
Therapeutic Use and Molecular Aspects of Ivabradine in Cardiac Remodeling: A Review


Abstract
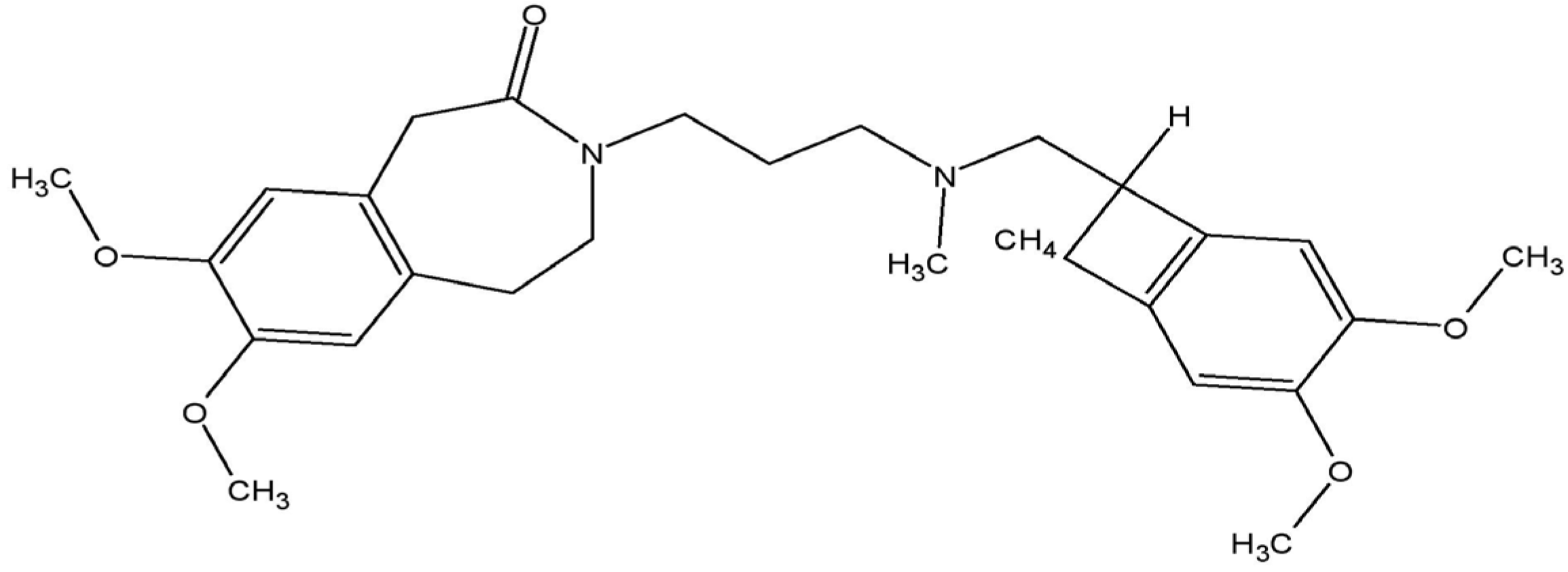
1. Introduction
2. Clinical Outcomes of Ivabradine Therapy
3. Effects on Cardiac Function
4. Effects on the Cardiac Electrical Activity and Neurohumoral Systems
5. Effects on Myocardial Fibrosis
6. Effects on Biogenesis, Autophagy, and Apoptosis
7. Effects on Inflammation and Oxidative Stress
8. Effects on Cardiac Structure
9. Conclusions and Aspects for Future Studies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Afzal, M. Recent updates on novel therapeutic targets of cardiovascular diseases. Mol. Cell. Biochem. 2021, 476, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Reveals Leading Causes of Death and Disability Worldwide: 2000–2019. 2020. Available online: https://www.who.int/news/item/09-12-2020-who-reveals-leading-causes-of-death-and-disability-worldwide-2000-2019 (accessed on 8 October 2022).
- Van De Bruaene, A.; Meier, L.; Droogne, W.; De Meester, P.; Troost, E.; Gewillig, M.; Budts, W. Management of acute heart failure in adult patients with congenital heart disease. Hear. Fail. Rev. 2018, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Rowin, E.J.; Udelson, J.E.; Maron, M.S. Clinical Spectrum and Management of Heart Failure in Hypertrophic Cardiomyopathy. JACC: Hear. Fail. 2018, 6, 353–363. [Google Scholar] [CrossRef]
- Canet, E.; Lerebours, G.; Vilaine, J.-P. Innovation in coronary artery disease and heart failure: Clinical benefits of pure heart rate reduction with ivabradine. Ann. N. Y. Acad. Sci. 2011, 1222, 90–99. [Google Scholar] [CrossRef]
- Thorup, L.; Simonsen, U.; Grimm, D.; Hedegaard, E.R. Ivabradine: Current and Future Treatment of Heart Failure. Basic Clin. Pharmacol. Toxicol. 2017, 121, 89–97. [Google Scholar] [CrossRef]
- Sciatti, E.; Vizzardi, E.; Bonadei, I.; Dallapellegrina, L.; Carubelli, V. The role of heart rate and ivabradine in acute heart failure. Monaldi Arch. Chest Dis. 2019, 89, 1091. [Google Scholar] [CrossRef]
- Chen, C.; Kaur, G.; Mehta, P.K.; Morrone, D.; Godoy, L.C.; Bangalore, S.; Sidhu, M.S. Ivabradine in Cardiovascular Disease Management Revisited: A Review. Cardiovasc. Drugs Ther. 2021, 35, 1045–1056. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Kakehi, K.; Iwanaga, Y.; Watanabe, H.; Sonobe, T.; Akiyama, T.; Shimizu, S.; Yamamoto, H.; Miyazaki, S. Modulation of Sympathetic Activity and Innervation with Chronic Ivabradine and β-Blocker Therapies: Analysis of Hypertensive Rats with Heart Failure. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 387–396. [Google Scholar] [CrossRef]
- Ma, D.; Xu, T.; Cai, G.; Wu, X.; Lei, Z.; Liu, X.; Li, J.; Yang, N. Effects of ivabradine hydrochloride combined with trimetazidine on myocardial fibrosis in rats with chronic heart failure. Exp. Ther. Med. 2019, 18, 1639–1644. [Google Scholar] [CrossRef]
- Zuo, G.; Ren, X.; Qian, X.; Ye, P.; Luo, J.; Gao, X.; Zhang, J.; Chen, S. Inhibition of JNK and p38 MAPK-mediated inflammation and apoptosis by ivabradine improves cardiac function in streptozotocin-induced diabetic cardiomyopathy. J. Cell. Physiol. 2018, 234, 1925–1936. [Google Scholar] [CrossRef]
- Ceconi, C.; Comini, L.; Suffredini, S.; Stillitano, F.; Bouly, M.; Cerbai, E.; Mugelli, A.; Ferrari, R. Heart rate reduction with ivabradine prevents the global phenotype of left ventricular remodeling. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H366–H373. [Google Scholar] [CrossRef]
- Yu, Y.; Hu, Z.; Li, B.; Wang, Z.; Chen, S. Ivabradine improved left ventricular function and pressure overload-induced cardiomyocyte apoptosis in a transverse aortic constriction mouse model. Mol. Cell. Biochem. 2018, 450, 25–34. [Google Scholar] [CrossRef]
- Al-Balushi, S.; Alam, M.F.; Abid, A.R.; Sharfi, A. The effect of ivabradine on hospitalization of heart failure patients: A retrospective cohort study. Hear. Views 2021, 22, 165–173. [Google Scholar] [CrossRef]
- Böhm, M.; Robertson, M.; Ford, I.; Borer, J.S.; Komajda, M.; Kindermann, I.; Maack, C.; Lainscak, M.; Swedberg, K.; Tavazzi, L. Influence of Cardiovascular and Noncardiovascular Co-morbidities on Outcomes and Treatment Effect of Heart Rate Reduction with Ivabradine in Stable Heart Failure (from the SHIFT Trial). Am. J. Cardiol. 2015, 116, 1890–1897. [Google Scholar] [CrossRef]
- Liu, Y.X.; Chen, W.; Lin, X.; Zhu, Y.L.; Lai, J.Z.; Li, J.Y.; Guo, X.X.; Yang, J.; Qian, H.; Zhu, Y.Y.; et al. Initiating ivabradine during hospitalization in patients with acute heart failure: A real-world experience in China. Clin. Cardiol. 2022, 45, 928–935. [Google Scholar] [CrossRef]
- Bouabdallaoui, N.; O’Meara, E.; Bernier, V.; Komajda, M.; Swedberg, K.; Tavazzi, L.; Borer, J.S.; Bohm, M.; Ford, I.; Tardif, J.C. Beneficial effects of ivabradine in patients with heart failure, low ejection fraction, and heart rate above 77 b.p.m. ESC Hear. Fail. 2019, 6, 1199–1207. [Google Scholar] [CrossRef]
