
Modeling SILAC Data to Assess Protein Turnover in a Cellular Model of Diabetic Nephropathy

 ,
, 
Abstract
1. Introduction
2. Results
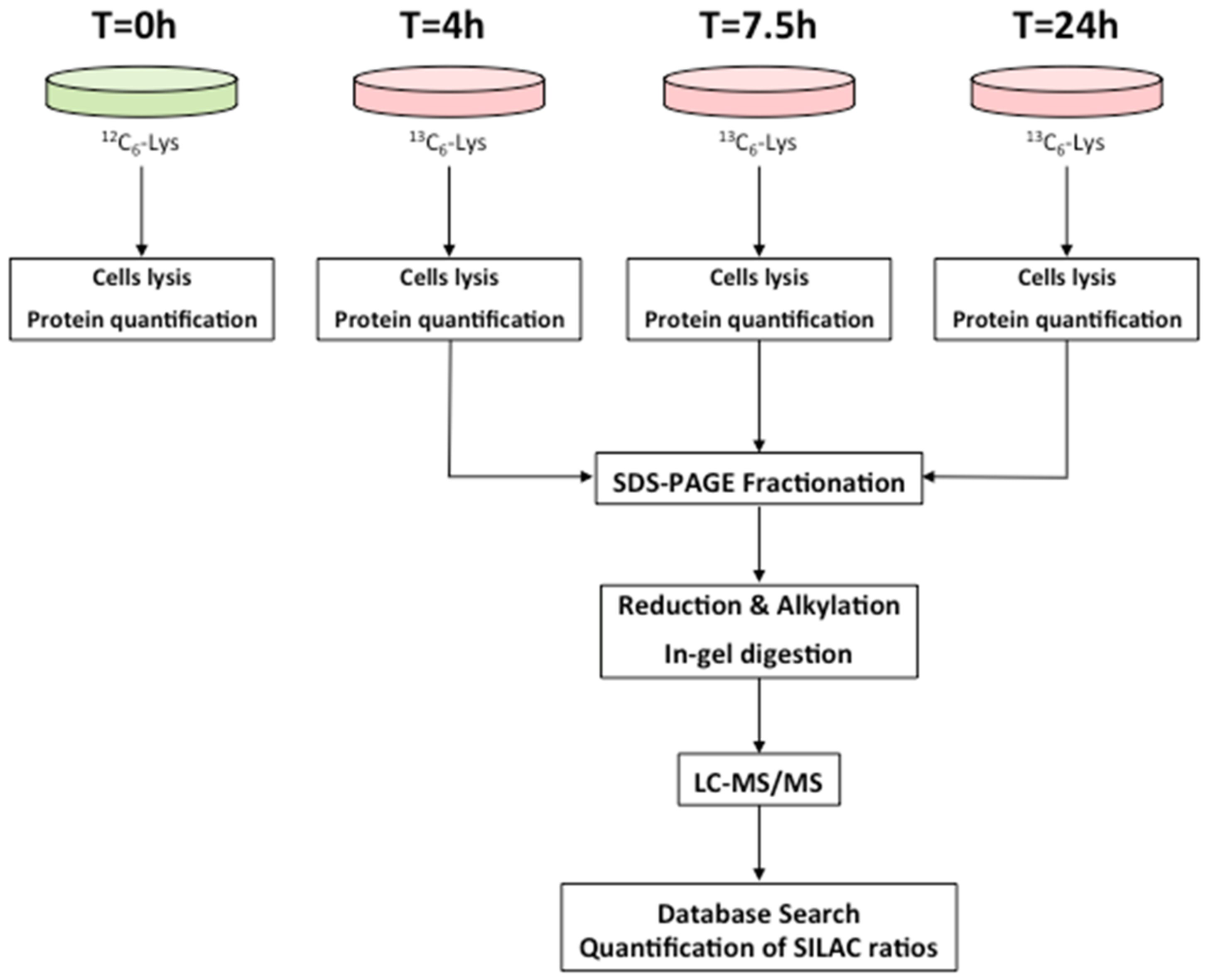
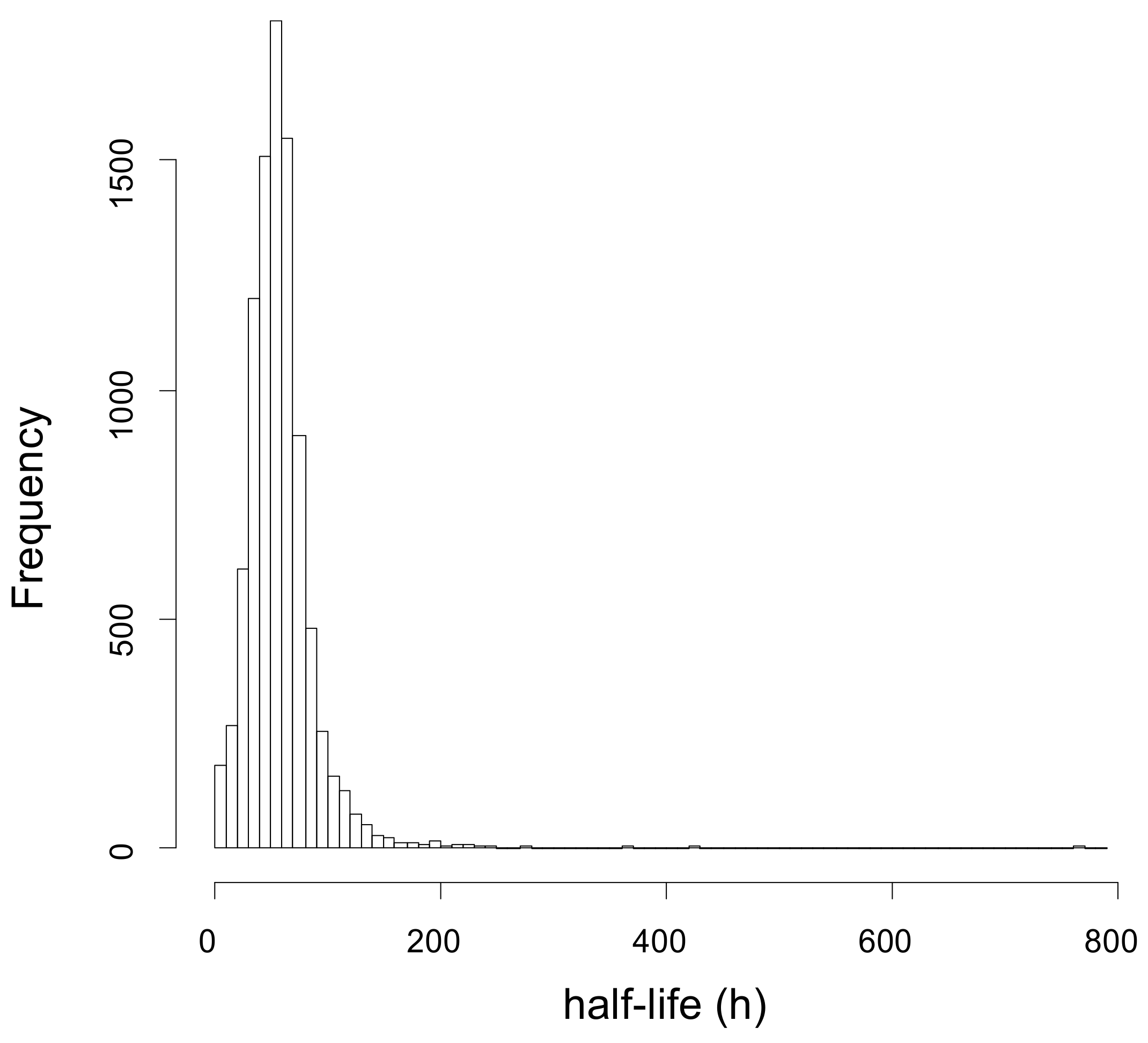
2.1. Dynamic-SILAC Experiment and Determination of prOtein Half-Lives
2.2. Protein Turnover in Diabetic-Controls vs. Diabetic-Nephropathic Subjects
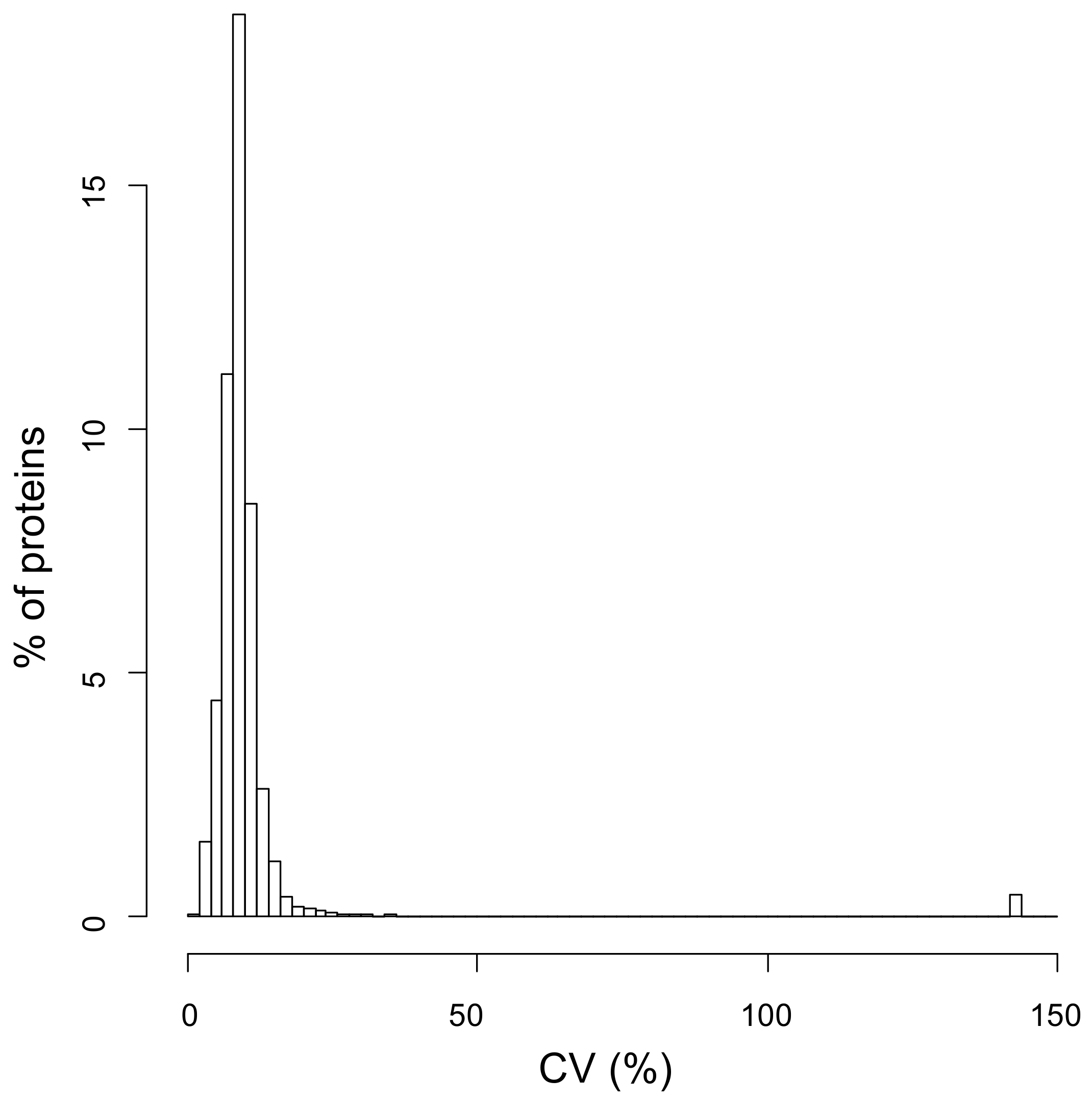
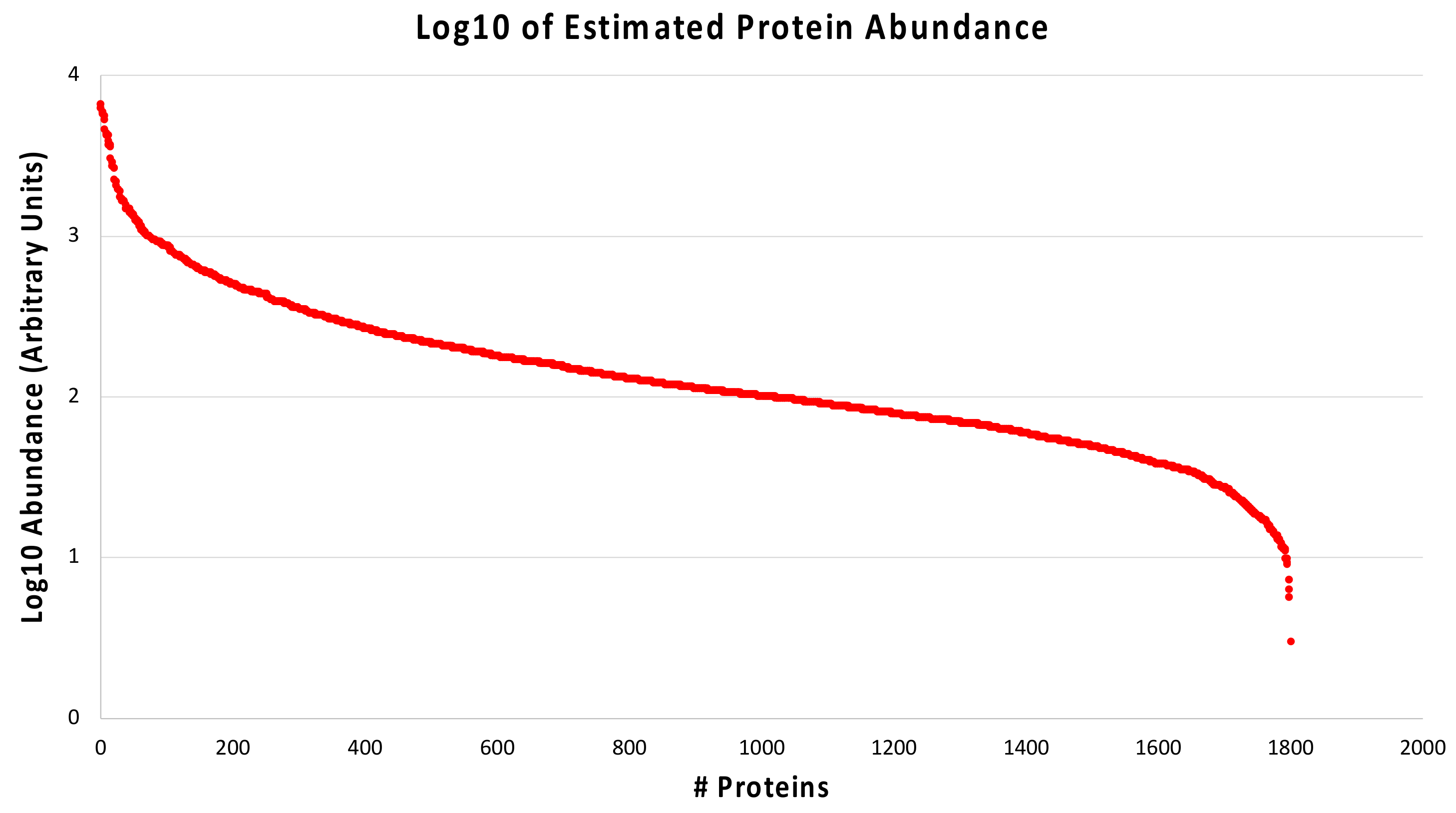
2.3. Protein Quantification in Diabetic-Controls vs. Diabetic-Nephropathic Subjects
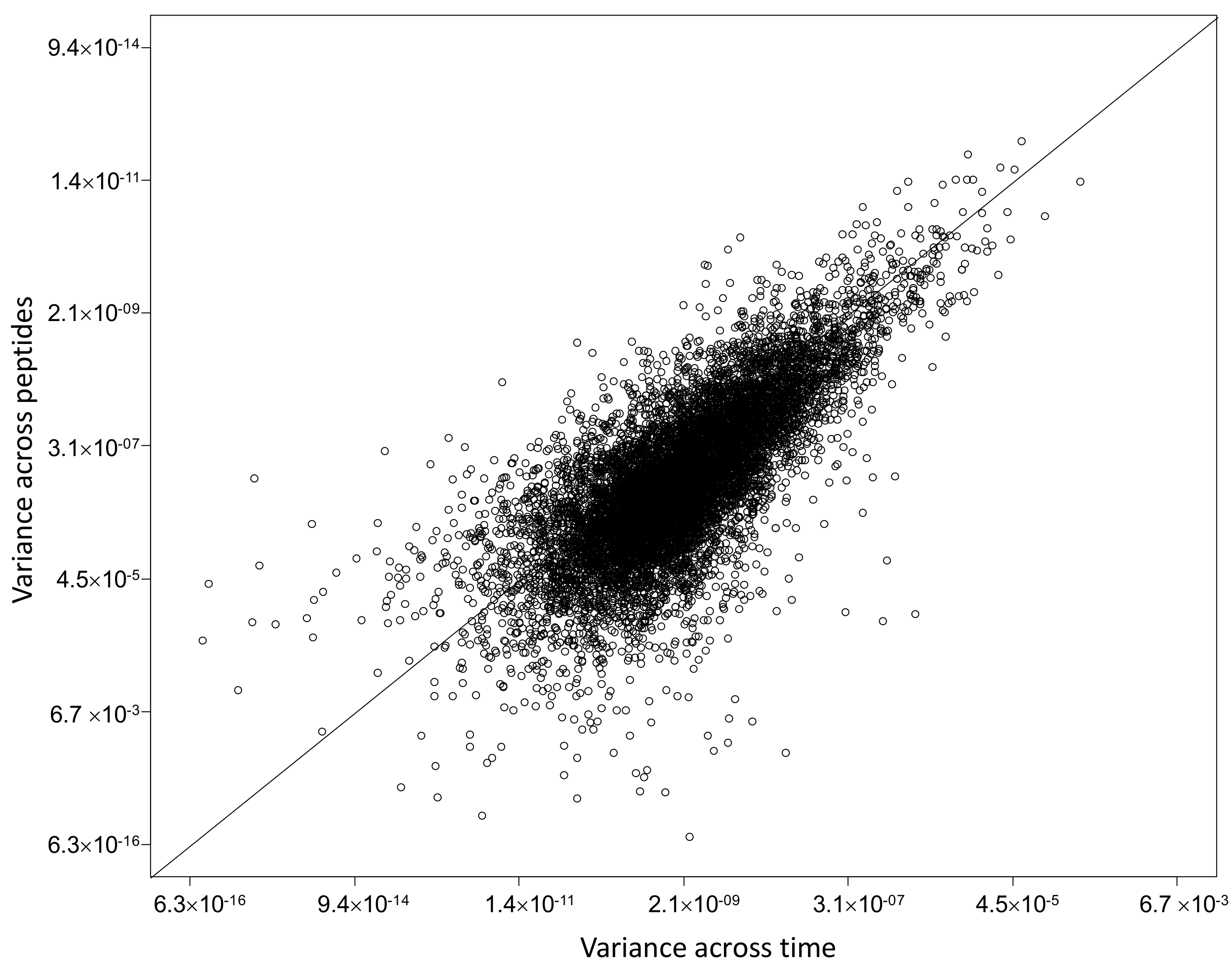
2.4. Characterization of Proteins with Similar Half-Life and Abundance
3. Discussion
3.1. Proteins with a Significantly Different Turnover in Nephropathic Subjects
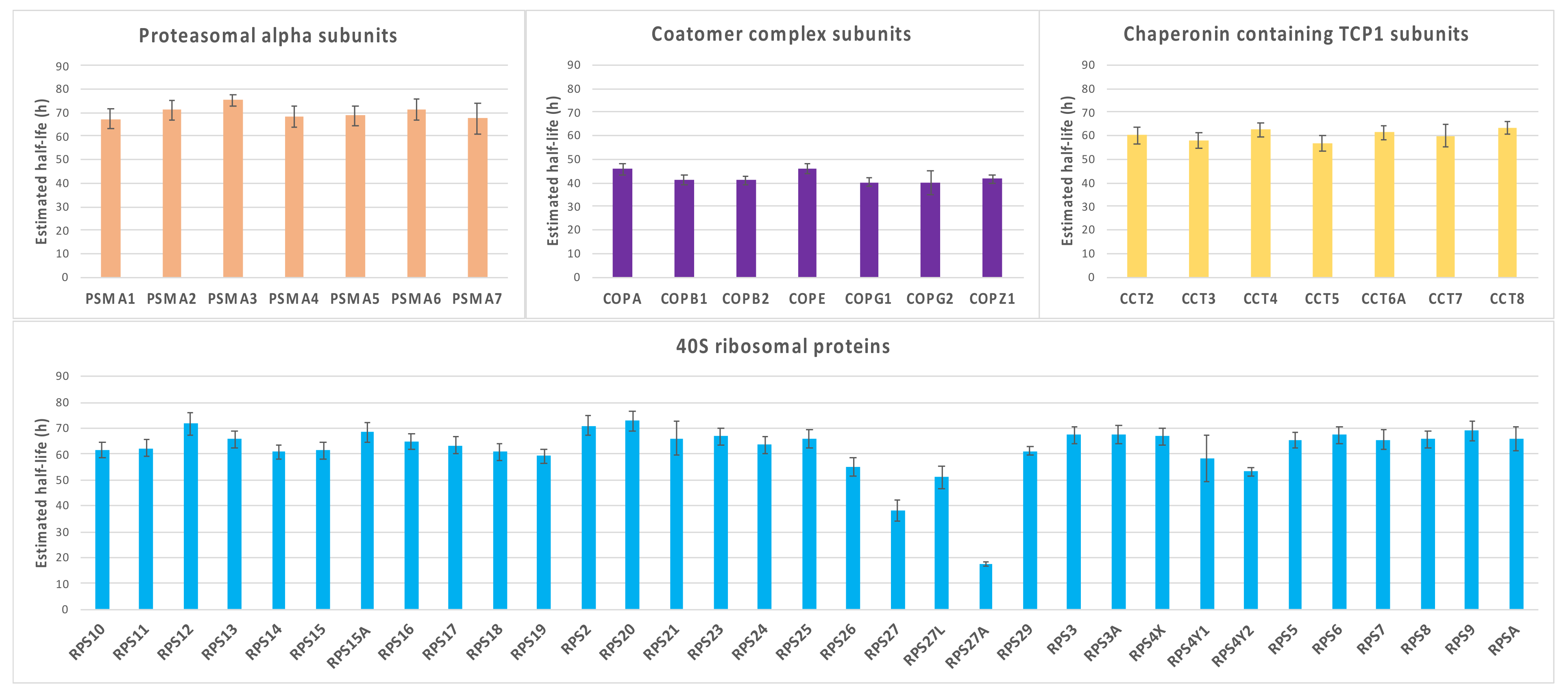
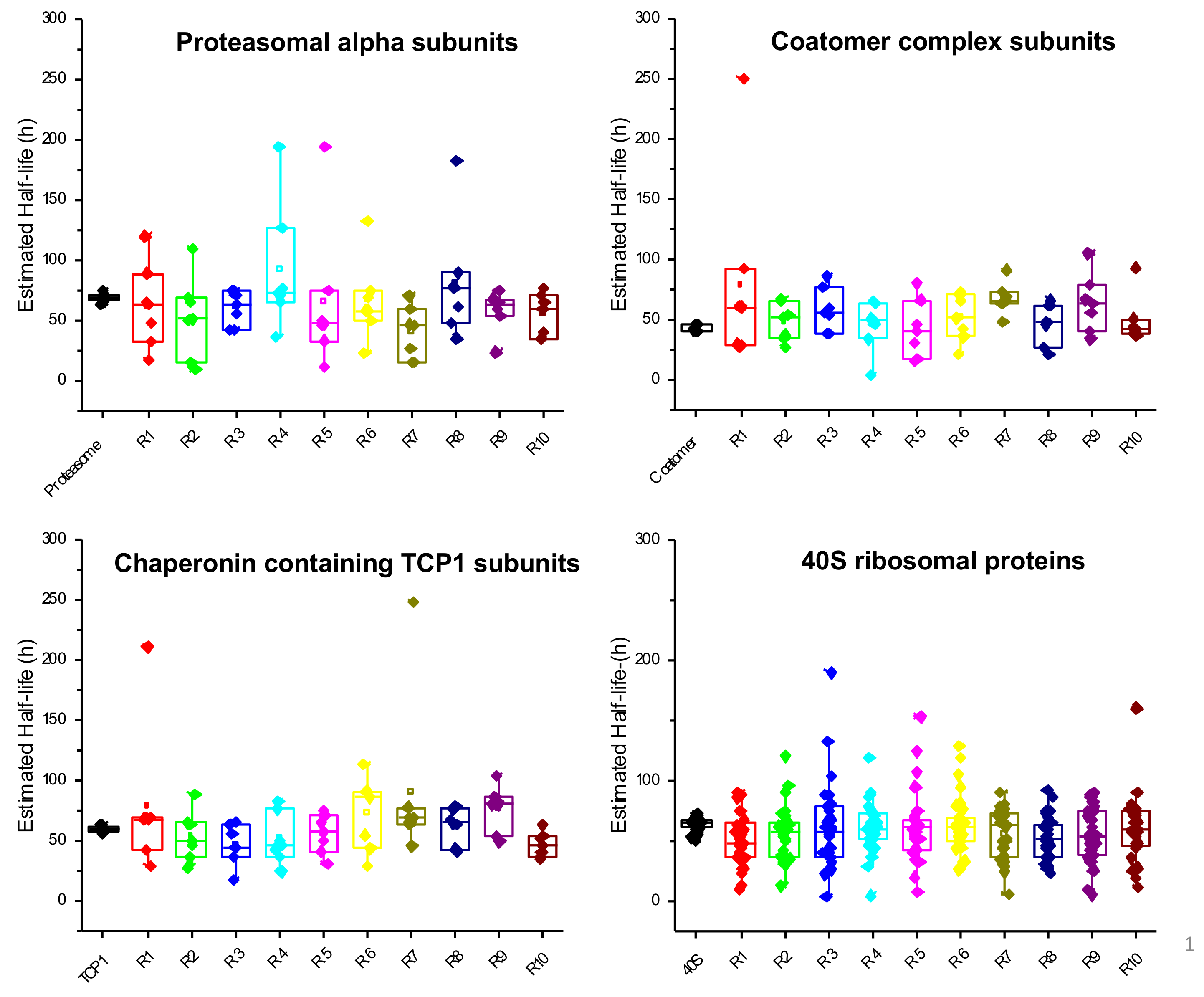
3.1.1. Mesangial Proteins
3.1.2. Chaperone and Cytoskeleton Proteins
3.1.3. Proteins Associated to Hydrogen Sulfide (H2S) Metabolism
3.1.4. Proteins Involved in Translation and Kidney Hypertrophy
3.1.5. Other Proteins with Altered Turnover Rate
3.1.6. GSEA Reveals That Proteasomal Proteins Have Longer Half-Lives in Nephropathic Subjects
3.2. Proteins with Different Abundance in Nephropathic Subjects
3.3. Proteins with Similar Half-Life and Abundance Are Functionally Related
4. Materials and Methods
4.1. Patients’ Selection and Enrolment
4.2. Cell Collection and Culture
4.3. Dynamic-SILAC Experiment, Sample Preparation and In-Gel Protein Digestion
4.4. LC-MS/MS Analysis
4.5. Protein Identification and Quantification
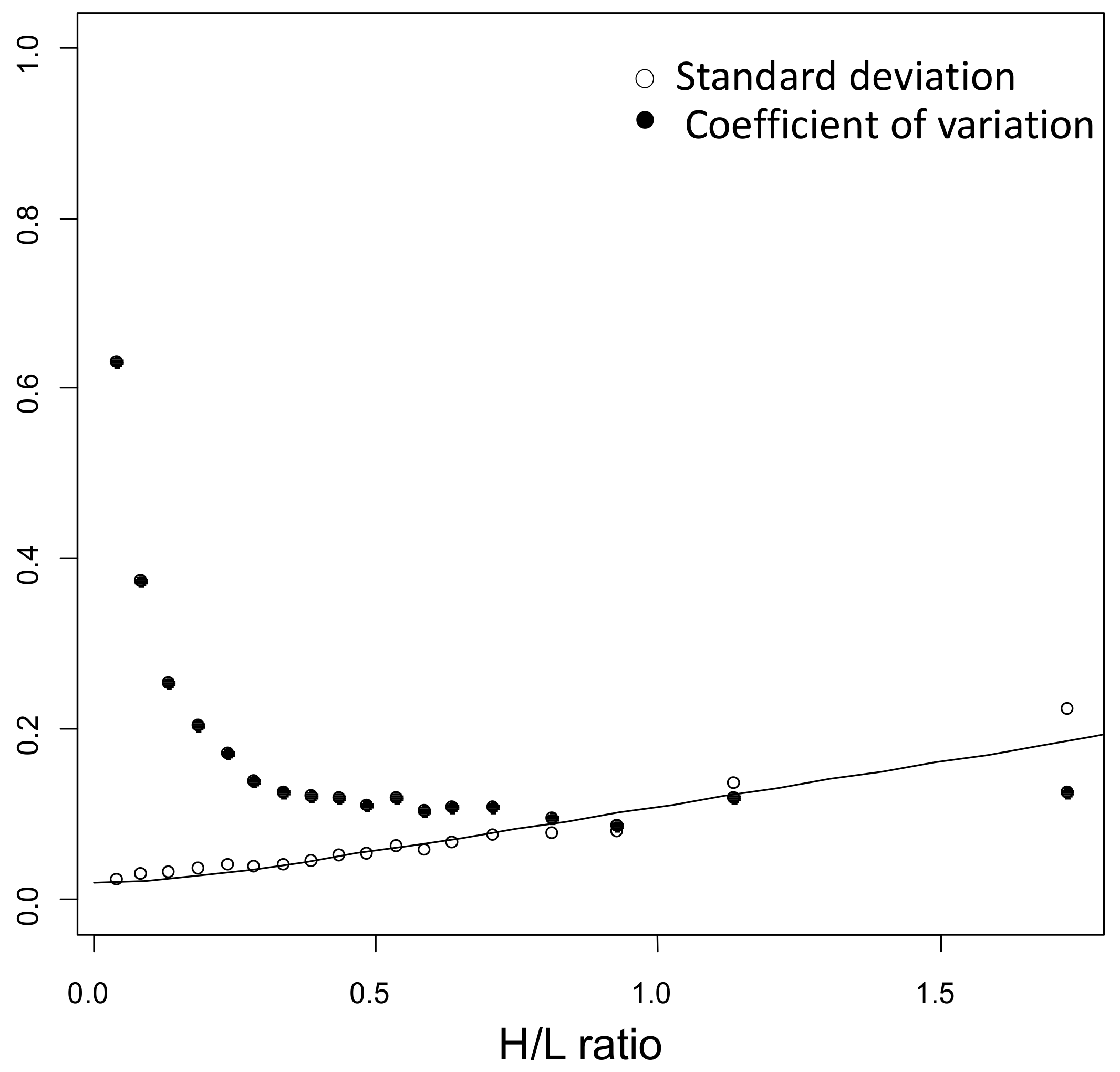
4.6. Kinetic Analyses
4.7. Protein Abundance
4.8. Bioinformatic and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rozanova, S.; Barkovits, K.; Nikolov, M.; Schmidt, C.; Urlaub, H.; Marcus, K. Quantitative Mass Spectrometry-Based Proteomics: An Overview. Methods Mol. Biol. 2021, 2228, 85–116. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.B.; Langer, J.D.; Jovanovic, M. Proteome Turnover in the Spotlight: Approaches, Applications, and Perspectives. Mol. Cell Proteom. 2021, 20, 100016. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef]
- Abu Ahmad, Y.; Oknin-Vaisman, A.; Bitman-Lotan, E.; Orian, A. From the Evasion of Degradation to Ubiquitin-Dependent Protein Stabilization. Cells. 2021, 10, 2374. [Google Scholar] [CrossRef]
- Kaur, N.; Raja, R.; Ruiz-Velasco, A.; Liu, W. Cellular Protein Quality Control in Diabetic Cardiomyopathy: From Bench to Bedside. Front. Cardiovasc. Med. 2020, 7, 585309. [Google Scholar] [CrossRef]
- Waterlow, J.C. Whole-body protein turnover in humans—Past, present, and future. Annu. Rev. Nutr. 1995, 15, 57–92. [Google Scholar] [CrossRef]
- Doherty, M.K.; Hammond, D.E.; Clague, M.J.; Gaskell, S.J.; Beynon, R.J. Turnover of the human proteome: Determination of protein intracellular stability by dynamic SILAC. J. Proteome. Res. 2009, 8, 104–112. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342, Erratum in Nature 2013, 495, 126–127. [Google Scholar] [CrossRef]
- Fornasiero, E.F.; Mandad, S.; Wildhagen, H.; Alevra, M.; Rammner, B.; Keihani, S.; Opazo, F.; Urban, I.; Ischebeck, T.; Sakib, M.S.; et al. Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat. Commun. 2018, 9, 4230. [Google Scholar] [CrossRef]
- McClatchy, D.B.; Martínez-Bartolomé, S.; Gao, Y.; Lavallée-Adam, M.; Yates, J.R., III. Quantitative analysis of global protein stability rates in tissues. Sci. Rep. 2020, 10, 15983. [Google Scholar] [CrossRef]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Mathieson, T.; Franken, H.; Kosinski, J.; Kurzawa, N.; Zinn, N.; Sweetman, G.; Poeckel, D.; Ratnu, V.S.; Schramm, M.; Becher, I.; et al. Systematic analysis of protein turnover in primary cells. Nat. Commun. 2018, 9, 689. [Google Scholar] [CrossRef]
- Lerman, O.Z.; Galiano, R.D.; Armour, M.; Levine, J.P.; Gurtner, G.C. Cellular dysfunction in the diabetic fibroblast: Impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am. J. Pathol. 2003, 162, 303–315. [Google Scholar] [CrossRef]
- Brem, H.; Golinko, M.S.; Stojadinovic, O.; Kodra, A.; Diegelmann, R.F.; Vukelic, S.; Entero, H.; Coppock, D.L.; Tomic-Canic, M. Primary cultured fibroblasts derived from patients with chronic wounds: A methodology to produce human cell lines and test putative growth factor therapy such as GMCSF. J. Transl. Med. 2008, 6, 75. [Google Scholar] [CrossRef]
- Jones, A.R.I.V.; Coleman, E.L.; Husni, N.R.; Deeney, J.T.; Raval, F.; Steenkamp, D.; Dooms, H.; Nikolajczyk, B.S.; Corkey, B.E. Type 1 diabetes alters lipid handling and metabolism in human fibroblasts and peripheral blood mononuclear cells. PLoS ONE 2017, 12, e0188474. [Google Scholar] [CrossRef]
- Al-Rikabi, A.H.A.; Tobin, D.J.; Riches-Suman, K.; Thornton, M.J. Dermal fibroblasts cultured from donors with type 2 diabetes mellitus retain an epigenetic memory associated with poor wound healing responses. Sci. Rep. 2021, 11, 1474. [Google Scholar] [CrossRef]
- LaPointe, M.S.; Batlle, D. Cultured skin fibroblasts as an in vitro model to assess phenotypic features in subjects with diabetic nephropathy. Am. J. Kidney Dis. 2001, 38, 646–648. [Google Scholar] [CrossRef]
- Caramori, M.L.; Kim, Y.; Fioretto, P.; Huang, C.; Rich, S.S.; Miller, M.E.; Russell, G.B.; Mauer, M. Cellular basis of diabetic nephropathy: IV. Antioxidant enzyme mRNA expression levels in skin fibroblasts of type 1 diabetic sibling pairs. Nephrol. Dial. Transplant. 2006, 21, 3122–3126. [Google Scholar] [CrossRef]
