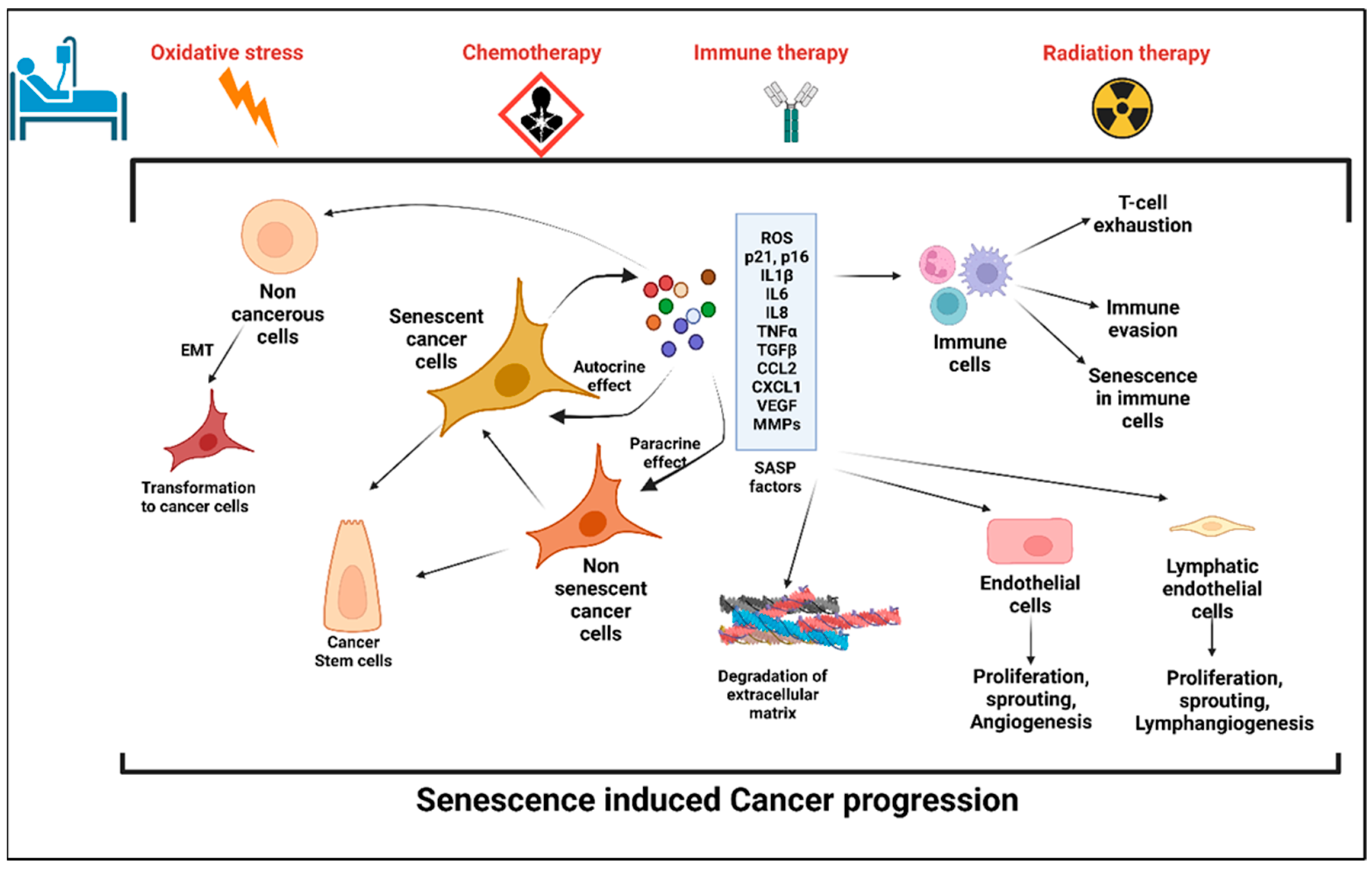
2. Senescence and Senescence-Associated Secretory Phenotype (SASP)
2.1. Senescence
Cellular Senescence: Double Edged Sword for Cancer
2.2. Anticancer Treatment: A Potential Trigger to Cellular Senescence
- A.
- Chemotherapy and senescence
- B.
- Radiation-induced senescence
- C.
- Immunotherapy-induced senescence
2.3. The Cellular and Molecular Mechanism of SIPS
2.3.1. Mitochondrial Dysfunction and SIPS
2.3.2. Molecular Pathways
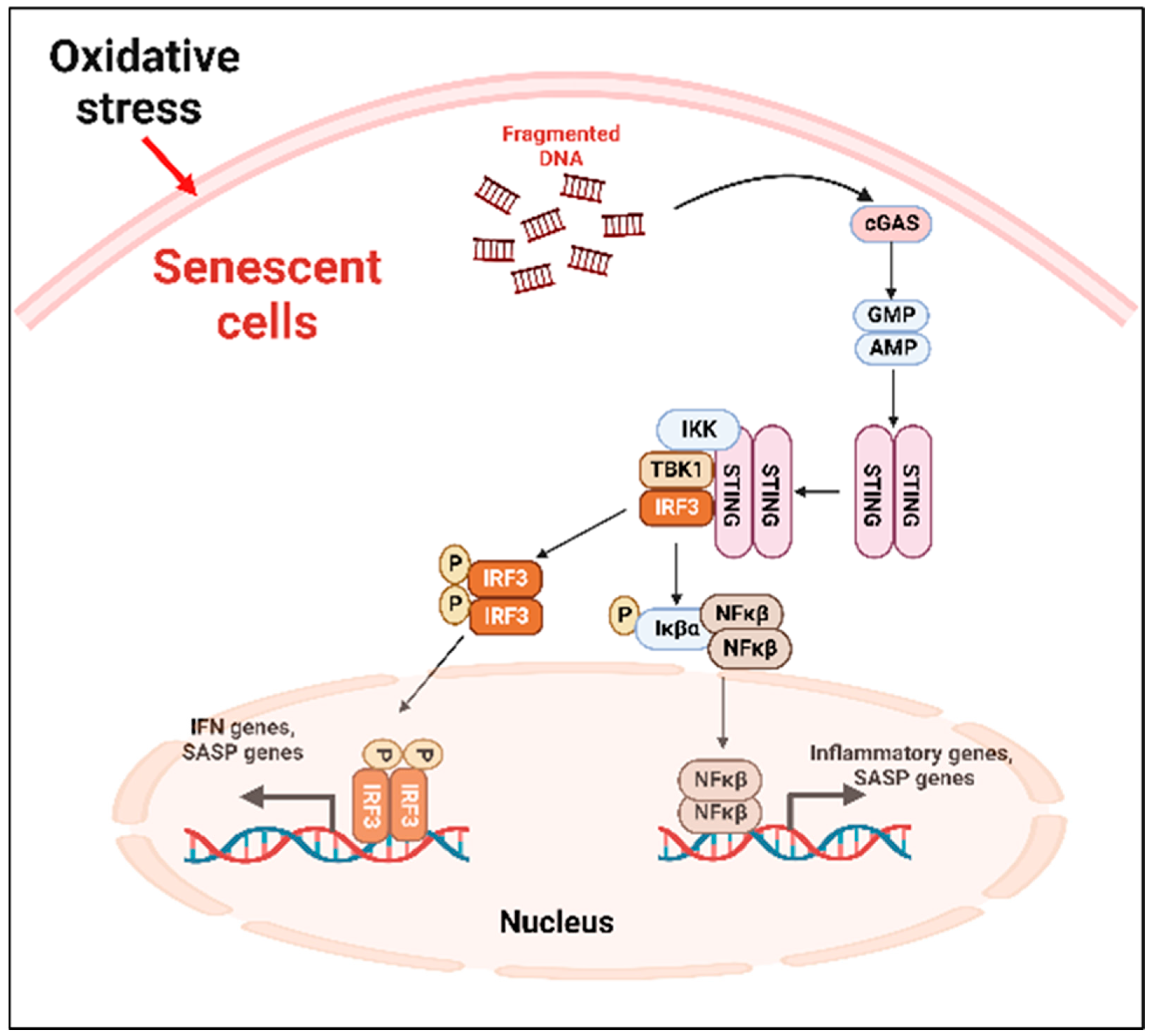
- A.
- Cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) (cGAS-STING) pathway
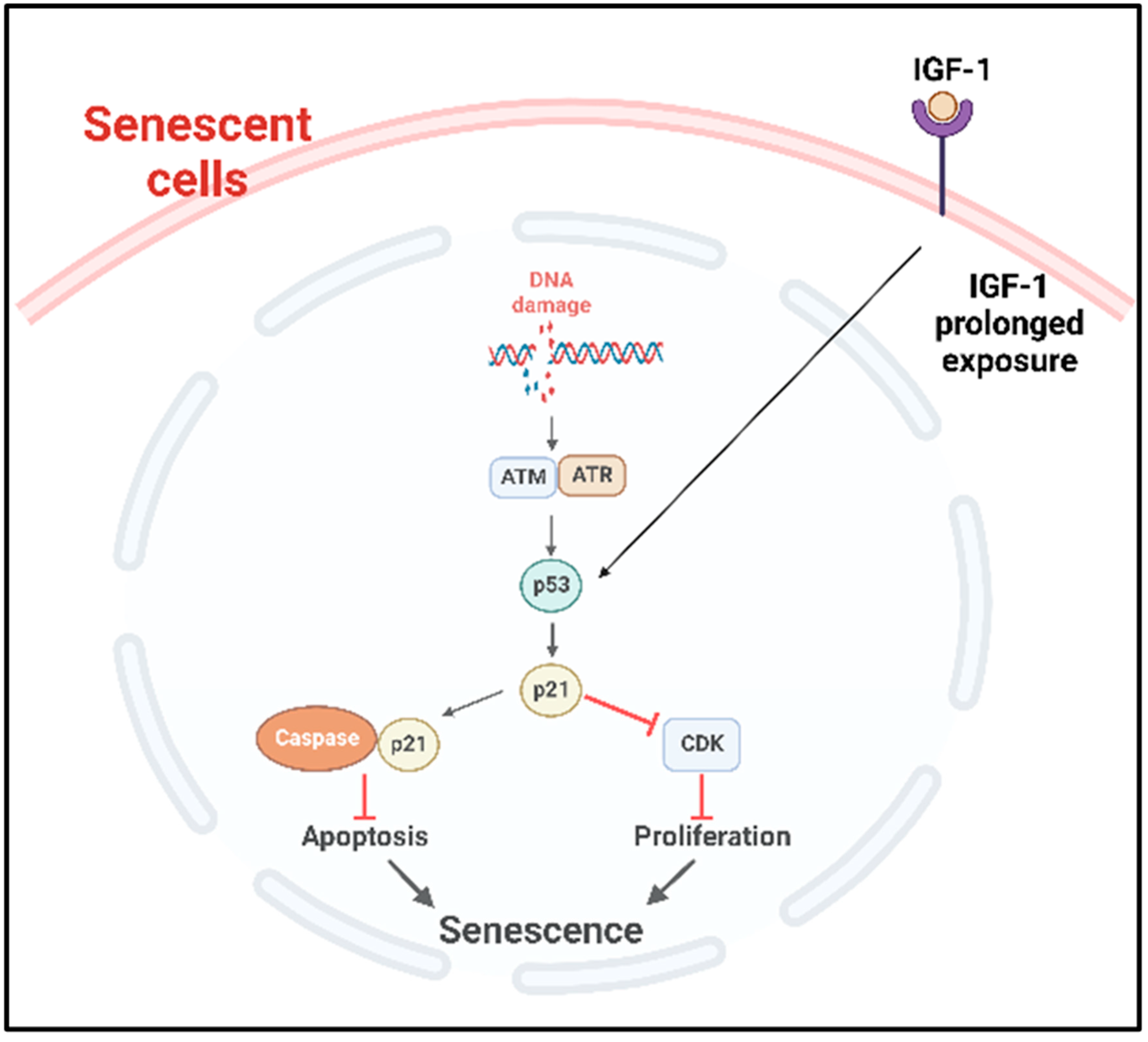
- B.