- Zugck, C.; Störk, S.; Stöckl, G.; RELIf-CHF Study Investigators. Long-term treatment with ivabradine over 12 months in patients with chronic heart failure in clinical practice: Effect on symptoms, quality of life and hospitalizations. Int. J. Cardiol. 2017, 240, 258–264. [Google Scholar] [CrossRef]
- Böhm, M.; Borer, J.; Ford, I.; Juanatey, J.R.G.; Komajda, M.; Lopez-Sendon, J.; Reil, J.-C.; Swedberg, K.; Tavazzi, L. Heart rate at baseline influences the effect of ivabradine on cardiovascular outcomes in chronic heart failure: Analysis from the SHIFT study. Clin. Res. Cardiol. 2013, 102, 11–22. [Google Scholar] [CrossRef]
- Komajda, M.; Tavazzi, L.; Swedberg, K.; Böhm, M.; Borer, J.S.; Moyne, A.; Ford, I.; SHIFT Investigators. Chronic Exposure to Ivabradine Reduces Readmissions in The Vulnerable Phase After Hospitalization for Worsening Systolic Heart Failure: A Post-Hoc Analysis of SHIFT. Eur. J. Heart Fail. 2016, 18, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, L.; Zhao, J.; Guo, Y.; Liu, J.; Shi, D.; Yang, J.; Liu, Y.; Lai, J.; Shen, Z. Early short-term ivabradine treatment in new-onset acute systolic heart failure and sinus tachycardia patients with inflammatory rheumatic disease. Exp. Ther. Med. 2019, 18, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Borer, J.S.; Swedberg, K.; Komajda, M.; Ford, I.; Tavazzi, L.; Böhm, M.; Depre, C.; Wu, Y.; Maya, J.; Dominjon, F. Efficacy Profile of Ivabradine in Patients with Heart Failure plus Angina Pectoris. Cardiology 2017, 136, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Komajda, M.; Böhm, M.; Borer, J.; Ford, I.; Krum, H.; Tase, A.; Tavazzi, L.; Swedberg, K. Influence of background treatment with mineralocorticoid receptor antagonists on ivabradine’s effects in patients with chronic heart failure. Eur. J. Hear. Fail. 2013, 15, 79–84. [Google Scholar] [CrossRef]
- Komajda, M.; Tavazzi, L.; Francq, B.G.; Böhm, M.; Borer, J.S.; Ford, I.; Swedberg, K.; SHIFT Investigators. Efficacy and safety of ivabradine in patients with chronic systolic heart failure and diabetes: An analysis from the SHIFT trial. Eur. J. Hear. Fail. 2015, 17, 1294–1301. [Google Scholar] [CrossRef]
- Komajda, M.; Isnard, R.; Cohen-Solal, A.; Metra, M.; Pieske, B.; Ponikowski, P.; Voors, A.A.; Dominjon, F.; Henon-Goburdhun, C.; Pannaux, M.; et al. Effect of ivabradine in patients with heart failure with preserved ejection fraction: The EDIFY randomized placebo-controlled trial. Eur. J. Hear. Fail. 2017, 19, 1495–1503. [Google Scholar] [CrossRef]
- Liao, C.T.; Huang, J.L.; Liang, H.W.; Chung, F.P.; Lee, Y.H.; Lin, P.L.; Chiou, W.R.; Lin, W.Y.; Hsu, C.Y.; Chang, H.Y. The association between ivabradine and adverse cardiovascular events in acute decompensated HFrEF patients. ESC Hear. Fail. 2021, 8, 4199–4210. [Google Scholar] [CrossRef]
- Ordu, S.; Yildiz, B.S.; Alihanoglu, Y.I.; Ozsoy, A.; Tosun, M.; Evrengul, H.; Kaftan, H.A.; Ozhan, H. Effects of ivabradine therapy on heart failure biomarkers. Cardiol. J. 2015, 22, 501–509. [Google Scholar] [CrossRef]
- Reil, J.-C.; Robertson, M.; Ford, I.; Borer, J.; Komajda, M.; Swedberg, K.; Tavazzi, L.; Böhm, M. Impact of left bundle branch block on heart rate and its relationship to treatment with ivabradine in chronic heart failure. Eur. J. Hear. Fail. 2013, 15, 1044–1052. [Google Scholar] [CrossRef]
- Riccioni, G.; Masciocco, L.; Benvenuto, A.; Saracino, P.; De Viti, D.; Massari, F.; Meliota, G.; Buta, F.; Speziale, G. Ivabradine Improves Quality of Life in Subjects with Chronic Heart Failure Compared to Treatment with β-Blockers: Results of a Multicentric Observational APULIA Study. Pharmacology 2013, 92, 276–280. [Google Scholar] [CrossRef]
- Rohm, I.; Kretzschmar, D.; Pistulli, R.; Franz, M.; Schulze, P.C.; Stumpf, C.; Yilmaz, A. Impact of Ivabradine on Inflammatory Markers in Chronic Heart Failure. J. Immunol. Res. 2016, 2016, 6949320. [Google Scholar] [CrossRef]
- Sargento, L.; Satendra, M.; Longo, S.; Lousada, N.; dos Reis, R.P. Heart Rate Reduction with Ivabradine in Patients with Acute Decompensated Systolic Heart Failure. Am. J. Cardiovasc. Drugs 2014, 14, 229–235. [Google Scholar] [CrossRef]
- Villacorta, A.S.; Villacorta, H.; Caldas, J.A.; Precht, B.C.; Porto, P.B.; Rodrigues, L.U.; Neves, M.; Xavier, A.R.; Kanaan, S.; Mesquita, C.T.; et al. Effects of Heart Rate Reduction with Either Pyridostigmine or Ivabradine in Patients with Heart Failure: A Randomized, Double-Blind Study. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 139–145. [Google Scholar] [CrossRef]
- Zugck, C.; Martinka, P.; Stöckl, G. Ivabradine Treatment in a Chronic Heart Failure Patient Cohort: Symptom Reduction and Improvement in Quality of Life in Clinical Practice. Adv. Ther. 2014, 31, 961–974. [Google Scholar] [CrossRef]
- Bonnet, D.; Berger, F.; Jokinen, E.; Kantor, P.; Daubeney, P.E. Ivabradine in Children with Dilated Cardiomyopathy and Symptomatic Chronic Heart Failure. J. Am. Coll. Cardiol. 2017, 70, 1262–1272. [Google Scholar] [CrossRef]