- Millioni, R.; Iori, E.; Puricelli, L.; Arrigoni, G.; Vedovato, M.; Trevisan, R.; James, P.; Tiengo, A.; Tessari, P. Abnormal cytoskeletal protein expression in cultured skin fibroblasts from type 1 diabetes mellitus patients with nephropathy: A proteomic approach. Proteomics. Clin. Appl. 2008, 2, 492–503. [Google Scholar] [CrossRef]
- Selby, N.M.; Taal, M.W. An updated overview of diabetic nephropathy: Diagnosis, prognosis, treatment goals and latest guidelines. Diabetes Obes. Metab. 2020, 22 (Suppl. S1), 3–15. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Toni, G.; Tascini, G.; Santi, E.; Berioli, M.G.; Principi, N. Environmental Factors Associated With Type 1 Diabetes. Front. Endocrinol. 2019, 10, 592. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.I.; Bostrom, M.; Daeihagh, P.; Bowden, D.W. Genetic factors in diabetic nephropathy. Clin. J. Am. Soc. Nephrol. 2007, 2, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Millioni, R.; Puricelli, L.; Iori, E.; Trevisan, R.; Tessari, P. Skin fibroblasts as a tool for identifying the risk of nephropathy in the type 1 diabetic population. Diabetes Metab. Res. Rev. 2012, 28, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Puricelli, L.; Iori, E.; Millioni, R.; Arrigoni, G.; James, P.; Vedovato, M.; Tessari, P. Proteome analysis of cultured fibroblasts from type 1 diabetic patients and normal subjects. J. Clin. Endocrinol. Metab. 2006, 91, 3507–3514. [Google Scholar] [CrossRef] [PubMed]
- Iori, E.; Millioni, R.; Puricelli, L.; Arrigoni, G.; Lenzini, L.; Trevisan, R.; James, P.; Rossi, G.P.; Pinna, L.A.; Tessari, P. Glycolytic enzyme expression and pyruvate kinase activity in cultured fibroblasts from type 1 diabetic patients with and without nephropathy. Biochim. Biophys. Acta 2008, 1782, 627–633. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic. Acids. Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic. Acids. Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Hwang, S.I.; Lundgren, D.H.; Mayya, V.; Rezaul, K.; Cowan, A.E.; Eng, J.K.; Han, D.K. Systematic characterization of nuclear proteome during apoptosis: A quantitative proteomic study by differential extraction and stable isotope labeling. Mol. Cell Proteom. 2006, 5, 1131–1145. [Google Scholar] [CrossRef]
- Blagoev, B.; Mann, M. Quantitative proteomics to study mitogen-activated protein kinases. Methods 2006, 40, 243–250. [Google Scholar] [CrossRef]
- Ong, S.E.; Kratchmarova, I.; Mann, M. Properties of 13C-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC). J. Proteome. Res. 2003, 2, 173–181. [Google Scholar] [CrossRef]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-binding proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117, Erratum in Nat. Rev. Mol. Cell Biol. 2001, 2, 216. [Google Scholar] [CrossRef]
- Morimoto, S.; Nishimura, N.; Terai, T.; Manabe, S.; Yamamoto, Y.; Shinahara, W.; Miyake, H.; Tashiro, S.; Shimada, M.; Sasaki, T. Rab13 mediates the continuous endocytic recycling of occludin to the cell surface. J. Biol. Chem. 2005, 280, 2220–2228. [Google Scholar] [CrossRef]
- Yamamura, R.; Nishimura, N.; Nakatsuji, H.; Arase, S.; Sasaki, T. The interaction of JRAB/MICAL-L2 with Rab8 and Rab13 coordinates the assembly of tight junctions and adherens junctions. Mol. Biol. Cell 2008, 19, 971–983. [Google Scholar] [CrossRef]
- Bethunaickan, R.; Berthier, C.C.; Ramanujam, M.; Sahu, R.; Zhang, W.; Sun, Y.; Bottinger, E.P.; Ivashkiv, L.; Kretzler, M.; Davidson, A. A unique hybrid renal mononuclear phagocyte activation phenotype in murine systemic lupus erythematosus nephritis. J. Immunol. 2011, 186, 4994–5003. [Google Scholar] [CrossRef]
- Huang, T.H.; Shui, H.A.; Ka, S.M.; Tang, B.L.; Chao, T.K.; Chen, J.S.; Lin, Y.F.; Chen, A. Rab 23 is expressed in the glomerulus and plays a role in the development of focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2009, 24, 743–754. [Google Scholar] [CrossRef]
- Huang, T.H.; Ka, S.M.; Hsu, Y.J.; Shui, H.A.; Tang, B.L.; Hu, K.Y.; Chang, J.L.; Chen, A. Rab23 plays a role in the pathophysiology of mesangial cells--a proteomic analysis. Proteomics 2011, 11, 380–394. [Google Scholar] [CrossRef]
- Yam, A.Y.; Xia, Y.; Lin, H.T.; Burlingame, A.; Gerstein, M.; Frydman, J. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat. Struct. Mol. Biol. 2008, 15, 1255–1262. [Google Scholar] [CrossRef]
- Frydman, J.; Nimmesgern, E.; Erdjument-Bromage, H.; Wall, J.S.; Tempst, P.; Hartl, F.U. Function in protein folding of TRiC, a cytosolic ring complex containing TCP-1 and structurally related subunits. EMBO J. 1992, 11, 4767–4778. [Google Scholar] [CrossRef]
- Gao, Y.; Thomas, J.O.; Chow, R.L.; Lee, G.H.; Cowan, N.J. A cytoplasmic chaperonin that catalyzes beta-actin folding. Cell 1992, 69, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, M.B.; Farr, G.W.; Miklos, D.; Horwich, A.L.; Sternlicht, M.L.; Sternlicht, H. TCP1 complex is a molecular chaperone in tubulin biogenesis. Nature 1992, 358, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Brackley, K.I.; Grantham, J. Activities of the chaperonin containing TCP-1 (CCT): Implications for cell cycle progression and cytoskeletal organisation. Cell Stress Chaperones. 2009, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Z.; Chang, L.C.; Lin, Y.F.; Hung, Y.J.; Pei, D.; Chen, J.S. Chaperonin-containing t-complex protein-1 subunit β as a possible biomarker for the phase of glomerular hyperfiltration of diabetic nephropathy. Dis. Markers. 2015, 2015, 548101. [Google Scholar] [CrossRef]