- p53 pathway
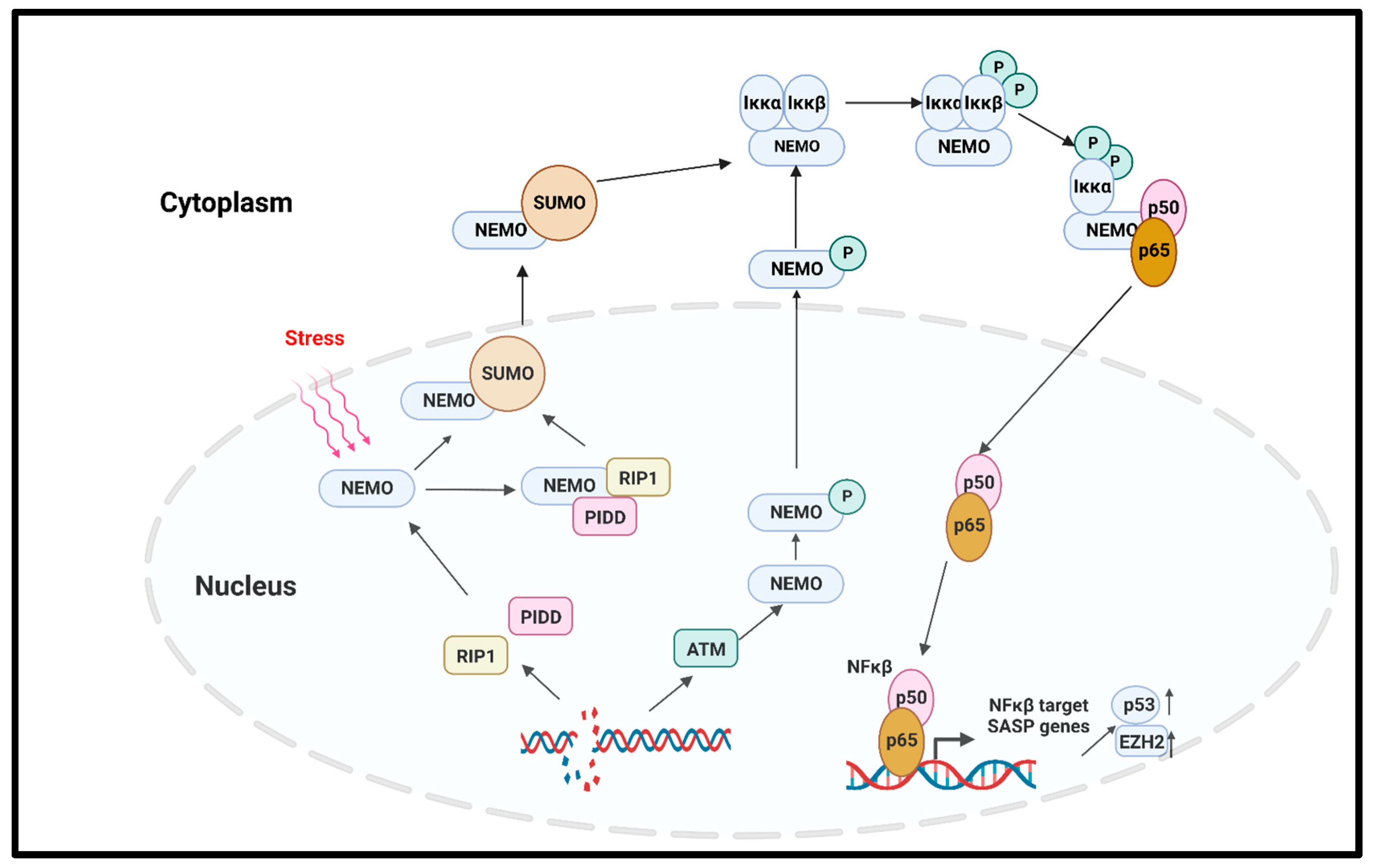
- C.
- NFκβ pathway
- D.
- Mammalian target of rapamycin (mTOR) pathway
- E.
- Transforming growth factor-β (TGFβ) pathway
- F.
- Mitogen-activated protein kinase (MAPK) pathway
2.4. Telomerase Activity Suppresses Senescence and Its Inhibition Enhances Senescence
3. Lymphatic System: A Critical Regulator of Fluid Homeostasis and Immune Response
3.1. Structural Components of Lymphatic System and Its Function
3.2. Aging and Effects on Lymphatic Function and Pathophysiology
3.2.1. Lymphatic Inflammation and Lymphangiogenesis
3.2.2. Lymphatic Contractility
3.2.3. Immunosuppression of Lymph Nodes in Tumor Microenvironment
4. Prosenescence Mechanisms in Different Cancer Treatments
5. Potential Senotherapies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Senotherapeutic Drugs | Class | Targeted Diseases | Status |
|---|---|---|---|
| Dasatinib+Quercetin | Senolytic | Alzheimer disease, aging | Clinical trial: NCT04063124, NCT05422885 |
| Quercetin | Senolytic, senomorphic | Coronary artery disease | Clinical trial: NCT04907253 |
| Navitovlax | Senolytic | Clearing senescent bone marrow hematopoietic stem cells (HSCs) and senescent muscle stem cells (MuSCs) from aged mice or mice under irradiation | [165] |
| Clearing senescent osteoarthritic chondrocytes in osteoarthritis | [166] | ||
| Cardiac Glycosides (Ouabain, Digoxin, and Proscillaridin A) | Senolytic | Lung fibrosis, elimination of apoptotic cells | [167] |
| Fisetin | Senolytic, senomorphic | Aging, progeroid mice model | [168] |
| UBX0101 | Senolytic | Osteoarthritis, knee, treating degenerative joint disease | Clinical trial: NCT03513016 [169,170] |
| UBX1967 | Senolytic | Pathological neovascularization (NV) | https://iovs.arvojournals.org/article.aspx?articleid=2774894 (accessed on 28 January 2023) |
| UBX1325 | Senolytic | Neovascular age-related macular degeneration | NCT04537884, NCT05275205 |
| Curcumin | Senolytic | Cardiovascular risk factor, vascular aging, aging | NCT04119752, NCT01968564 [171] |
| Curcumin Analog EF24 | Senolytic | Senolytic elimination of senescent endothelial cells, senescent fibroblast | [172] |
| A1331852 | Senolytic | Eliminate senescent cells (HUVEC) and IMR90 | [173] |
| A1155463 | Senolytic | Eliminate senescent cells (HUVEC) and IMR90 | [173] |
| Hsp90 inhibitors (Geldanamycin, Tanespimycin, Alvespimycin) | Senolytic | Elimination of senescent cells in vitro | [174] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Vaziri, H.; Counter, C.M.; Allsopp, R.C. The telomere hypothesis of cellular aging. Exp. Gerontol. 1992, 27, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Invest. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence and apoptosis: How cellular responses might influence aging phenotypes. Exp. Gerontol. 2003, 38, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Kotla, S.; Reddy Velatooru, L.; Abe, R.J.; Davis, E.A.; Cooke, J.P.; Schadler, K.; Deswal, A.; Herrmann, J.; Lin, S.H.; et al. Senescence-Associated Secretory Phenotype as a Hinge Between Cardiovascular Diseases and Cancer. Front. Cardiovasc. Med 2021, 8, 763930. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Lawrenson, K.; Grun, B.; Benjamin, E.; Jacobs, I.J.; Dafou, D.; Gayther, S.A. Senescent fibroblasts promote neoplastic transformation of partially transformed ovarian epithelial cells in a three-dimensional model of early stage ovarian cancer. Neoplasia 2010, 12, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Liu, G.; Coleman, I.; Nelson, P.S.; Zhang, M.; Dash, R.; Fisher, P.B.; Plymate, S.R.; Wu, J.D. IL-6 promotes prostate tumorigenesis and progression through autocrine cross-activation of IGF-IR. Oncogene 2011, 30, 2345–2355. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Di, G.H.; Liu, Y.; Lu, Y.; Liu, J.; Wu, C.; Duan, H.F. IL-6 secreted from senescent mesenchymal stem cells promotes proliferation and migration of breast cancer cells. PLoS ONE 2014, 9, e113572. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Rawal, B.; Nemeth, J.A.; Haura, E.B. JAK1 activates STAT3 activity in non-small-cell lung cancer cells and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling. Mol. Cancer Ther. 2011, 10, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Moon, S.; Kim, D.K.; Zhang, X.; Kim, J. CXCL1 induces senescence of cancer-associated fibroblasts via autocrine loops in oral squamous cell carcinoma. PLoS ONE 2018, 13, e0188847. [Google Scholar] [CrossRef]
- Li, H.; Qiu, L.; Liu, Q.; Ma, Z.; Xie, X.; Luo, Y.; Wu, X. Senescent Fibroblasts Generate a CAF Phenotype through the Stat3 Pathway. Genes 2022, 13, 1579. [Google Scholar] [CrossRef]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278. [Google Scholar] [CrossRef]
- Freitas-Rodriguez, S.; Folgueras, A.R.; Lopez-Otin, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Hassona, Y.; Cirillo, N.; Heesom, K.; Parkinson, E.K.; Prime, S.S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 2014, 111, 1230–1237. [Google Scholar] [CrossRef]
- Liu, D.; Hornsby, P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tuxhorn, J.A.; Ressler, S.J.; McAlhany, S.J.; Dang, T.D.; Rowley, D.R. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res. 2005, 65, 8887–8895. [Google Scholar] [CrossRef]
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beausejour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef] [PubMed]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef]