- Koruth, J.S.; Lala, A.; Pinney, S.; Reddy, V.Y.; Dukkipati, S.R. The Clinical Use of Ivabradine. J. Am. Coll. Cardiol. 2017, 70, 1777–1784. [Google Scholar] [CrossRef]
- Flannery, G.; Gehrig-Mills, R.; Billah, B.; Krum, H. Analysis of Randomized Controlled Trials on the Effect of Magnitude of Heart Rate Reduction on Clinical Outcomes in Patients with Systolic Chronic Heart Failure Receiving Beta-Blockers. Am. J. Cardiol. 2008, 101, 865–869. [Google Scholar] [CrossRef]
- Lee, W.-C.; Fang, H.-Y. Ivabradine for the Treatment of Acute Mitral-Regurgitation-Related Decompensated Heart Failure. Cardiology 2019, 144, 97–100. [Google Scholar] [CrossRef]
- De Ferrari, G.M.; Mazzuero, A.; Agnesina, L.; Bertoletti, A.; Lettino, M.; Campana, C.; Schwartz, P.J.; Tavazzi, L. Favourable Effects of Heart Rate Reduction with Intravenous Administration of Ivabradine in Patients with Advanced Heart Failure. Eur. J. Heart Fail. 2008, 10, 550–555. [Google Scholar] [CrossRef]
- Bonadei, I.; Sciatti, E.; Vizzardi, E.; Fabbricatore, D.; Pagnoni, M.; Rossi, L.; Carubelli, V.; Lombardi, C.M.; Metra, M. Effects of ivabradine on endothelial function, aortic properties and ventricular-arterial coupling in chronic systolic heart failure patients. Cardiovasc. Ther. 2018, 36, e12323. [Google Scholar] [CrossRef]
- Böhm, M.; Borer, J.S.; Camm, J.; Ford, I.; Lloyd, S.M.; Komajda, M.; Tavazzi, L.; Talajic, M.; Lainscak, M.; Reil, J.-C.; et al. Twenty-four-hour heart rate lowering with ivabradine in chronic heart failure: Insights from the SHIFT Holter substudy. Eur. J. Hear. Fail. 2015, 17, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Mert, K.U.; Dural, M.; Mert, G.Ö.; Iskenderov, K.; Özen, A. Effects of heart rate reduction with ivabradine on the international ındex of erectile function (IIEF-5) in patients with heart failure. Aging Male 2017, 21, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, S.; Öztürk, S.; Erdem, F.H.; Erdem, A.; Ayhan, S.; Dönmez, I.; Yazıcı, M. The effects of ivabradine on left atrial electromechanical function in patients with systolic heart failure. J. Interv. Card. Electrophysiol. 2016, 46, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Reil, J.-C.; Tardif, J.-C.; Ford, I.; Lloyd, S.M.; O’Meara, E.; Komajda, M.; Borer, J.S.; Tavazzi, L.; Swedberg, K.; Böhm, M. Selective Heart Rate Reduction with Ivabradine Unloads the Left Ventricle in Heart Failure Patients. J. Am. Coll. Cardiol. 2013, 62, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; O’Meara, E.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Tavazzi, L.; Swedberg, K.; SHIFT Investi-gators. Effects of selective heart rate reduction with ivabradine on left ventricular remodelling and function: Results from the SHIFT echocardiography substudy. Eur. Hear. J. 2011, 32, 2507–2515. [Google Scholar] [CrossRef]
- Gul, M.; Inci, S.; Aksan, G.; Sigirci, S.; Keskin, P. Using Tissue Doppler and Speckle Tracking Echocardiography to Assess if Ivabradine Improves Right Ventricular Function. Cureus 2021, 13, e12920. [Google Scholar] [CrossRef]
- Fang, F.; Lee, A.P.; Yu, C.-M. Left atrial function in heart failure with impaired and preserved ejection fraction. Curr. Opin. Cardiol. 2014, 29, 430–436. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Li, H.; Zhang, A.; Han, Y.; Wang, J.; Hou, Y. Ivabradine and Atrial Fibrillation: A Meta-Analysis of Randomized Controlled Trials. J. Cardiovasc. Pharmacol. 2022, 79, 549–557. [Google Scholar] [CrossRef]
- Amstetter, D.; Badt, F.; Rubi, L.; Bittner, R.E.; Ebner, J.; Uhrin, P.; Hilber, K.; Koenig, X.; Todt, H. The bradycardic agent ivabradine decreases conduction velocity in the AV node and in the ventricles in-vivo. Eur. J. Pharmacol. 2021, 893, 173818. [Google Scholar] [CrossRef]
- Jozwiak, M.; Melka, J.; Rienzo, M.; Bizé, A.; Sambin, L.; Hittinger, L.; Berdeaux, A.; Su, J.B.; Bouhemad, B.; Ghaleh, B. Ivabradine improves left ventricular twist and untwist during chronic hypertension. Int. J. Cardiol. 2018, 252, 175–180. [Google Scholar] [CrossRef]
- Melka, J.; Rienzo, M.; Bizé, A.; Jozwiak, M.; Sambin, L.; Hittinger, L.; Su, J.B.; Berdeaux, A.; Ghaleh, B. Improvement of left ventricular filling by ivabradine during chronic hypertension: Involvement of contraction-relaxation coupling. Basic Res. Cardiol. 2016, 111, 30. [Google Scholar] [CrossRef]
- Simko, F.; Baka, T.; Stanko, P.; Repova, K.; Krajcirovicova, K.; Aziriova, S.; Domenig, O.; Zorad, S.; Adamcova, M.; Paulis, L. Sacubitril/Valsartan and Ivabradine Attenuate Left Ventricular Remodelling and Dysfunction in Spontaneously Hypertensive Rats: Different Interactions with the Renin–Angiotensin–Aldosterone System. Biomedicines 2022, 10, 1844. [Google Scholar] [CrossRef]
- Simko, F.; Baka, T.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Paulis, L.; Adamcova, M. Ivabradine improves survival and attenuates cardiac remodeling in isoproterenol-induced myocardial injury. Fundam. Clin. Pharmacol. 2021, 35, 744–748. [Google Scholar] [CrossRef]