- Wang, X.; Wilkinson, R.; Kildey, K.; Potriquet, J.; Mulvenna, J.; Lobb, R.J.; Möller, A.; Cloonan, N.; Mukhopadhyay, P.; Kassianos, A.J.; et al. Unique molecular profile of exosomes derived from primary human proximal tubular epithelial cells under diseased conditions. J. Extracell. Vesicles. 2017, 6, 1314073. [Google Scholar] [CrossRef]
- Sanai, T.; Sobka, T.; Johnson, T.; el-Essawy, M.; Muchaneta-Kubara, E.C.; Ben Gharbia, O.; el Oldroyd, S.; Nahas, A.M. Expression of cytoskeletal proteins during the course of experimental diabetic nephropathy. Diabetologia 2000, 43, 91–100. [Google Scholar] [CrossRef]
- Shimoni, Y.; Rattner, J.B. Type 1 diabetes leads to cytoskeleton changes that are reflected in insulin action on rat cardiac K(+) currents. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E575–E585. [Google Scholar] [CrossRef]
- Tessari, P.; Puricelli, L.; Iori, E.; Arrigoni, G.; Vedovato, M.; James, P.; Coracina, A.; Millioni, R. Altered chaperone and protein turnover regulators expression in cultured skin fibroblasts from type 1 diabetes mellitus with nephropathy. J. Proteome. Res. 2007, 6, 976–986. [Google Scholar] [CrossRef]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. [Google Scholar] [CrossRef]
- Kabil, O.; Vitvitsky, V.; Banerjee, R. Sulfur as a signaling nutrient through hydrogen sulfide. Annu. Rev. Nutr. 2014, 34, 171–205. [Google Scholar] [CrossRef]
- Rose, P.; Moore, P.K.; Zhu, Y.Z. H2S biosynthesis and catabolism: New insights from molecular studies. Cell Mol. Life Sci. 2017, 74, 1391–1412. [Google Scholar] [CrossRef]
- Olson, K.R. Mitochondrial adaptations to utilize hydrogen sulfide for energy and signaling. J. Comp. Physiol. B 2012, 182, 881–897. [Google Scholar] [CrossRef]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef]
- Kleiner, G.; Barca, E.; Ziosi, M.; Emmanuele, V.; Xu, Y.; Hidalgo-Gutierrez, A.; Qiao, C.; Tadesse, S.; Area-Gomez, E.; Lopez, L.C.; et al. CoQ10 supplementation rescues nephrotic syndrome through normalization of H2S oxidation pathway. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864, 3708–3722. [Google Scholar] [CrossRef]
- Zhou, X.; Feng, Y.; Zhan, Z.; Chen, J. Hydrogen sulfide alleviates diabetic nephropathy in a streptozotocin-induced diabetic rat model. J. Biol. Chem. 2014, 289, 28827–28834. [Google Scholar] [CrossRef]
- Yu, Y.T.; Yang, J.W. Protective Effect of Hydrogen Sulfide in the Secondary Renal Disease and Renal Transplantation. Sheng Li Ke Xue Jin Zhan. 2016, 47, 93–96. [Google Scholar]
- Dugbartey, G.J. Diabetic nephropathy: A potential savior with ‘rotten-egg’ smell. Pharmacol. Rep. 2017, 69, 331–339. [Google Scholar] [CrossRef]
- Hostetter, T.H. Progression of renal disease and renal hypertrophy. Annu. Rev. Physiol. 1995, 57, 263–278. [Google Scholar] [CrossRef]
- Kasinath, B.S.; Feliers, D.; Sataranatarajan, K.; Ghosh Choudhury, G.; Lee, M.J.; Mariappan, M.M. Regulation of mRNA translation in renal physiology and disease. Am. J. Physiol. Renal. Physiol. 2009, 297, F1153–F1165. [Google Scholar] [CrossRef]
- Marintchev, A.; Edmonds, K.A.; Marintcheva, B.; Hendrickson, E.; Oberer, M.; Suzuki, C.; Herdy, B.; Sonenberg, N.; Wagner, G. Topology and regulation of the human eIF4A/4G/4H helicase complex in translation initiation. Cell 2009, 136, 447–460. [Google Scholar] [CrossRef]
- Kasinath, B.S.; Mariappan, M.M.; Sataranatarajan, K.; Lee, M.J.; Ghosh Choudhury, G.; Feliers, D. Novel mechanisms of protein synthesis in diabetic nephropathy--role of mRNA translation. Rev. Endocr. Metab. Disord. 2008, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Grice, B. What I didn’t see before. Nursing 1991, 21, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Xin, G.; Zhang, W.; Zhang, X. Biomarker prediction for membranous nephropathy prognosis by microarray analysis. Nephrology 2019, 24, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Miyamoto, Y. Urinary cystatin C as a biomarker for diabetic nephropathy and its immunohistochemical localization in kidney in Zucker diabetic fatty (ZDF) rats. Exp. Toxicol. Pathol. 2013, 65, 615–622. [Google Scholar] [CrossRef]
- Brennan, E.P.; Ehrich, M.; O’Donovan, H.; Brazil, D.P.; Crean, J.K.; Murphy, M.; Sadlier, D.M.; Martin, F.; Godson, C.; van den Boom, D.; et al. DNA methylation profiling in cell models of diabetic nephropathy. Epigenetics 2010, 5, 396–401. [Google Scholar] [CrossRef]
- Lo, C.S.; Shi, Y.; Chenier, I.; Ghosh, A.; Wu, C.H.; Cailhier, J.F.; Ethier, J.; Lattouf, J.B.; Filep, J.G.; Ingelfinger, J.R.; et al. Heterogeneous Nuclear Ribonucleoprotein F Stimulates Sirtuin-1 Gene Expression and Attenuates Nephropathy Progression in Diabetic Mice. Diabetes 2017, 66, 1964–1978. [Google Scholar] [CrossRef]
- Ghosh, A.; Abdo, S.; Zhao, S.; Wu, C.H.; Shi, Y.; Lo, C.S.; Chenier, I.; Alquier, T.; Filep, J.G.; Ingelfinger, J.R.; et al. Insulin Inhibits Nrf2 Gene Expression via Heterogeneous Nuclear Ribonucleoprotein F/K in Diabetic Mice. Endocrinology 2017, 158, 903–919. [Google Scholar] [CrossRef]
- Lo, C.S.; Chang, S.Y.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Heterogeneous nuclear ribonucleoprotein F suppresses angiotensinogen gene expression and attenuates hypertension and kidney injury in diabetic mice. Diabetes 2012, 61, 2597–2608. [Google Scholar] [CrossRef]