- Wu, S.G.; He, Z.Y.; Li, Q.; Sun, J.Y.; Li, F.Y.; Lin, Q.; Lin, H.X.; Guan, X.X. Prognostic value of metastatic axillary lymph node ratio for Chinese breast cancer patients. PLoS ONE 2013, 8, e61410. [Google Scholar] [CrossRef]
- Wang, L.; Dou, X.; Liu, T.; Lu, W.; Ma, Y.; Yang, Y. Tumor size and lymph node metastasis are prognostic markers of small cell lung cancer in a Chinese population. Medicine 2018, 97, e11712. [Google Scholar] [CrossRef]
- Taghizadeh-Kermani, A.; Yahouiyan, S.Z.; AliAkbarian, M.; Seilanian Toussi, M. Prognostic significance of metastatic lymph node ratio in patients with gastric cancer: An evaluation in north-East of iran. Iran. J. Cancer Prev. 2014, 7, 73–79. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.H.; Park, T.J. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 2017, 8, 15208. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence-Stemness Alliance—A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Khadka, D.B.; Cho, W.J. Topoisomerase inhibitors as anticancer agents: A patent update. Expert. Opin. Ther. Pat. 2013, 23, 1033–1056. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, M.L. Topoisomerase I inhibitors: Review and update. Ann. Oncol. 1997, 8, 837–855. [Google Scholar] [CrossRef]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Karabicici, M.; Alptekin, S.; Firtina Karagonlar, Z.; Erdal, E. Doxorubicin-induced senescence promotes stemness and tumorigenicity in EpCAM-/CD133- nonstem cell population in hepatocellular carcinoma cell line, HuH-7. Mol. Oncol. 2021, 15, 2185–2202. [Google Scholar] [CrossRef] [PubMed]
- Mitry, M.A.; Laurent, D.; Keith, B.L.; Sira, E.; Eisenberg, C.A.; Eisenberg, L.M.; Joshi, S.; Gupte, S.; Edwards, J.G. Accelerated cardiomyocyte senescence contributes to late-onset doxorubicin-induced cardiotoxicity. Am. J. Physiol. Cell Physiol. 2020, 318, C380–C391. [Google Scholar] [CrossRef]
- Beltzig, L.; Schwarzenbach, C.; Leukel, P.; Frauenknecht, K.B.M.; Sommer, C.; Tancredi, A.; Hegi, M.E.; Christmann, M.; Kaina, B. Senescence Is the Main Trait Induced by Temozolomide in Glioblastoma Cells. Cancers 2022, 14, 2233. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.; Chiu, C.C.; Li, C.H.; Chang, Y.T.; Hwang, H.T. Cisplatin-induced senescence and growth inhibition in human non-small cell lung cancer cells with ectopic transfer of p16INK4a. Oncol. Res. 2007, 16, 479–488. [Google Scholar] [CrossRef]
- Zhao, W.; Lin, Z.X.; Zhang, Z.Q. Cisplatin-induced premature senescence with concomitant reduction of gap junctions in human fibroblasts. Cell Res. 2004, 14, 60–66. [Google Scholar] [CrossRef]
- Sun, X.; Shi, B.; Zheng, H.; Min, L.; Yang, J.; Li, X.; Liao, X.; Huang, W.; Zhang, M.; Xu, S.; et al. Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis. 2018, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, W.; Dong, H.; Li, Y.; Li, L.; Han, L.; Han, Z.; Wang, S.; Ma, D.; Wang, H. Cisplatin-induced senescence in ovarian cancer cells is mediated by GRP78. Oncol. Rep. 2014, 31, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, H. Doxorubicin-Induced Cancer Cell Senescence Shows a Time Delay Effect and Is Inhibited by Epithelial-Mesenchymal Transition (EMT). Med. Sci. Monit 2019, 25, 3617–3623. [Google Scholar] [CrossRef]
- Tamamori-Adachi, M.; Koga, A.; Susa, T.; Fujii, H.; Tsuchiya, M.; Okinaga, H.; Hisaki, H.; Iizuka, M.; Kitajima, S.; Okazaki, T. DNA damage response induced by Etoposide promotes steroidogenesis via GADD45A in cultured adrenal cells. Sci. Rep. 2018, 8, 9636. [Google Scholar] [CrossRef]
- Teng, Y.N.; Chang, H.C.; Chao, Y.Y.; Cheng, H.L.; Lien, W.C.; Wang, C.Y. Etoposide Triggers Cellular Senescence by Inducing Multiple Centrosomes and Primary Cilia in Adrenocortical Tumor Cells. Cells 2021, 10, 1466. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Barth, K. Bleomycin and its role in inducing apoptosis and senescence in lung cells-modulating effects of caveolin-1. Curr. Cancer Drug Targets 2009, 9, 341–353. [Google Scholar] [CrossRef]
- Wang, X.; Wong, S.C.; Pan, J.; Tsao, S.W.; Fung, K.H.; Kwong, D.L.; Sham, J.S.; Nicholls, J.M. Evidence of cisplatin-induced senescent-like growth arrest in nasopharyngeal carcinoma cells. Cancer Res. 1998, 58, 5019–5022. [Google Scholar]
- Seifrtova, M.; Havelek, R.; Soukup, T.; Filipova, A.; Mokry, J.; Rezacova, M. Mitoxantrone ability to induce premature senescence in human dental pulp stem cells and human dermal fibroblasts. J. Physiol. Pharmacol. 2013, 64, 255–266. [Google Scholar]
- Han, L.; Long, Q.; Li, S.; Xu, Q.; Zhang, B.; Dou, X.; Qian, M.; Jiramongkol, Y.; Guo, J.; Cao, L.; et al. Senescent Stromal Cells Promote Cancer Resistance through SIRT1 Loss-Potentiated Overproduction of Small Extracellular Vesicles. Cancer Res. 2020, 80, 3383–3398. [Google Scholar] [CrossRef]
- Aasland, D.; Gotzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Mhaidat, N.M.; Zhang, X.D.; Allen, J.; Avery-Kiejda, K.A.; Scott, R.J.; Hersey, P. Temozolomide induces senescence but not apoptosis in human melanoma cells. Br. J. Cancer 2007, 97, 1225–1233. [Google Scholar] [CrossRef]
- Mohiuddin, M.; Kasahara, K. The Mechanisms of the Growth Inhibitory Effects of Paclitaxel on Gefitinib-resistant Non-small Cell Lung Cancer Cells. Cancer Genom. Proteom. 2021, 18, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Milczarek, M. The Premature Senescence in Breast Cancer Treatment Strategy. Cancers 2020, 12, 1815. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, M.; Mosieniak, G.; Skierski, J.; Sikora, E.; Rode, W. Methotrexate-induced senescence in human adenocarcinoma cells is accompanied by induction of p21(waf1/cip1) expression and lack of polyploidy. Cancer Lett. 2009, 284, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.W.; Zhang, S.S.; Song, J.R.; Sun, K.; Zong, C.; Zhao, Q.D.; Liu, W.T.; Li, R.; Wu, M.C.; Wei, L.X. Autophagy inhibition switches low-dose camptothecin-induced premature senescence to apoptosis in human colorectal cancer cells. Biochem. Pharmacol. 2014, 90, 265–275. [Google Scholar] [CrossRef]