- Noel, R.; Ali, M.N.A.K. Effect of Ivabradine on Cardiac Remodeling in Experimentally Induced Heart Failure in Rats. Iraqi J. Commun. Med. 2019, 32, 1–7. [Google Scholar]
- Xie, H.; Shen, X.-Y.; Zhao, N.; Ye, P.; Ge, Z.; Hu, Z.-Y. Ivabradine Ameliorates Cardiac Diastolic Dysfunction in Diabetic Mice Independent of Heart Rate Reduction. Front. Pharmacol. 2021, 12, 696635. [Google Scholar] [CrossRef]
- Kim, H.B.; Hong, Y.J.; Park, H.J.; Ahn, Y.; Jeong, M.H. Effects of Ivabradine on Left Ventricular Systolic Function and Cardiac Fibrosis in Rat Myocardial Ischemia-Reperfusion Model. Chonnam Med. J. 2018, 54, 167–172. [Google Scholar] [CrossRef]
- Shao, S.; Zhang, Y.; Gong, M.; Yang, Q.; Yuan, M.; Yuan, M.; Suo, Y.; Wang, X.; Li, Y.; Bao, Q.; et al. Ivabradine Ameliorates Cardiac Function in Heart Failure with Preserved and Reduced Ejection Fraction via Upregulation of miR-133a. Oxidative Med. Cell. Longev. 2021, 2021, 1257283. [Google Scholar] [CrossRef]
- Paterek, A.; Sochanowicz, B.; Oknińska, M.; Śmigielski, W.; Kruszewski, M.; Mackiewicz, U.; Mączewski, M.; Leszek, P. Ivabradine prevents deleterious effects of dopamine therapy in heart failure: No role for HCN4 overexpression. Biomed. Pharmacother. 2021, 136, 111250. [Google Scholar] [CrossRef]
- Pascual Izco, M.; Ramírez-Carracedo, R.; Hernández Navarro, I.; Osorio Ruiz, Á.; Castejón Navarro, B.; Cuadrado Berrocal, I.; Largo Aramburu, C.; Alonso Salinas, G.L.; Díez, J.; Saura Redondo, M.; et al. Ivabradine in Acute Heart Failure: Effects on Heart Rate and Hemodynamic Parameters in a Randomized and Controlled Swine Trial. Cardiol. J. 2020, 27, 62–71. [Google Scholar] [CrossRef]
- Mączewski, M.; Mackiewicz, U. Effect of metoprolol and ivabradine on left ventricular remodelling and Ca2+ handling in the post-infarction rat heart. Cardiovasc. Res. 2008, 79, 42–51. [Google Scholar] [CrossRef]
- Simko, F.; Baka, T.; Poglitsch, M.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Zorad, S.; Adamcova, M.; Paulis, L. Effect of Ivabradine on a Hypertensive Heart and the Renin-Angiotensin-Aldosterone System in L-NAME-Induced Hypertension. Int. J. Mol. Sci. 2018, 19, 3017. [Google Scholar] [CrossRef] [PubMed]
- Milliez, P.; Messaoudi, S.; Nehme, J.; Rodriguez, C.; Samuel, J.-L.; Delcayre, C. Beneficial effects of delayed ivabradine treatment on cardiac anatomical and electrical remodeling in rat severe chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H435–H441. [Google Scholar] [CrossRef] [PubMed]
- Bakkehaug, J.P.; Naesheim, T.; Torgersen Engstad, E.; Kildal, A.B.; Myrmel, T.; How, O.-J. Reversing dobutamine-induced tachycardia using ivabradine increases stroke volume with neutral effect on cardiac energetics in left ventricular post-ischaemia dysfunction. Acta Physiol. 2016, 218, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Sun, Z.; Zhang, B.; Li, X.; Xia, H. The Effects of Ivabradine on Cardiac Function after Myocardial Infarction are Weaker in Diabetic Rats. Cell. Physiol. Biochem. 2016, 39, 2055–2064. [Google Scholar] [CrossRef]
- Christensen, L.P.; Zhang, R.-L.; Zheng, W.; Campanelli, J.J.; Dedkov, E.I.; Weiss, R.M.; Tomanek, R.J. Postmyocardial infarction remodeling and coronary reserve: Effects of ivabradine and beta blockade therapy. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H322–H330. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, Y.; Wei, G.; Zha, L.; Li, X. Ivabradine protects rats against myocardial infarction through reinforcing autophagy via inhibiting PI3K/AKT/mTOR/p70S6K pathway. Bioengineered 2021, 12, 1826–1837. [Google Scholar] [CrossRef]
- El-Naggar, A.E.; El-Gowilly, S.M.; Sharabi, F.M. Possible Ameliorative Effect of Ivabradine on the Autonomic and Left Ventricular Dysfunction Induced by Doxorubicin in Male Rats. J. Cardiovasc. Pharmacol. 2018, 72, 22–31. [Google Scholar] [CrossRef]
- Gómez, O.; Okumura, K.; Honjo, O.; Sun, M.; Ishii, R.; Bijnens, B.; Friedberg, M.K. Heart Rate Reduction Improves Biventricular Function and Interactions in Experimental Pulmonary Hypertension. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H542–H551. [Google Scholar] [CrossRef]
- Gomes, F.A.; Noronha, S.I.; Silva, S.C.; Machado-Júnior, P.A.; Ostolin, T.L.; Chírico, M.T.; Ribeiro, M.C.; Reis, A.B.; Cangussú, S.D.; Montano, N.; et al. Ivabradine treatment lowers blood pressure and promotes cardiac and renal protection in spontaneously hypertensive rats. Life Sci. 2022, 308, 120919. [Google Scholar] [CrossRef]
- Hernandez, I.; Tesoro, L.; Ramirez-Carracedo, R.; Diez-Mata, J.; Sanchez, S.; Saura, M.; Zamorano, J.; Zaragoza, C.; Botana, L. Ivabradine Induces Cardiac Protection against Myocardial Infarction by Preventing Cyclophilin-A Secretion in Pigs under Coronary Ischemia/Reperfusion. Int. J. Mol. Sci. 2021, 22, 2902. [Google Scholar] [CrossRef]