- Moura, I.C.; Centelles, M.N.; Arcos-Fajardo, M.; Malheiros, D.M.; Collawn, J.F.; Cooper, M.D.; Monteiro, R.C. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J. Exp. Med. 2001, 194, 417–425. [Google Scholar] [CrossRef]
- Berthelot, L.; Papista, C.; Maciel, T.T.; Biarnes-Pelicot, M.; Tissandie, E.; Wang, P.H.; Tamouza, H.; Jamin, A.; Bex-Coudrat, J.; Gestin, A.; et al. Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J. Exp. Med. 2012, 209, 793–806. [Google Scholar] [CrossRef]
- Zhou, L.T.; Qiu, S.; Lv, L.L.; Li, Z.L.; Liu, H.; Tang, R.N.; Ma, K.L.; Liu, B.C. Integrative Bioinformatics Analysis Provides Insight into the Molecular Mechanisms of Chronic Kidney Disease. Kidney Blood Press Res. 2018, 43, 568–581. [Google Scholar] [CrossRef]
- Grill, B.; Wilson, G.M.; Zhang, K.X.; Wang, B.; Doyonnas, R.; Quadroni, M.; Schrader, J.W. Activation/division of lymphocytes results in increased levels of cytoplasmic activation/proliferation-associated protein-1: Prototype of a new family of proteins. J. Immunol. 2004, 172, 2389–2400. [Google Scholar] [CrossRef]
- Jaffa, M.A.; Kobeissy, F.; Al Hariri, M.; Chalhoub, H.; Eid, A.; Ziyadeh, F.N.; Jaffa, A.A. Global renal gene expression profiling analysis in B2-kinin receptor null mice: Impact of diabetes. PLoS ONE 2012, 7, e44714. [Google Scholar] [CrossRef]
- Matsuo, S.; López-Guisa, J.M.; Cai, X.; Okamura, D.M.; Alpers, C.E.; Bumgarner, R.E.; Peters, M.A.; Zhang, G.; Eddy, A.A. Multifunctionality of PAI-1 in fibrogenesis: Evidence from obstructive nephropathy in PAI-1-overexpressing mice. Kidney Int. 2005, 67, 2221–2238, Erratum in Kidney Int. 2005, 68, 910. [Google Scholar] [CrossRef]
- Zhi, W.; Sharma, A.; Purohit, S.; Miller, E.; Bode, B.; Anderson, S.W.; Reed, J.C.; Steed, R.D.; Steed, L.; Hopkins, D.; et al. Discovery and validation of serum protein changes in type 1 diabetes patients using high throughput two dimensional liquid chromatography-mass spectrometry and immunoassays. Mol. Cell Proteom. 2011, 10, M111.012203. [Google Scholar] [CrossRef]
- Deb, D.K.; Chen, Y.; Sun, J.; Wang, Y.; Li, Y.C. ATP-citrate lyase is essential for high glucose-induced histone hyperacetylation and fibrogenic gene upregulation in mesangial cells. Am. J. Physiol. Renal. Physiol. 2017, 313, F423–F429. [Google Scholar] [CrossRef]
- Chen, Y.; Deb, D.K.; Fu, X.; Yi, B.; Liang, Y.; Du, J.; He, L.; Li, Y.C. ATP-citrate lyase is an epigenetic regulator to promote obesity-related kidney injury. FASEB J. 2019, 33, 9602–9615. [Google Scholar] [CrossRef]
- Imamura, M.; Moon, J.S.; Chung, K.P.; Nakahira, K.; Muthukumar, T.; Shingarev, R.; Ryter, S.W.; Choi, A.M.; Choi, M.E. RIPK3 promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight. 2018, 3, e94979. [Google Scholar] [CrossRef]
- Gao, C.; Huang, W.; Kanasaki, K.; Xu, Y. The role of ubiquitination and sumoylation in diabetic nephropathy. Biomed. Res. Int. 2014, 2014, 160692. [Google Scholar] [CrossRef]
- Goru, S.K.; Kadakol, A.; Gaikwad, A.B. Hidden targets of ubiquitin proteasome system: To prevent diabetic nephropathy. Pharmacol. Res. 2017, 120, 170–179. [Google Scholar] [CrossRef]
- Huang, W.; Yang, C.; Nan, Q.; Gao, C.; Feng, H.; Gou, F.; Chen, G.; Zhang, Z.; Yan, P.; Peng, J.; et al. The proteasome inhibitor, MG132, attenuates diabetic nephropathy by inhibiting SnoN degradation in vivo and in vitro. Biomed. Res. Int. 2014, 2014, 684765. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Qi, W.; Mu, J.; Wei, Y.; Yang, L.L.; Zhang, Q.; Wu, Q.; Tang, J.Y.; Feng, B. MG132 protects against renal dysfunction by regulating Akt-mediated inflammation in diabetic nephropathy. Sci. Rep. 2019, 9, 2049. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.F.; Qi, W.; Feng, B.; Mu, J.; Zeng, W.; Guo, Y.H.; Pang, Q.; Ye, Z.L.; Liu, L.; Yuan, F.H. Prevention of diabetic nephropathy in rats through enhanced renal antioxidative capacity by inhibition of the proteasome. Life Sci. 2011, 88, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Yadranji Aghdam, S.; Mahmoudpour, A. Proteasome Activators, PA28α and PA28β, Govern Development of Microvascular Injury in Diabetic Nephropathy and Retinopathy. Int. J. Nephrol. 2016, 2016, 3846573. [Google Scholar] [CrossRef]
- Queisser, M.A.; Yao, D.; Geisler, S.; Hammes, H.P.; Lochnit, G.; Schleicher, E.D.; Brownlee, M.; Preissner, K.T. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes 2010, 59, 670–678. [Google Scholar] [CrossRef]
- Tashiro, K.; Tamada, S.; Kuwabara, N.; Komiya, T.; Takekida, K.; Asai, T.; Iwao, H.; Sugimura, K.; Matsumura, Y.; Takaoka, M.; et al. Attenuation of renal fibrosis by proteasome inhibition in rat obstructive nephropathy: Possible role of nuclear factor kappaB. Int. J. Mol. Med. 2003, 12, 587–592. [Google Scholar] [CrossRef]
- Pujols, L.; Fernández-Bertolín, L.; Fuentes-Prado, M.; Alobid, I.; Roca-Ferrer, J.; Agell, N.; Mullol, J.; Picado, C. Proteasome inhibition reduces proliferation, collagen expression, and inflammatory cytokine production in nasal mucosa and polyp fibroblasts. J. Pharmacol. Exp. Ther. 2012, 343, 184–197. [Google Scholar] [CrossRef]
- Neubert, K.; Meister, S.; Moser, K.; Weisel, F.; Maseda, D.; Amann, K.; Wiethe, C.; Winkler, T.H.; Kalden, J.R.; Manz, R.A.; et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat. Med. 2008, 14, 748–755. [Google Scholar] [CrossRef]