- Aguado-Flor, E.; Fuentes-Raspall, M.J.; Gonzalo, R.; Alonso, C.; Ramon, Y.C.T.; Fisas, D.; Seoane, A.; Sanchez-Pla, A.; Giralt, J.; Diez, O.; et al. Cell Senescence-Related Pathways Are Enriched in Breast Cancer Patients With Late Toxicity After Radiotherapy and Low Radiation-Induced Lymphocyte Apoptosis. Front. Oncol. 2022, 12, 825703. [Google Scholar] [CrossRef]
- Meng, J.; Li, Y.; Wan, C.; Sun, Y.; Dai, X.; Huang, J.; Hu, Y.; Gao, Y.; Wu, B.; Zhang, Z.; et al. Targeting senescence-like fibroblasts radiosensitizes non-small cell lung cancer and reduces radiation-induced pulmonary fibrosis. JCI Insight 2021, 6, e146334. [Google Scholar] [CrossRef]
- Dabritz, J.H.; Yu, Y.; Milanovic, M.; Schonlein, M.; Rosenfeldt, M.T.; Dorr, J.R.; Kaufmann, A.M.; Dorken, B.; Schmitt, C.A. CD20-Targeting Immunotherapy Promotes Cellular Senescence in B-Cell Lymphoma. Mol. Cancer Ther. 2016, 15, 1074–1081. [Google Scholar] [CrossRef]
- Chibaya, L.; Snyder, J.; Ruscetti, M. Senescence and the tumor-immune landscape: Implications for cancer immunotherapy. Semin. Cancer Biol. 2022, 86, 827–845. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, K.; Xia, Y.; Li, Y.; Hou, Y.; Wang, L.; Li, L.; Chang, L.; Li, W. Cellular senescence in ionizing radiation (Review). Oncol. Rep. 2019, 42, 883–894. [Google Scholar] [CrossRef]
- Wang, Y.; Boerma, M.; Zhou, D. Ionizing Radiation-Induced Endothelial Cell Senescence and Cardiovascular Diseases. Radiat. Res. 2016, 186, 153–161. [Google Scholar] [CrossRef]
- Nishimura, T.; Iwakabe, K.; Sekimoto, M.; Ohmi, Y.; Yahata, T.; Nakui, M.; Sato, T.; Habu, S.; Tashiro, H.; Sato, M.; et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J. Exp. Med. 1999, 190, 617–627. [Google Scholar] [CrossRef]
- Qin, S.; Schulte, B.A.; Wang, G.Y. Role of senescence induction in cancer treatment. World J. Clin. Oncol. 2018, 9, 180–187. [Google Scholar] [CrossRef]
- Muller-Hermelink, N.; Braumuller, H.; Pichler, B.; Wieder, T.; Mailhammer, R.; Schaak, K.; Ghoreschi, K.; Yazdi, A.; Haubner, R.; Sander, C.A.; et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 2008, 13, 507–518. [Google Scholar] [CrossRef]
- Braumuller, H.; Wieder, T.; Brenner, E.; Assmann, S.; Hahn, M.; Alkhaled, M.; Schilbach, K.; Essmann, F.; Kneilling, M.; Griessinger, C.; et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013, 494, 361–365. [Google Scholar] [CrossRef]
- Rosemblit, C.; Datta, J.; Lowenfeld, L.; Xu, S.; Basu, A.; Kodumudi, K.; Wiener, D.; Czerniecki, B.J. Oncodriver inhibition and CD4(+) Th1 cytokines cooperate through Stat1 activation to induce tumor senescence and apoptosis in HER2+ and triple negative breast cancer: Implications for combining immune and targeted therapies. Oncotarget 2018, 9, 23058–23077. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4337–4341. [Google Scholar] [CrossRef] [PubMed]
- Kotla, S.; Zhang, A.; Imanishi, M.; Ko, K.A.; Lin, S.H.; Gi, Y.J.; Moczygemba, M.; Isgandarova, S.; Schadler, K.L.; Chung, C.; et al. Nucleus-mitochondria positive feedback loop formed by ERK5 S496 phosphorylation-mediated poly (ADP-ribose) polymerase activation provokes persistent pro-inflammatory senescent phenotype and accelerates coronary atherosclerosis after chemo-radiation. Redox. Biol. 2021, 47, 102132. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.; Heidel, F.H. Molecular Mechanisms of Senescence and Implications for the Treatment of Myeloid Malignancies. Cancers 2021, 13, 612. [Google Scholar] [CrossRef] [PubMed]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Benslimane, Y.; Sanchez-Osuna, M.; Coulombe-Huntington, J.; Bertomeu, T.; Henry, D.; Huard, C.; Bonneil, E.; Thibault, P.; Tyers, M.; Harrington, L. A novel p53 regulator, C16ORF72/TAPR1, buffers against telomerase inhibition. Aging Cell 2021, 20, e13331. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef] [PubMed]
- de Ostrovich, K.K.; Lambertz, I.; Colby, J.K.; Tian, J.; Rundhaug, J.E.; Johnston, D.; Conti, C.J.; DiGiovanni, J.; Fuchs-Young, R. Paracrine overexpression of insulin-like growth factor-1 enhances mammary tumorigenesis in vivo. Am. J. Pathol. 2008, 173, 824–834. [Google Scholar] [CrossRef]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef]
- De Donatis, G.M.; Le Pape, E.; Pierron, A.; Cheli, Y.; Hofman, V.; Hofman, P.; Allegra, M.; Zahaf, K.; Bahadoran, P.; Rocchi, S.; et al. NF-kB2 induces senescence bypass in melanoma via a direct transcriptional activation of EZH2. Oncogene 2016, 35, 2735–2745. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Fan, T.; Jiang, S.; Chung, N.; Alikhan, A.; Ni, C.; Lee, C.C.; Hornyak, T.J. EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol. Cancer Res. 2011, 9, 418–429. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Wu, Z.H.; Miyamoto, S. Many faces of NF-kappaB signaling induced by genotoxic stress. J. Mol. Med. 2007, 85, 1187–1202. [Google Scholar] [CrossRef] [PubMed]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Tong, F.; Qian, C.; Zhang, R.; Dong, J.; Wu, G.; Hu, Y. NEMO modulates radiation-induced endothelial senescence of human umbilical veins through NF-kappaB signal pathway. Radiat. Res. 2015, 183, 82–93. [Google Scholar] [CrossRef]