- Ishii, R.; Okumura, K.; Akazawa, Y.; Malhi, M.; Ebata, R.; Sun, M.; Fujioka, T.; Kato, H.; Honjo, O.; Kabir, G.; et al. Heart Rate Reduction Improves Right Ventricular Function and Fibrosis in Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 63, 843–855. [Google Scholar] [CrossRef]
- Kim, B.H.; Cho, K.I.; Kim, S.M.; Kim, N.; Han, J.; Kim, J.Y.; Kim, I.J. Heart rate reduction with ivabradine prevents thyroid hormone-induced cardiac remodeling in rat. Hear. Vessel. 2012, 28, 524–535. [Google Scholar] [CrossRef]
- Tesoro, L.; Ramirez-Carracedo, R.; Hernandez, I.; Diez-Mata, J.; Pascual, M.; Saura, M.; Sanmartin, M.; Zamorano, J.L.; Zaragoza, C. Ivabradine induces cardiac protection by preventing cardiogenic shock-induced extracellular matrix degradation. Rev. Esp. Cardiol. 2021, 74, 1062–1071. [Google Scholar] [CrossRef]
- Sadeghpour, A.; Alizadehasl, A. Echocardiography. In Practical Cardiology; Maleki, M., Alizadehasl, A., Haghjoo, M., Eds.; Elsevier: St Louis, MO, USA, 2018; pp. 67–111. [Google Scholar] [CrossRef]
- Kranias, E.G.; Hajjar, R.J. Modulation of Cardiac Contractility by the Phopholamban/SERCA2a Regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef]
- Ramli, F.F.; Hashim, S.A.S.; Raman, B.; Mahmod, M.; Kamisah, Y. Role of Trientine in Hypertrophic Cardiomyopathy: A Review of Mechanistic Aspects. Pharmaceuticals 2022, 15, 1145. [Google Scholar] [CrossRef]
- Frank, K.; Kranias, E.G. Phospholamban and cardiac contractility. Ann. Med. 2000, 32, 572–578. [Google Scholar] [CrossRef]
- Salim, S.; Yunos, N.; Jauri, M.; Kamisah, Y. Cardiotonic Effects of Cardiac Glycosides from Plants of Apocynaceae Family. Chula. Med. J. 2020, 64, 449–456. [Google Scholar] [CrossRef]
- Lai, L.; Qiu, H. The Physiological and Pathological Roles of Mitochondrial Calcium Uptake in Heart. Int. J. Mol. Sci. 2020, 21, 7689. [Google Scholar] [CrossRef]
- Zhang, T. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc. Res. 2004, 63, 476–486. [Google Scholar] [CrossRef]
- Mengesha, H.G.; Tafesse, T.B.; Bule, M.H. If Channel as an Emerging Therapeutic Target for Cardiovascular Diseases: A Review of Current Evidence and Controversies. Front. Pharmacol. 2017, 8, 874. [Google Scholar] [CrossRef]
- Kennedy, A.; Finlay, D.D.; Guldenring, D.; Bond, R.; Moran, K.; McLaughlin, J. The Cardiac Conduction System: Generation and Conduction of the Cardiac Impulse. Crit. Care Nurs. Clin. North Am. 2016, 28, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Rivolta, I.; Binda, A.; Masi, A.; DiFrancesco, J.C. Cardiac and neuronal HCN channelopathies. Pflugers Arch. 2020, 472, 931–951. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D.; Borer, J.S. The Funny Current: Cellular Basis for The Control of Heart Rate. Drugs 2007, 67, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Sartiani, L.; Mannaioni, G.; Masi, A.; Novella Romanelli, M.; Cerbai, E. The Hyperpolarization-Activated Cyclic Nucleotide–Gated Channels: From Biophysics to Pharmacology of a Unique Family of Ion Channels. Pharmacol. Rev. 2017, 69, 354–395. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Zhang, K.; Rastogi, S. Heart rate reduction with ivabradine improves left ventricular function and reverses multiple pathological maladaptations in dogs with chronic heart failure. ESC Hear. Fail. 2014, 1, 94–102. [Google Scholar] [CrossRef]
- DiFrancesco, M.L.; Mesirca, P.; Bidaud, I.; Isbrandt, D.; Mangoni, M.E. The funny current in genetically modified mice. Prog. Biophys. Mol. Biol. 2021, 166, 39–50. [Google Scholar] [CrossRef]
- Verrier, R.L.; Bonatti, R.; Silva, A.F.; Batatinha, J.A.; Nearing, B.D.; Liu, G.; Rajamani, S.; Zeng, D.; Belardinelli, L. If inhibition in the atrioventricular node by ivabradine causes rate-dependent slowing of conduction and reduces ventricular rate during atrial fibrillation. Hear. Rhythm. 2014, 11, 2288–2296. [Google Scholar] [CrossRef]
- Fontenla, A.; López-Gil, M.; Tamargo-Menéndez, J.; Matía-Francés, R.; Salgado-Aranda, R.; Rey-Blas, J.R.; Miracle-Blanco, Á.; Mejía-Martínez, E.; Pastor-Fuentes, A.; Toquero-Ramos, J.; et al. Ivabradine for Chronic Heart Rate Control in Persistent Atrial Fibrillation. Design of the BRAKE-AF Project. Rev. Esp. Cardiol. 2020, 73, 368–375. [Google Scholar] [CrossRef]
- Tomasoni, D.; Adamo, M.; Lombardi, C.M.; Metra, M. Highlights in heart failure. ESC Hear. Fail. 2019, 6, 1105–1127. [Google Scholar] [CrossRef]
- Dickson, P.W.; Briggs, G.D. Tyrosine Hydroxylase: Regulation by Feedback Inhibition and Phosphorylation. Adv. Pharmacol. 2013, 68, 13–21. [Google Scholar] [CrossRef]
- Dunkley, P.R.; Dickson, P.W. Tyrosine hydroxylase phosphorylation in vivo. J. Neurochem. 2019, 149, 706–728. [Google Scholar] [CrossRef]
- Alam, M.J.; Gupta, R.; Mahapatra, N.R.; Goswami, S.K. Catestatin reverses the hypertrophic effects of norepinephrine in H9c2 cardiac myoblasts by modulating the adrenergic signaling. Mol. Cell. Biochem. 2019, 464, 205–219. [Google Scholar] [CrossRef]