- Huang, S.; Liu, F.; Niu, Q.; Li, Y.; Liu, C.; Zhang, L.; Ni, D.; Pu, X. GLIPR-2 overexpression in HK-2 cells promotes cell EMT and migration through ERK1/2 activation. PLoS ONE. 2013, 8, e58574. [Google Scholar] [CrossRef]
- Zhao, D.; Jia, J.; Shao, H. miR-30e targets GLIPR-2 to modulate diabetic nephropathy: In vitro and in vivo experiments. J. Mol. Endocrinol. 2017, 59, 181–190. [Google Scholar] [CrossRef]
- El-Meanawy, A.; Schelling, J.R.; Iyengar, S.K.; Hayden, P.; Barathan, S.; Goddard, K.; Pozuelo, F.; Elashi, E.; Nair, V.; Kretzler, M.; et al. Identification of nephropathy candidate genes by comparing sclerosis-prone and sclerosis-resistant mouse strain kidney transcriptomes. BMC Nephrol. 2012, 13, 61. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, X.; Xin, G. Investigation of mechanisms of mesenchymal stem cells for treatment of diabetic nephropathy via construction of a miRNA-TF-mRNA network. Ren. Fail. 2018, 40, 136–145. [Google Scholar] [CrossRef]
- Gu, Y.; Gong, Y.; Zhang, H.; Dong, X.; Zhao, T.; Burczynski, F.J.; Wang, G.; Sun, S.; Zhu, B.; Han, W.; et al. Regulation of transforming growth factor beta 1 gene expression by dihydropteridine reductase in kidney 293T cells. Biochem. Cell Biol. 2013, 91, 187–193. [Google Scholar] [CrossRef]
- Si, Q.; Sun, S.; Gu, Y. A278C mutation of dihydropteridine reductase decreases autophagy via mTOR signaling. Acta Biochim. Biophys. Sin. 2017, 49, 706–712. [Google Scholar] [CrossRef]
- Sharma, M.; Mehndiratta, M.; Gupta, S.; Kalra, O.P.; Shukla, R.; Gambhir, J.K. Genetic association of NAD(P)H quinone oxidoreductase (NQO1*2) polymorphism with NQO1 levels and risk of diabetic nephropathy. Biol. Chem. 2016, 397, 725–730. [Google Scholar] [CrossRef]
- Finsterer, J.; Scorza, F.A. Renal manifestations of primary mitochondrial disorders. Biomed. Rep. 2017, 6, 487–494. [Google Scholar] [CrossRef]
- Emma, F.; Montini, G.; Parikh, S.M.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 267–280. [Google Scholar] [CrossRef]
- Cox, S.N.; Serino, G.; Sallustio, F.; Blasi, A.; Rossini, M.; Pesce, F.; Schena, F.P. Altered monocyte expression and expansion of non-classical monocyte subset in IgA nephropathy patients. Nephrol. Dial. Transplant. 2015, 30, 1122–1232. [Google Scholar] [CrossRef]
- Mafi, A.; Aghadavod, E.; Mirhosseini, N.; Mobini, M.; Asemi, Z. The effects of expression of different microRNAs on insulin secretion and diabetic nephropathy progression. J. Cell Physiol. 2018, 234, 42–50. [Google Scholar] [CrossRef]
- Higuchi, C.; Nakatsuka, A.; Eguchi, J.; Teshigawara, S.; Kanzaki, M.; Katayama, A.; Yamaguchi, S.; Takahashi, N.; Murakami, K.; Ogawa, D.; et al. Identification of circulating miR-101, miR-375 and miR-802 as biomarkers for type 2 diabetes. Metabolism 2015, 64, 489–497. [Google Scholar] [CrossRef]
- Scott, L.J.; Mohlke, K.L.; Bonnycastle, L.L.; Willer, C.J.; Li, Y.; Duren, W.L.; Erdos, M.R.; Stringham, H.M.; Chines, P.S.; Jackson, A.U.; et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007, 316, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Liljedahl, L.; Pedersen, M.H.; Norlin, J.; McGuire, J.N.; James, P. N-glycosylation proteome enrichment analysis in kidney reveals differences between diabetic mouse models. Clin. Proteomics. 2016, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Wei, X.; Liu, J.; Mi, N. Bioinformatic analysis of specific genes in diabetic nephropathy. Ren. Fail. 2015, 37, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Yesil-Devecioglu, T.; Dayan, A.; Demirtunc, R.; Sardas, S. Role of DNA repair genes XRCC3 and XRCC1 in predisposition to type 2 diabetes mellitus and diabetic nephropathy. Endocrinol. Diabetes Nutr. 2019, 66, 90–98, (In English, Spanish). [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Chen, J.S.; Huang, C.F.; Chen, C.C.; Lu, K.C.; Chu, P.; Sytwu, H.K.; Lin, Y.F. Approaching biomarkers of membranous nephropathy from a murine model to human disease. J. Biomed. Biotechnol. 2011, 2011, 581928. [Google Scholar] [CrossRef]
- Iacobini, C.; Amadio, L.; Oddi, G.; Ricci, C.; Barsotti, P.; Missori, S.; Sorcini, M.; Di Mario, U.; Pricci, F.; Pugliese, G. Role of galectin-3 in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14 (Suppl. S3), S264–S270. [Google Scholar] [CrossRef]
- Li, Y.; Li, T.; Zhou, Z.; Xiao, Y. Emerging roles of Galectin-3 in diabetes and diabetes complications: A snapshot. Rev. Endocr. Metab. Disord. 2022, 23, 569–577. [Google Scholar] [CrossRef]
- Nicholas, S.B.; Aguiniga, E.; Ren, Y.; Kim, J.; Wong, J.; Govindarajan, N.; Noda, M.; Wang, W.; Kawano, Y.; Collins, A.; et al. Plasminogen activator inhibitor-1 deficiency retards diabetic nephropathy. Kidney Int. 2005, 67, 1297–1307. [Google Scholar] [CrossRef]
- Guo, J.; Rackham, O.J.L.; Sandholm, N.; He, B.; Österholm, A.M.; Valo, E.; Harjutsalo, V.; Forsblom, C.; Toppila, I.; Parkkonen, M.; et al. Whole-Genome Sequencing of Finnish Type 1 Diabetic Siblings Discordant for Kidney Disease Reveals DNA Variants associated with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2020, 31, 309–323. [Google Scholar] [CrossRef]
- Jacobson, S.L.; Piper, H.M. Cell cultures of adult cardiomyocytes as models of the myocardium. J. Mol. Cell Cardiol. 1986, 18, 661–678. [Google Scholar] [CrossRef]