- Fang, L.; Choudhary, S.; Zhao, Y.; Edeh, C.B.; Yang, C.; Boldogh, I.; Brasier, A.R. ATM regulates NF-kappaB-dependent immediate-early genes via RelA Ser 276 phosphorylation coupled to CDK9 promoter recruitment. Nucleic Acids Res. 2014, 42, 8416–8432. [Google Scholar] [CrossRef]
- Pandita, T.K. ATM function and telomere stability. Oncogene 2002, 21, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Smilenov, L.B.; Morgan, S.E.; Mellado, W.; Sawant, S.G.; Kastan, M.B.; Pandita, T.K. Influence of ATM function on telomere metabolism. Oncogene 1997, 15, 2659–2665. [Google Scholar] [CrossRef] [PubMed]
- Pandita, T.K.; Pathak, S.; Geard, C.R. Chromosome end associations, telomeres and telomerase activity in ataxia telangiectasia cells. Cytogenet. Cell Genet. 1995, 71, 86–93. [Google Scholar] [CrossRef]
- Smilenov, L.B.; Dhar, S.; Pandita, T.K. Altered telomere nuclear matrix interactions and nucleosomal periodicity in ataxia telangiectasia cells before and after ionizing radiation treatment. Mol. Cell Biol. 1999, 19, 6963–6971. [Google Scholar] [CrossRef]
- Wood, L.D.; Halvorsen, T.L.; Dhar, S.; Baur, J.A.; Pandita, R.K.; Wright, W.E.; Hande, M.P.; Calaf, G.; Hei, T.K.; Levine, F.; et al. Characterization of ataxia telangiectasia fibroblasts with extended life-span through telomerase expression. Oncogene 2001, 20, 278–288. [Google Scholar] [CrossRef]
- Robles, S.J.; Adami, G.R. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 1998, 16, 1113–1123. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Cai, Y.; Wei, Y. mTOR Signaling from Cellular Senescence to Organismal Aging. Aging Dis. 2014, 5, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bartolome, R.; Patel, V.; Cotrim, A.; Leelahavanichkul, K.; Molinolo, A.A.; Mitchell, J.B.; Gutkind, J.S. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell 2012, 11, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Lerner, C.; Bitto, A.; Pulliam, D.; Nacarelli, T.; Konigsberg, M.; Van Remmen, H.; Torres, C.; Sell, C. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell 2013, 12, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology 2010, 52, 966–974. [Google Scholar] [CrossRef]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Borlon, C.; Pascal, T.; Royer, V.; Eliaers, F.; Ninane, N.; Carrard, G.; Friguet, B.; de Longueville, F.; Boffe, S.; et al. Repeated exposure of human skin fibroblasts to UVB at subcytotoxic level triggers premature senescence through the TGF-beta1 signaling pathway. J. Cell Sci. 2005, 118, 743–758. [Google Scholar] [CrossRef]
- Papageorgis, P. Complex Interplay Between Aging and Cancer: Role of TGF-beta Signaling. Crit. Rev. Oncog. 2017, 22, 313–321. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-beta Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Lee, J.H.; Hwang, S.C.; Choi, K.S.; Yoon, G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Albright, C.D.; Salganik, R.I.; Craciunescu, C.N.; Mar, M.H.; Zeisel, S.H. Mitochondrial and microsomal derived reactive oxygen species mediate apoptosis induced by transforming growth factor-beta1 in immortalized rat hepatocytes. J. Cell Biochem. 2003, 89, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef]
- Rapisarda, V.; Borghesan, M.; Miguela, V.; Encheva, V.; Snijders, A.P.; Lujambio, A.; O’Loghlen, A. Integrin Beta 3 Regulates Cellular Senescence by Activating the TGF-beta Pathway. Cell Rep. 2017, 18, 2480–2493. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef]
- Harley, C.B.; Sherwood, S.W. Telomerase, checkpoints and cancer. Cancer Surv. 1997, 29, 263–284. [Google Scholar]
- Greider, C.W. Telomere length regulation. Annu. Rev. Biochem. 1996, 65, 337–365. [Google Scholar] [CrossRef]
- Harley, C.B.; Sherwood, S.W. Aging of cultured human skin fibroblasts. Methods Mol. Biol. 1997, 75, 23–30. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef]
- Fu, W.; Killen, M.; Culmsee, C.; Dhar, S.; Pandita, T.K.; Mattson, M.P. The catalytic subunit of telomerase is expressed in developing brain neurons and serves a cell survival-promoting function. J. Mol. Neurosci. 2000, 14, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Hahn, W.C.; Ino, Y.; Ronfard, V.; Wu, J.Y.; Weinberg, R.A.; Louis, D.N.; Li, F.P.; Rheinwald, J.G. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell Biol. 2000, 20, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, T.; Foster, S.A.; Koop, J.I.; McDougall, J.K.; Galloway, D.A.; Klingelhutz, A.J. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998, 396, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Baranwal, G.; Rutkowski, J.M. Reduced lymphatic function contributes to age-related disease. Aging 2019, 11, 9969–9970. [Google Scholar] [CrossRef]
- Shang, T.; Liang, J.; Kapron, C.M.; Liu, J. Pathophysiology of aged lymphatic vessels. Aging 2019, 11, 6602–6613. [Google Scholar] [CrossRef]
- Banerjee, P.; Roy, S.; Chakraborty, S. Recent advancement of imaging strategies of the lymphatic system: Answer to the decades old questions. Microcirculation 2022, 29, e12780. [Google Scholar] [CrossRef] [PubMed]
- Jakic, B.; Kerjaschki, D.; Wick, G. Lymphatic Capillaries in Aging. Gerontology 2020, 66, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Loyola, A.; Petrova, T.V. Development and aging of the lymphatic vascular system. Adv. Drug Deliv. Rev. 2021, 169, 63–78. [Google Scholar] [CrossRef]
- Shimada, R.; Tatara, Y.; Kibayashi, K. Gene expression in meningeal lymphatic endothelial cells following traumatic brain injury in mice. PLoS ONE 2022, 17, e0273892. [Google Scholar] [CrossRef]