- Boengler, K.; Rohrbach, S.; Weissmann, N.; Schulz, R. Importance of Cx43 for Right Ventricular Function. Int. J. Mol. Sci. 2021, 22, 987. [Google Scholar] [CrossRef]
- Garcia-Garduño, T.C.; Padilla-Gutierrez, J.R.; Cambrón-Mora, D.; Valle, Y. RAAS: A Convergent Player in Ischemic Heart Failure and Cancer. Int. J. Mol. Sci. 2021, 22, 7106. [Google Scholar] [CrossRef]
- Kamisah, Y.; Periyah, V.; Lee, K.T.; Noor-Izwan, N.; Nurul-Hamizah, A.; Nurul-Iman, B.S.; Subermaniam, K.; Jaarin, K.; Azman, A.; Faizah, O.; et al. Cardioprotective effect of virgin coconut oil in heated palm oil diet-induced hypertensive rats. Pharm. Biol. 2015, 53, 1243–1249. [Google Scholar] [CrossRef]
- Kamisah, Y.; Zuhair, J.S.F.; Juliana, A.H.; Jaarin, K. Parkia speciosa empty pod prevents hypertension and cardiac damage in rats given N(G)-nitro-l-arginine methyl ester. Biomed. Pharmacother. 2017, 96, 291–298. [Google Scholar] [CrossRef]
- Lehmann, L.H.; Rostosky, J.S.; Buss, S.J.; Kreusser, M.M.; Krebs, J.; Mier, W.; Enseleit, F.; Spiger, K.; Hardt, S.E.; Wieland, T.; et al. Essential role of sympathetic endothelin A receptors for adverse cardiac remodeling. Proc. Natl. Acad. Sci. USA 2014, 111, 13499–13504. [Google Scholar] [CrossRef]
- Miyauchi, T.; Sakai, S. Endothelin and the heart in health and diseases. Peptides 2019, 111, 77–88. [Google Scholar] [CrossRef]
- Siti, H.N.; Jalil, J.; Asmadi, A.Y.; Kamisah, Y. Rutin Modulates MAPK Pathway Differently from Quercetin in Angiotensin II-Induced H9c2 Cardiomyocyte Hypertrophy. Int. J. Mol. Sci. 2021, 22, 5063. [Google Scholar] [CrossRef]
- Wang, H.X.; Zhang, Q.F.; Zeng, X.J.; Wang, W.; Tang, C.S.; Zhang, L.K. Effects of Angiotensin III on Protein, DNA, and Collagen Synthesis of Neonatal Cardiomyocytes and Cardiac Fibroblasts In Vitro. J. Cardiovasc. Pharmacol. Ther. 2010, 15, 393–402. [Google Scholar] [CrossRef]
- Park, B.M.; Cha, S.A.; Lee, S.H.; Kim, S.H. Angiotensin IV protects cardiac reperfusion injury by inhibiting apoptosis and inflammation via AT4R in rats. Peptides 2016, 79, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Park, B.M.; Li, W.; Kim, S.H. Cardio-protective effects of angiotensin-(1–5) via mas receptor in rats against ischemic-perfusion injury. Peptides 2021, 139, 170516. [Google Scholar] [CrossRef] [PubMed]
- Lelis, D.F.; Freitas, D.F.; Machado, A.S.; Crespo, T.S.; Santos, S.H.S. Angiotensin-(1–7), Adipokines and Inflammation. Metabolism 2019, 95, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, C.; Hong, X.; Miao, J.; Liao, Y.; Hou, F.F.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling mediates both heart and kidney injury in type 2 cardiorenal syndrome. Kidney Int. 2019, 95, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Zuo, G.-F.; Ren, X.-M.; Ge, Q.; Luo, J.; Ye, P.; Wang, F.; Wu, W.; Chao, Y.-L.; Gu, Y.; Gao, X.-F.; et al. Activation of the PP2A catalytic subunit by ivabradine attenuates the development of diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 130, 170–183. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Patel, K.D. Matrix metalloproteinase-2 (mmp-2) and mmp-9 in pulmonary pathology. Exp. Lung Res. 2005, 31, 599–621. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68-69, 490–506. [Google Scholar] [CrossRef]
- Nikolov, A.; Popovski, N. Extracellular Matrix in Heart Disease: Focus on Circulating Collagen Type I and III Derived Peptides as Biomarkers of Myocardial Fibrosis and Their Potential in the Prognosis of Heart Failure: A Concise Review. Metabolites 2022, 12, 297. [Google Scholar] [CrossRef]
- Frangogiannis, N. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Zeng, Z.; Wang, Q.; Yang, X.; Ren, Y.; Jiao, S.; Zhu, Q.; Guo, D.; Xia, K.; Wang, Y.; Li, C.; et al. Qishen granule attenuates cardiac fibrosis by regulating TGF-β/Smad3 and GSK-3β pathway. Phytomedicine 2019, 62, 152949. [Google Scholar] [CrossRef]
- Kumar, S.; Wang, G.; Zheng, N.; Cheng, W.; Ouyang, K.; Lin, H.; Liao, Y.; Liu, J. HIMF (Hypoxia-Induced Mitogenic Factor)-IL (Interleukin)-6 Signaling Mediates Cardiomyocyte-Fibroblast Crosstalk to Promote Cardiac Hypertrophy and Fibrosis. Hypertension 2019, 73, 1058–1070. [Google Scholar] [CrossRef]
- Vallée, A.; LeCarpentier, Y. TGF-β in fibrosis by acting as a conductor for contractile properties of myofibroblasts. Cell Biosci. 2019, 9, 98. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Invest. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Balestrino, M. Role of Creatine in the Heart: Health and Disease. Nutrients 2021, 13, 1215. [Google Scholar] [CrossRef]
- Yeh, J.-N.; Yue, Y.; Chu, Y.-C.; Huang, C.-R.; Yang, C.-C.; Chiang, J.Y.; Yip, H.-K.; Guo, J. Entresto protected the cardiomyocytes and preserved heart function in cardiorenal syndrome rat fed with high-protein diet through regulating the oxidative stress and Mfn2-mediated mitochondrial functional integrity. Biomed. Pharmacother. 2021, 144, 112244. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Sun, Y.; Yao, X.; Zhang, Q.-J.; Zhu, M.; Liu, Z.-P.; Ci, B.; Xie, Y.; Carlson, D.; Rothermel, B.A.; Sun, Y.; et al. Beclin-1-Dependent Autophagy Protects the Heart During Sepsis. Circulation 2018, 138, 2247–2262. [Google Scholar] [CrossRef]