- Du, F.; Wang, T.; Li, S.; Meng, X.; Zhang, H.Y.; Li, D.T.; Du, Z.X.; Wang, H.Q. Cathepsin D protects renal tubular cells from damage induced by high glucose independent of its enzymatic activity. Am. J. Transl. Res. 2017, 9, 5528–5537. [Google Scholar]
- Limonte, C.P.; Valo, E.; Drel, V.; Natarajan, L.; Darshi, M.; Forsblom, C.; Henderson, C.M.; Hoofnagle, A.N.; Ju, W.; Kretzler, M.; et al. Urinary Proteomics Identifies Cathepsin D as a Biomarker of Rapid eGFR Decline in Type 1 Diabetes. Diabetes Care 2022, 45, 1416–1427. [Google Scholar] [CrossRef]
- Li, S.Y.; Huang, P.H.; Tarng, D.C.; Lin, T.P.; Yang, W.C.; Chang, Y.H.; Yang, A.H.; Lin, C.C.; Yang, M.H.; Chen, J.W.; et al. Four-and-a-Half LIM Domains Protein 2 Is a Coactivator of Wnt Signaling in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2015, 26, 3072–3084. [Google Scholar] [CrossRef]
- Cai, T.; Sun, D.; Duan, Y.; Qiu, Y.; Dai, C.; Yang, J.; He, W. FHL2 promotes tubular epithelial-to-mesenchymal transition through modulating β-catenin signalling. J. Cell Mol. Med. 2018, 22, 1684–1695. [Google Scholar] [CrossRef]
- Adhikary, L.; Chow, F.; Nikolic-Paterson, D.J.; Stambe, C.; Dowling, J.; Atkins, R.C.; Tesch, G.H. Abnormal p38 mitogen-activated protein kinase signalling in human and experimental diabetic nephropathy. Diabetologia. 2004, 47, 1210–1222. [Google Scholar] [CrossRef]
- Toyoda, M.; Suzuki, D.; Honma, M.; Uehara, G.; Sakai, T.; Umezono, T.; Sakai, H. High expression of PKC-MAPK pathway mRNAs correlates with glomerular lesions in human diabetic nephropathy. Kidney Int. 2004, 66, 1107–1114. [Google Scholar] [CrossRef]
- Price, J.C.; Guan, S.; Burlingame, A.; Prusiner, S.B.; Ghaemmaghami, S. Analysis of proteome dynamics in the mouse brain. Proc. Natl. Acad. Sci. USA 2010, 107, 14508–14513, Erratum in Proc. Natl. Acad. Sci. USA 2014, 111, 3645. [Google Scholar] [CrossRef]
- Fernandez-Pol, J.A.; Hamilton, P.D.; Klos, D.J. Essential viral and cellular zinc and iron containing metalloproteins as targets for novel antiviral and anticancer agents: Implications for prevention and therapy of viral diseases and cancer. Anticancer Res. 2001, 21, 931–957. [Google Scholar]
- Fernandez-Pol, J.A.; Klos, D.J.; Hamilton, P.D. Metallopanstimulin gene product produced in a baculovirus expression system is a nuclear phosphoprotein that binds to DNA. Cell Growth Differ. 1994, 5, 811–825. [Google Scholar]
- Welle, K.A.; Zhang, T.; Hryhorenko, J.R.; Shen, S.; Qu, J.; Ghaemmaghami, S. Time-resolved Analysis of Proteome Dynamics by Tandem Mass Tags and Stable Isotope Labeling in Cell Culture (TMT-SILAC) Hyperplexing. Mol. Cell Proteom. 2016, 15, 3551–3563. [Google Scholar] [CrossRef]
- Resmini, G.; Rizzo, S.; Franchin, C.; Zanin, R.; Penzo, C.; Pegoraro, S.; Ciani, Y.; Piazza, S.; Arrigoni, G.; Sgarra, R.; et al. HMGA1 regulates the Plasminogen activation system in the secretome of breast cancer cells. Sci. Rep. 2017, 7, 11768. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic. Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic. Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011, 6, e21800. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Point | Average Cell Number |
|---|---|
| 1 h | 5.26 × 106 |
| 2 h | 5.68 × 106 |
| 4 h | 5.66 × 106 |
| 7.5 h | 5.56 × 106 |
| 24 h | 5.98 × 106 |
| Gene Name | Protein Description | Average Half-Lives in h (SD) | p-Value | Valid Values | ||
|---|---|---|---|---|---|---|
| Diabetic | Nephropathic | Diabetic | Nephropathic | |||
| RAB13 | RAB13, member RAS oncogene family | 26.4 (1.5) | 32.6 (0.1) | 1.90 × 10−3 | 5 | 2 |
| RAB23 | RAB23, member RAS oncogene family | 33.0 (2.8) | 42.9 (4.3) | 1.10 × 10−2 | 5 | 3 |
| SQRDL | sulfide quinone reductase-like (yeast) | 74.6 (12.7) | 123.6 (16.5) | 1.54 × 10−2 | 4 | 2 |
| PABPC1 | poly(A) binding protein, cytoplasmic 1 | 41.2 (3.9) | 49.1 (4.4) | 1.85 × 10−2 | 5 | 5 |
| IPO7 | importin 7 | 28.7 (1.8) | 34.7 (3.9) | 2.02 × 10−2 | 4 | 5 |
| NPM1 | nucleophosmin (nucleolar phosphoprotein B23, numatrin) | 48.2 (3.4) | 55.6 (4.5) | 2.10 × 10−2 | 5 | 5 |
| TCP1 | t-complex 1 | 58.0 (6.0) | 73.7 (12.1) | 2.71 × 10−2 | 5 | 5 |
| EIF4A1 | eukaryotic translation initiation factor 4A1 | 29.8 (1.8) | 34.7 (4.0) | 3.31 × 10−2 | 5 | 5 |
| CCT8 | chaperonin containing TCP1, subunit 8 (theta) | 57.8 (5.2) | 68.9 (9.3) | 3.34 × 10−2 | 5 | 5 |
| EIF4G1 | eukaryotic translation initiation factor 4 gamma, 1 | 26.8 (1.3) | 32.2 (3.8) | 3.66 × 10−2 | 4 | 4 |
| TFRC | transferrin receptor | 23.3 (2.9) | 29.2 (3.2) | 3.72 × 10−2 | 4 | 5 |
| TOP2B | topoisomerase (DNA) II beta 180kDa | 33.0 (3.3) | 25.9 (0.3) | 3.93 × 10−2 | 3 | 2 |
| ACTB | actin, beta | 65.3 (5.3) | 89.8 (20.9) | 4.12 × 10−2 | 5 | 4 |
| ACLY | ATP citrate lyase | 35.2 (4.7) | 43.5 (4.9) | 4.25 × 10−2 | 5 | 5 |
| ST13P4 | Suppression of tumorigenicity 13 pseudogene 4 | 21.8 (2.5) | 37.6 (7.3) | 4.36 × 10−2 | 3 | 2 |
| GSTM5 | glutathione S-transferase mu 5 | 53.5 (5.9) | 104.9 (18.5) | 4.38 × 10−2 | 2 | 2 |
| HNRNPF | heterogeneous nuclear ribonucleoprotein F | 29.8 (4.0) | 38.1 (6.0) | 4.80 × 10−2 | 5 | 4 |
| PA2G4 | proliferation-associated 2G4, 38kDa | 49.4 (3.7) | 62.2 (12.5) | 4.85 × 10−2 | 5 | 5 |
| CAPRIN1 | cell cycle associated protein 1 | 14.7 (0.9) | 16.2 (1.1) | 4.89 × 10−2 | 5 | 5 |
| EIF4H | eukaryotic translation initiation factor 4H | 28.9 (1.9) | 37.6 (3.6) | 4.90 × 10−2 | 2 | 3 |
| Gene Name | Protein Description | Fold Change (Nephropathic vs. Diabetic) | p-Value | Valid Values | |
|---|---|---|---|---|---|