- Lin, F.J.; Chen, X.; Qin, J.; Hong, Y.K.; Tsai, M.J.; Tsai, S.Y. Direct transcriptional regulation of neuropilin-2 by COUP-TFII modulates multiple steps in murine lymphatic vessel development. J. Clin. Invest. 2010, 120, 1694–1707. [Google Scholar] [CrossRef]
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362. [Google Scholar] [CrossRef] [PubMed]
- Oliver, G.; Kipnis, J.; Randolph, G.J.; Harvey, N.L. The Lymphatic Vasculature in the 21(st) Century: Novel Functional Roles in Homeostasis and Disease. Cell 2020, 182, 270–296. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Shen, Q.; Pivetti, C.D.; Lee, E.S.; Wu, M.H.; Yuan, S.Y. Molecular mechanisms of endothelial hyperpermeability: Implications in inflammation. Expert. Rev. Mol. Med. 2009, 11, e19. [Google Scholar] [CrossRef] [PubMed]
- Bruunsgaard, H.; Pedersen, M.; Pedersen, B.K. Aging and proinflammatory cytokines. Curr. Opin. Hematol. 2001, 8, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; von der Weid, P.Y. Inflammation-induced lymphangiogenesis and lymphatic dysfunction. Angiogenesis 2014, 17, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Roy, S.; Kumaravel, S.; Banerjee, P.; White, T.K.; O’Brien, A.; Seelig, C.; Chauhan, R.; Ekser, B.; Bayless, K.J.; Alpini, G.; et al. Tumor Lymphatic Interactions Induce CXCR2-CXCL5 Axis and Alter Cellular Metabolism and Lymphangiogenic Pathways to Promote Cholangiocarcinoma. Cells 2021, 10, 3093. [Google Scholar] [CrossRef]
- Kumaravel, S.; Singh, S.; Roy, S.; Venkatasamy, L.; White, T.K.; Sinha, S.; Glaser, S.S.; Safe, S.H.; Chakraborty, S. CXCL11-CXCR3 Axis Mediates Tumor Lymphatic Cross Talk and Inflammation-Induced Tumor, Promoting Pathways in Head and Neck Cancers. Am. J. Pathol. 2020, 190, 900–915. [Google Scholar] [CrossRef]
- Maini, R.; Nagalli, S. Lymphadenopathy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Chakraborty, S.; Davis, M.J.; Muthuchamy, M. Emerging trends in the pathophysiology of lymphatic contractile function. Semin. Cell Dev. Biol. 2015, 38, 55–66. [Google Scholar] [CrossRef]
- Zawieja, D.C. Contractile physiology of lymphatics. Lymphat. Res. Biol. 2009, 7, 87–96. [Google Scholar] [CrossRef]
- Zheng, W.; Aspelund, A.; Alitalo, K. Lymphangiogenic factors, mechanisms, and applications. J. Clin. Invest. 2014, 124, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Zolla, V.; Nizamutdinova, I.T.; Scharf, B.; Clement, C.C.; Maejima, D.; Akl, T.; Nagai, T.; Luciani, P.; Leroux, J.C.; Halin, C.; et al. Aging-related anatomical and biochemical changes in lymphatic collectors impair lymph transport, fluid homeostasis, and pathogen clearance. Aging Cell 2015, 14, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Bridenbaugh, E.A.; Gashev, A.A. Aging-associated alterations in contractility of rat mesenteric lymphatic vessels. Microcirculation 2011, 18, 463–473. [Google Scholar] [CrossRef]
- Bridenbaugh, E.A.; Nizamutdinova, I.T.; Jupiter, D.; Nagai, T.; Thangaswamy, S.; Chatterjee, V.; Gashev, A.A. Lymphatic muscle cells in rat mesenteric lymphatic vessels of various ages. Lymphat. Res. Biol. 2013, 11, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Gashev, A.A.; Zawieja, D.C. Hydrodynamic regulation of lymphatic transport and the impact of aging. Pathophysiology 2010, 17, 277–287. [Google Scholar] [CrossRef]
- Chatterjee, V.; Gashev, A.A. Aging-associated shifts in functional status of mast cells located by adult and aged mesenteric lymphatic vessels. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H693–H702. [Google Scholar] [CrossRef]
- Girard, J.P.; Moussion, C.; Forster, R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012, 12, 762–773. [Google Scholar] [CrossRef]
- Cakala-Jakimowicz, M.; Kolodziej-Wojnar, P.; Puzianowska-Kuznicka, M. Aging-Related Cellular, Structural and Functional Changes in the Lymph Nodes: A Significant Component of Immunosenescence? An Overview. Cells 2021, 10, 3148. [Google Scholar] [CrossRef]
- Bousso, P. T-cell activation by dendritic cells in the lymph node: Lessons from the movies. Nat. Rev. Immunol. 2008, 8, 675–684. [Google Scholar] [CrossRef]
- Shen, J.; Luo, X.; Wu, Q.; Huang, J.; Xiao, G.; Wang, L.; Yang, B.; Li, H.; Wu, C. A Subset of CXCR5(+)CD8(+) T Cells in the Germinal Centers From Human Tonsils and Lymph Nodes Help B Cells Produce Immunoglobulins. Front. Immunol. 2018, 9, 2287. [Google Scholar] [CrossRef]
- He, R.; Hou, S.; Liu, C.; Zhang, A.; Bai, Q.; Han, M.; Yang, Y.; Wei, G.; Shen, T.; Yang, X.; et al. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature 2016, 537, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Ramello, M.C.; Nunez, N.G.; Tosello Boari, J.; Bossio, S.N.; Canale, F.P.; Abrate, C.; Ponce, N.; Del Castillo, A.; Ledesma, M.; Viel, S.; et al. Polyfunctional KLRG-1(+)CD57(+) Senescent CD4(+) T Cells Infiltrate Tumors and Are Expanded in Peripheral Blood From Breast Cancer Patients. Front. Immunol. 2021, 12, 713132. [Google Scholar] [CrossRef] [PubMed]
- Shankwitz, K.; Pallikkuth, S.; Sirupangi, T.; Kirk Kvistad, D.; Russel, K.B.; Pahwa, R.; Gama, L.; Koup, R.A.; Pan, L.; Villinger, F.; et al. Compromised steady-state germinal center activity with age in nonhuman primates. Aging Cell 2020, 19, e13087. [Google Scholar] [CrossRef] [PubMed]
- Demirci, D.; Dayanc, B.; Mazi, F.A.; Senturk, S. The Jekyll and Hyde of Cellular Senescence in Cancer. Cells 2021, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Exosomal vesicles enhance immunosuppression in chronic inflammation: Impact in cellular senescence and the aging process. Cell Signal. 2020, 75, 109771. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, J.; Legge, K.; Perlman, S. Age-related increases in PGD(2) expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J. Clin. Invest. 2011, 121, 4921–4930. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef]