- Rusten, T.E.; Stenmark, H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–279. [Google Scholar] [CrossRef]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef]
- Ye, J.; Wang, Y.; Wang, Z.; Liu, L.; Yang, Z.; Ye, D.; Wang, M.; Xu, Y.; Zhang, J.; Zhao, M.; et al. Interleukin-12p35 deficiency enhances mitochondrial dysfunction and aggravates cardiac remodeling in aging mice. Aging 2020, 12, 193–203. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Vahl, T.P.; Goliasch, G.; Picatoste, B.; Arias, T.; Ishikawa, K.; Njerve, I.U.; Sanz, J.; Narula, J.; Sengupta, P.P.; et al. Sphingosine-1-Phosphate Receptor Agonist Fingolimod Increases Myocardial Salvage and Decreases Adverse Postinfarction Left Ventricular Remodeling in a Porcine Model of Ischemia/Reperfusion. Circulation 2016, 133, 954–966. [Google Scholar] [CrossRef] [PubMed]
- da Silva, D.M.; Langer, H.; Graf, T. Inflammatory and Molecular Pathways in Heart Failure—Ischemia, HFpEF and Transthyretin Cardiac Amyloidosis. Int. J. Mol. Sci. 2019, 20, 2322. [Google Scholar] [CrossRef] [PubMed]
- Jirak, P.; Fejzic, D.; Paar, V.; Wernly, B.; Pistulli, R.; Rohm, I.; Jung, C.; Hoppe, U.C.; Schulze, P.C.; Lichtenauer, M.; et al. Influences of Ivabradine treatment on serum levels of cardiac biomarkers sST2, GDF-15, suPAR and H-FABP in patients with chronic heart failure. Acta Pharmacol. Sin. 2018, 39, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.-H.; Chen, J.-W.; Jen, H.-L.; Chiang, M.-C.; Huang, W.-P.; Feng, A.-N.; Young, M.S.; Lin, S.-J. Independent prognostic value of elevated high-sensitivity C-reactive protein in chronic heart failure. Am. Hear. J. 2004, 147, 931–938. [Google Scholar] [CrossRef]
- Guindolet, D.; Gabison, E.E. Role of CD147 (EMMPRIN/Basigin) in Tissue Remodeling. Anat. Rec. 2020, 303, 1584–1589. [Google Scholar] [CrossRef]
- Puah, B.-P.; Jalil, J.; Attiq, A.; Kamisah, Y. New Insights into Molecular Mechanism behind Anti-Cancer Activities of Lycopene. Molecules 2021, 26, 3888. [Google Scholar] [CrossRef]
- Siti, H.N.; Jalil, J.; Asmadi, A.Y.; Kamisah, Y. Parkia speciosa Hassk. Empty Pod Extract Alleviates Angiotensin II-Induced Cardiomyocyte Hypertrophy in H9c2 Cells by Modulating the Ang II/ROS/NO Axis and MAPK Pathway. Front. Pharmacol. 2021, 12, 741623. [Google Scholar] [CrossRef]
- Raut, G.K.; Manchineela, S.; Chakrabarti, M.; Bhukya, C.K.; Naini, R.; Venkateshwari, A.; Reddy, V.D.; Mendonza, J.J.; Suresh, Y.; Nallari, P.; et al. Imine stilbene analog ameliorate isoproterenol-induced cardiac hypertrophy and hydrogen peroxide-induced apoptosis. Free. Radic. Biol. Med. 2020, 153, 80–88. [Google Scholar] [CrossRef]
- Gui, J.S.; Jalil, J.; Jubri, Z.; Kamisah, Y. Parkia speciosa empty pod extract exerts anti-inflammatory properties by modulating NFκB and MAPK pathways in cardiomyocytes exposed to tumor necrosis factor-α. Cytotechnology 2019, 71, 79–89. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF-kappa B Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Ren, B.; Feng, J.; Yang, N.; Guo, Y.; Chen, C.; Qin, Q. Ginsenoside Rg3 attenuates angiotensin II-induced myocardial hypertrophy through repressing NLRP3 inflammasome and oxidative stress via modulating SIRT1/NF-κB pathway. Int. Immunopharmacol. 2021, 98, 107841. [Google Scholar] [CrossRef] [PubMed]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca2+/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef] [PubMed]
- Hou, N.; Li, L.-R.; Shi, Y.-Y.; Yuan, W.-C.; Zhao, G.-J.; Liu, X.-W.; Cai, S.-A.; Huang, Y.; Zhan, H.-X.; Pan, W.-B.; et al. Azilsartan ameliorates ventricular hypertrophy in rats suffering from pressure overload-induced cardiac hypertrophy by activating the Keap1–Nrf2 signalling pathway. J. Pharm. Pharmacol. 2021, 73, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Jia, Y.; Zhu, B. BNP and NT-proBNP as Diagnostic Biomarkers for Cardiac Dysfunction in Both Clinical and Forensic Medicine. Int. J. Mol. Sci. 2019, 20, 1820. [Google Scholar] [CrossRef]
- Morillas, P.; Castillo, J.; Quiles, J.; Núñez, D.; Guillén, S.; Maceira, A.; Rivera, M.; Bertomeu, V. Usefulness of NT-proBNP Level for Diagnosing Left Ventricular Hypertrophy in Hypertensive Patients. A Cardiac Magnetic Resonance Study. Rev. Espanola Cardiol. 2008, 61, 972–975. [Google Scholar] [CrossRef]
- Reid, A.; Miller, C.; Farrant, J.P.; Polturi, R.; Clark, D.; Ray, S.; Cooper, G.; Schmitt, M. Copper chelation in patients with hypertrophic cardiomyopathy. Open Hear. 2022, 9, e001803. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Subjects | Dose of Ivabradine | Type of Study | Findings | Reference |
|---|---|---|---|---|
| Patients with HF (LVEF < 40%, HR > 70 bpm) (n = 37) | 2.5–7.5 mg, b.i.d. for >12 months | Retrospective cohort study | ↓ risk of hospitalization ↓ number of hospitalizations ↔ length of hospitalization ↔ death rate | [16] |