| Diabetic | Nephropathic | ||||
| GLIPR2 | GLI pathogenesis-related 2 | −1.8 | 3.30 × 10−3 | 4 | 4 |
| RPS3A | ribosomal protein S3A | −1.6 | 3.90 × 10−3 | 5 | 5 |
| TRIM25 | tripartite motif containing 25 | −1.5 | 6.41 × 10−3 | 3 | 4 |
| SLC25A6 | solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 6 | −1.7 | 6.95 × 10−3 | 5 | 5 |
| TUBB4A | tubulin, beta 4A class IVa | 2.1 | 9.41 × 10−3 | 5 | 5 |
| NDUFB10 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 10, 22kDa | −1.7 | 1.21 × 10−2 | 5 | 4 |
| NPC2 | Niemann-Pick disease, type C2 | 2.5 | 1.47 × 10−2 | 2 | 4 |
| SYNE1 | spectrin repeat containing, nuclear envelope 1 | −2.2 | 1.47 × 10−2 | 5 | 5 |
| SEC31A | SEC31 homolog A (S. cerevisiae) | 1.6 | 1.55 × 10−2 | 3 | 4 |
| TUBB4B | tubulin, beta 4B class IVb | 3.3 | 1.70 × 10−2 | 2 | 4 |
| SLC25A3 | solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 3 | −1.7 | 2.40 × 10−2 | 5 | 5 |
| DNAJC8 | DnaJ (Hsp40) homolog, subfamily C, member 8 | −1.8 | 2.40 × 10−2 | 5 | 4 |
| COX4I1 | cytochrome c oxidase subunit IV isoform 1 | −1.7 | 2.42 × 10−2 | 5 | 5 |
| LGALS3 | lectin, galactoside-binding, soluble, 3 | −1.4 | 2.57 × 10−2 | 5 | 5 |
| NQO1 | NAD(P)H dehydrogenase, quinone 1 | −2.1 | 2.64 × 10−2 | 5 | 5 |
| SERPINB2 | serpin peptidase inhibitor, clade B (ovalbumin), member 2 | 1.6 | 2.76 × 10−2 | 2 | 4 |
| XRCC6 | X-ray repair complementing defective repair in Chinese hamster cells 6 | −1.4 | 2.80 × 10−2 | 5 | 5 |
| SDHB | succinate dehydrogenase complex, subunit B, iron sulfur (Ip) | −1.6 | 2.85 × 10−2 | 4 | 5 |
| FHL2 | four and a half LIM domains 2 | −1.6 | 2.87 × 10−2 | 4 | 4 |
| RBMX | RNA binding motif protein, X-linked | −1.6 | 3.03 × 10−2 | 2 | 3 |
| PSMA7 | proteasome (prosome, macropain) subunit, alpha type, 7 | −2.1 | 3.18 × 10−2 | 3 | 3 |
| XRCC5 | X-ray repair complementing defective repair in Chinese hamster cells 5 (double-strand-break rejoining) | −1.4 | 3.21 × 10−2 | 5 | 5 |
| SLC25A5 | solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 5 | −1.9 | 3.34 × 10−2 | 5 | 5 |
| RNH1 | ribonuclease/angiogenin inhibitor 1 | 1.4 | 3.43 × 10−2 | 5 | 5 |
| TUBA1C | tubulin, alpha 1c | 1.7 | 3.57 × 10−2 | 2 | 5 |
| CAPNS1 | calpain, small subunit 1 | −1.8 | 3.71 × 10−2 | 4 | 5 |
| RRAS | related RAS viral (r-ras) oncogene homolog | −1.7 | 3.82 × 10−2 | 5 | 5 |
| CYB5A | cytochrome b5 type A (microsomal) | −1.8 | 3.90 × 10−2 | 4 | 5 |
| CTSD | cathepsin D | −1.4 | 3.95 × 10−2 | 5 | 5 |
| EEF1B2 | eukaryotic translation elongation factor 1 beta 2 | −1.6 | 4.08 × 10−2 | 5 | 5 |
| MAPK14 | mitogen-activated protein kinase 14 | −1.6 | 4.09 × 10−2 | 2 | 3 |
| PSMB2 | proteasome (prosome, macropain) subunit, beta type, 2 | −1.6 | 4.22 × 10−2 | 2 | 3 |
| MTPN | myotrophin | −1.6 | 4.30 × 10−2 | 5 | 5 |
| MYO1B | myosin IB | 1.6 | 4.35 × 10−2 | 5 | 5 |
| APPL2 | adaptor protein, phosphotyrosine interaction, PH domain and leucine zipper containing 2 | −1.3 | 4.51 × 10−2 | 5 | 5 |
| RPS3 | ribosomal protein S3 | −1.7 | 4.55 × 10−2 | 5 | 5 |
| FKBP7 | FK506 binding protein 7 | −1.7 | 4.59 × 10−2 | 5 | 5 |
| QDPR | quinoid dihydropteridine reductase | −2.8 | 4.72 × 10−2 | 4 | 3 |
| HM13 | histocompatibility (minor) 13 | −2.0 | 4.74 × 10−2 | 2 | 4 |
| IMPA1 | inositol(myo)-1(or 4)-monophosphatase 1 | −2.6 | 4.99 × 10−2 | 3 | 3 |
| T1/2 Range (h) | # of Proteins | Biological Processes | Cellular Components | Molecular Functions | KEGG Pathways |
|---|---|---|---|---|---|
| <20 | 64 | Extracellular matrix organization; collagen metabolism; cell adhesion and motility | Extracellular matrix; vesicle; collagen trimer | Extracellular matrix structural constituent; Receptor binding; Protein binding | ECM-receptor interaction; Focal adhesion |
| 20–30 | 82 | Small GTPase mediated signal transduction; mRNA splicing; localization | Cytoplasm; Spliceosomal complex | RNA binding; GTPase activity | |
| 30–40 | 157 | Intracellular transport; Translation; RNA splicing; localization | Spliceosomal complex; Nuclear part; Cytoplasmic part; Ribonucleoprotein complex; EIF3 and EIF4F | Protein binding; RNA binding; Nucleotide binding; Nucleoside-triphosphatase activity | Spliceosome |
| 40–50 | 193 | Protein transport; Golgi vesicle transport; RNA splicing; Translation | Cytoplasm; Vesicle; Golgi; Cytoskeleton; EIF3; Ribonucleoprotein complex; Plasma membrane part | Protein binding; Nucleotide binding; RNA binding; actin binding; GTPase activity; Translation initiation factor activity | Endocytosis |
| 50–60 | 227 | Vesicle mediated transport; Cell cycle progress; Membrane and cytoskeleton organization; Translation elongation; Protein folding | Cytoplasm; Ribosome; Cytoskeleton; Endoplasmic reticulum; | Protein binding; Nucleotide binding; RNA binding; GTPase activity; Structural constituent of ribosome | Aminoacyl-tRNA biosynthesis; Ribosome |
| 60–70 | 272 | RNA processing; Translation; Protein metabolism; Ribosome biogenesis; Protein folding; Ras protein signal transduction | Cytosol; Mitochondria; Nuclear part; Ribosome; Cytoskeleton; Proteasome complex | Structural constituent of ribosome; actin and cytoskeletal protein binding; RNA binding; Protein binding; Threonine-type endopeptidase activity. | Ribosome; Proteasome |