- Taschner-Mandl, S.; Schwarz, M.; Blaha, J.; Kauer, M.; Kromp, F.; Frank, N.; Rifatbegovic, F.; Weiss, T.; Ladenstein, R.; Hohenegger, M.; et al. Metronomic topotecan impedes tumor growth of MYCN-amplified neuroblastoma cells in vitro and in vivo by therapy induced senescence. Oncotarget 2016, 7, 3571–3586. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Robbins, P.D. Senotherapeutics for healthy ageing. Nat. Rev. Drug Discov. 2018, 17, 377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular senescence: A key therapeutic target in aging and diseases. J. Clin. Invest. 2022, 132, e158450. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Kim, S.R.; Jiang, K.; Ogrodnik, M.; Chen, X.; Zhu, X.Y.; Lohmeier, H.; Ahmed, L.; Tang, H.; Tchkonia, T.; Hickson, L.J.; et al. Increased renal cellular senescence in murine high-fat diet: Effect of the senolytic drug quercetin. Transl. Res. 2019, 213, 112–123. [Google Scholar] [CrossRef]
- Shao, Z.; Wang, B.; Shi, Y.; Xie, C.; Huang, C.; Chen, B.; Zhang, H.; Zeng, G.; Liang, H.; Wu, Y.; et al. Senolytic agent Quercetin ameliorates intervertebral disc degeneration via the Nrf2/NF-kappaB axis. Osteoarthritis. Cartilage 2021, 29, 413–422. [Google Scholar] [CrossRef]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. Senolytics for Cancer Therapy: Is All That Glitters Really Gold? Cancers 2021, 13, 723. [Google Scholar] [CrossRef]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; Deursen, J.V.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Adamczyk-Grochala, J.; Lewinska, A. Nano-Based Theranostic Tools for the Detection and Elimination of Senescent Cells. Cells 2020, 9, 2659. [Google Scholar] [CrossRef] [PubMed]
- Nagesh, P.K.B.; Chowdhury, P.; Hatami, E.; Kumari, S.; Kashyap, V.K.; Tripathi, M.K.; Wagh, S.; Meibohm, B.; Chauhan, S.C.; Jaggi, M.; et al. Cross-Linked Polyphenol-Based Drug Nano-Self-Assemblies Engineered to Blockade Prostate Cancer Senescence. ACS Appl. Mater. Interfaces 2019, 11, 38537–38554. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Rovira, M.; Galiana, I.; Gimenez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Munoz-Martin, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Yang, H.; Chen, C.; Chen, H.; Duan, X.; Li, J.; Zhou, Y.; Zeng, W.; Yang, L. Navitoclax (ABT263) reduces inflammation and promotes chondrogenic phenotype by clearing senescent osteoarthritic chondrocytes in osteoarthritis. Aging 2020, 12, 12750–12770. [Google Scholar] [CrossRef]
- Triana-Martinez, F.; Picallos-Rabina, P.; Da Silva-Alvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreiros, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Zhang, X.X.; He, S.H.; Liang, X.; Li, W.; Li, T.F.; Li, D.F. Aging, Cell Senescence, the Pathogenesis and Targeted Therapies of Osteoarthritis. Front. Pharmacol. 2021, 12, 728100. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Bielak-Zmijewska, A.; Grabowska, W.; Ciolko, A.; Bojko, A.; Mosieniak, G.; Bijoch, L.; Sikora, E. The Role of Curcumin in the Modulation of Ageing. Int. J. Mol. Sci. 2019, 20, 1239. [Google Scholar] [CrossRef]
- Li, W.; He, Y.; Zhang, R.; Zheng, G.; Zhou, D. The curcumin analog EF24 is a novel senolytic agent. Aging 2019, 11, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]





| Class | Name of the Drug | Cancer Types | Reference |
|---|---|---|---|
| Chemotherapy | Doxorubicin | Cervical cancer (HeLa cells), hepatocellular carcinoma (HuH7), colorectal carcinoma, breast cancer | [34,43] |
| Etoposide | Adrenocortical H295R cells, epithelial carcinoma (A549), adrenocortical tumor cells | [44,45] | |
| Bleomycin | Pulmonary fibrosis, alveolar epithelial cells | [46] | |
| Cisplatin | Ovarian cancer, nasopharyngeal carcinoma cells, lung cancer | [40,47] | |
| Mitoxantrone | Dermal fibroblasts, prostate cancer | [48,49] | |
| Temozolomide | Glioma, melanoma | [50,51] | |
| Paclitaxel | Non-small-cell lung cancer cells, breast cancer | [52,53] | |
| Methotrexate | Breast cancer, colon cancer, adenocarcinoma | [53,54] | |
| Camptothecin | Colorectal cancer | [55] | |
| Radiation therapy | Breast cancer, glioblastoma, non-small-cell lung cancer | [56,57] | |
| Immune therapy | Rituximab | B-cell lymphoma | [58,59] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, P.; Gaddam, N.; Pandita, T.K.; Chakraborty, S. Cellular Senescence as a Brake or Accelerator for Oncogenic Transformation and Role in Lymphatic Metastasis. Int. J. Mol. Sci. 2023, 24, 2877. https://doi.org/10.3390/ijms24032877
Banerjee P, Gaddam N, Pandita TK, Chakraborty S. Cellular Senescence as a Brake or Accelerator for Oncogenic Transformation and Role in Lymphatic Metastasis. International Journal of Molecular Sciences. 2023; 24(3):2877. https://doi.org/10.3390/ijms24032877
Chicago/Turabian StyleBanerjee, Priyanka, Niyanshi Gaddam, Tej K. Pandita, and Sanjukta Chakraborty. 2023. "Cellular Senescence as a Brake or Accelerator for Oncogenic Transformation and Role in Lymphatic Metastasis" International Journal of Molecular Sciences 24, no. 3: 2877. https://doi.org/10.3390/ijms24032877
APA StyleBanerjee, P., Gaddam, N., Pandita, T. K., & Chakraborty, S. (2023). Cellular Senescence as a Brake or Accelerator for Oncogenic Transformation and Role in Lymphatic Metastasis. International Journal of Molecular Sciences, 24(3), 2877. https://doi.org/10.3390/ijms24032877