| 70–80 | 170 | Carbohydrate and protein metabolism; Oxidation-reduction process; Cellular respiration; Protein folding; Translation elongation; Acetyl-CoA metabolism; | Cytoplasm; Mitochondrion; Cytoskeleton; Endoplasmic reticulum part; Proteasome complex; Melanosome | Peroxiredoxin activity; Oxidoreductase activity; endopeptidase activity; Isomerase activity; Cytoskeletal protein binding; Coenzyme binding; | Glycolysis/Gluconeogenesis; Proteasome; Pyruvate metabolism; TCA cycle; |
| 80–90 | 76 | Carbohydrate metabolism; Generation of energy; Carboxylic acid, alcohol, and ketone metabolism; | Cytoplasm; Mitochondrion; | Catalytic activity; Monosaccharide binding; Oxidoreductase activity; Isomerase activity | Glycolysis/Gluconeogenesis |
| >90 | 97 | Generation of energy; Oxidative phosphorylation; Carboxylic acid, and ketone metabolism; Chromatin organization and DNA packaging | Cytoplasm; ATP synthase complex; Mitochondrion; Nucleosome; Nuclear membrane; Protein-DNA complex |
| Relative Abundance (Log2 A.U.) | # of Proteins | Biological Processes | Cellular Component | Molecular Function | KEGG Pathways |
|---|---|---|---|---|---|
| >9.0 | 202 | Translation elongation; Protein folding; DNA packaging; Cytoskeleton organization; Cell redox homeostasis, Glycolytic process | Cytosol; Nucleus; Protein-DNA complex; Cytoskeleton; Vesicle; Large ribosomal subunit | RNA binding; Structural constituent of ribosome; GTP binding; Protein binding; Cytoskeletal and actin binding | Ribosome; Systemic lupus erythematosus; Pathogenic Escherichia coli infection |
| 8.0–9.0 | 224 | Translational elongation; RNA splicing; Cytoskeleton organization; Protein transport; Small GTPase mediated signal transduction; Generation of precursor metabolites and energy | Cytoplasm; Actin cytoskeleton; Small ribosomal subunit; Spliceosomal complex; Vesicle | Protein binding; GTP binding; RNA binding; Cytoskeletal protein and actin binding; Structural constituent of ribosome; Hydrogen ion transmembrane transporter activity | Ribosome; Spliceosome; Parkinson’s disease |
| 7.5–8.0 | 180 | Intracellular transport; Vesicle mediated transport; Localization; Vesicle and membrane organization; Small GTPase mediated signal transduction | Cytoplasm; Vesicle; Endoplasmic reticulum; Cytoskeleton; Arp2/3 protein complex; Proteasome core complex; Ribonucleoprotein complex | Protein binding; Actin binding; RNA binding; GTP binding; GTPase activity; Threonine-type peptidase activity | Proteasome; Pathogenic Escherichia coli infection |
| 7.0–7.5 | 220 | Intracellular transport; RNA processing and splicing; Translation; Membrane organization; Protein folding; Energy derivation by oxidation of organic compounds | Cytosol; Endoplasmic reticulum; Vesicles; Endomembrane system; Spliceosomal complex; EIF3 complex | Protein binding; RNA binding; Translation initiation factory activity; | |
| 6.5–7.0 | 283 | Nucleotide Metabolism; Heterocycle metabolism; Protein metabolism; Protein folding; RNA splicing; Redox processes; Cellular respiration; Response to oxidative stress | Cytoplasm; Intracellular organelle part; Macromolecular complex | RNA binding; Nucleotide binding; NADH dehydrogenase activity; Oxidoreductase activity; Protein binding; Cofactor binding | Aminoacyl-tRNA biosynthesis; Proteasome; Oxidative phosphorylation; Huntington’s disease; Amino sugar and nucleotide sugar metabolism; Alzheimer’s disease |
| 6.0–6.5 | 261 | Protein transport; Protein metabolism; Carboxylic acid metabolism; Amine metabolism; Mitotic cell cycle; regulation of ligase activity | Cytoplasm; Mitochondrion; Proteasome complex; Ribonucleoprotein complex; Organelle membrane | Protein binding; RNA binding; Nucleotide binding; Cytoskeletal proteins and actin binding; Translation initiation factor activity | Proteasome |
| 5.0–6.0 | 298 | Acetyl-CoA metabolism; Heterocycle metabolism; Nucleotide biosynthesis; carboxylic acid metabolism; Cellular respiration; Protein transport | Cytoplasm; Mitochondrion; Golgi apparatus part; Envelope | Catalytic activity; Hydrolase activity; Protein binding; Purine nucleotide binding; Pyrophosphatase activity | TCA cycle |
| <5.0 | 135 | mRNA transport; Protein transport; Cellular localization; Oxidation-reduction process | Cytoplasm; Nuclear part; Envelope | Purine nucleotide binding; Catalytic activity; Electron carrier activity | Valine, leucine, and isoleucine degradation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Camillo, B.; Puricelli, L.; Iori, E.; Toffolo, G.M.; Tessari, P.; Arrigoni, G. Modeling SILAC Data to Assess Protein Turnover in a Cellular Model of Diabetic Nephropathy. Int. J. Mol. Sci. 2023, 24, 2811. https://doi.org/10.3390/ijms24032811
Di Camillo B, Puricelli L, Iori E, Toffolo GM, Tessari P, Arrigoni G. Modeling SILAC Data to Assess Protein Turnover in a Cellular Model of Diabetic Nephropathy. International Journal of Molecular Sciences. 2023; 24(3):2811. https://doi.org/10.3390/ijms24032811
Chicago/Turabian StyleDi Camillo, Barbara, Lucia Puricelli, Elisabetta Iori, Gianna Maria Toffolo, Paolo Tessari, and Giorgio Arrigoni. 2023. "Modeling SILAC Data to Assess Protein Turnover in a Cellular Model of Diabetic Nephropathy" International Journal of Molecular Sciences 24, no. 3: 2811. https://doi.org/10.3390/ijms24032811
APA StyleDi Camillo, B., Puricelli, L., Iori, E., Toffolo, G. M., Tessari, P., & Arrigoni, G. (2023). Modeling SILAC Data to Assess Protein Turnover in a Cellular Model of Diabetic Nephropathy. International Journal of Molecular Sciences, 24(3), 2811. https://doi.org/10.3390/ijms24032811