| Moderate-to-severe HF patients with HR > 70 bpm (n = 3241) (SHIFT study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | ↓ event rates in patients with 0 or 3+ comorbidities ↓ HF hospitalization | [17] |
| Hemodynamically stable acute HF patients (n = 63) | Started at 5 mg daily, followed by 10 mg daily for >90 days | Retrospective cohort | ↓ length of hospitalization ↓ rehospitalization ↓ high dose of β-blockers ↓ NYHA class | [18] |
| Moderate-to-severe HF patients with HR > 77 bpm (n = 208) (SHIFT study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 31–35 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | ↓ NYHA class ↑ Global self-assessment improvement ↑ Global assessment improvement (physician perspective) ↑ Health-related quality of life ↓ all-cause cardiovascular death ↓ all-cause hospitalization ↓ all-cause mortality | [19] |
| Patients with chronic HF (n = 767) (RELIf-CHF study) | 5 mg b.i.d. and titrated to 7.5 mg or 2.5 mg b.i.d. for 12 months | Observational follow-up study | ↓ NYHA class ↓ decompensation ↓ HF hospitalizations ↑ general health ↑ QoL | [20] |
| Moderate-to-severe HF patients with HR < 75 (n = 1188) and >75 bpm (n = 2052) (SHIFT study) | 5 mg b.i.d. titrated to 7.5 mg b.i.d. for a median follow-up of 22.5 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | In HR > 75 bpm group: ↓ cardiovascular death ↓ death from HF ↓ hospitalization In HR < 75 bpm group: ↔ cardiovascular death ↔ death from HF ↔ hospitalization | [21] |
| Hospitalized HF patients in the SHIFT study (n = 514) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 3 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | ↓ all-cause hospitalization at 1, 2, and 3 months ↔ hospitalization due to cardiovascular causes at all time-points ↔ death rate | [22] |
| Acute HF patients with inflammatory rheumatic disease (n = 12) | 2.5 mg/d b.i.d. titrated to 5 mg/d b.i.d. for 2 weeks | Retrospective observational study | ↓ NYHA class | [23] |
| Moderate-to-severe HF patients with HR > 70 bpm plus angina pectoris (n = 1085) (SHIFT and SIGNIFY studies) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 31-35 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | SHIFT study: ↔ Composite primary end point ↔ Cardiovascular death ↔ First hospitalization due to worsening HF SIGNIFY study: ↔ Composite primary end point ↔ Cardiovascular death ↔ non-fatal MI | [24] |
| Moderate-to-severe HF patients (HR > 70 bpm) with prior mineralocorticoid receptor antagonist (MRA) (n = 1981) (SHIFT study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 31–35 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | Compared to the MRA group at baseline: ↔ Composite primary end point ↔ Cardiovascular death ↔ HF death | [25] |
| Moderate-to-severe HF patients (HR > 70 bpm) with diabetes (n = 973) (SHIFT study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 31–35 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | ↔ Outcomes of different treatments (ivabradine vs. placebo; insulin vs. non-insulin) In diabetic and non-diabetic patients: ↓ hospitalization for worsening HF ↓ cardiovascular hospitalization In non-diabetic patients: ↓ all-cause hospitalization | [26] |
| Patients with HFpEF (n = 84) (EDIFY study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 8 months | Randomized, double-blind, placebo-controlled, multicenter clinical trial | ↔ 6MWT | [27] |
| Acute decompensated HFrEF patients (n = 292) | Not given. Follow-up for 1 year after discharge | Retrospective study | ↓ cardiovascular death ↓ all-cause mortality ↓ rehospitalization ↓ NYHA class | [28] |
| Patients with systolic chronic HF (n = 98) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 6 months | Open-label, blinded, parallel-group, interventional, prospective-cohort study | ↓ NYHA class | [29] |
| Moderate-to-severe HF patients (HR > 70 bpm) with left bundle branch block (n = 467) (SHIFT study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 31-35 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | ↔ primary end point ↔ cardiovascular death ↔ HF hospitalization ↔ all-cause death | [30] |
| Patients with chronic HF (n = 110) (APULIA study) | 5 mg b.i.d. for a month | Multicentric observational study | ↓ HR ↑ physical functioning ↑ physical role functioning ↑ emotional role functioning ↑ mental health scale | [31] |
| Patients with cardiomyopathy (n = 33) | 5 mg b.i.d. for 3 months and 7.5 mg b.i.d. for 3 months | Observational study | ↓ NYHA class ↑ general health ↑ social activity ↑ physical health ↑ emotional health | [32] |
| Hospitalized patients with acute decompensated systolic heart failure (n = 10) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. until discharged | Observational, open-label, longitudinal, and retrospective study | ↓ NYHA class | [33] |
| Patients with HF (n = 10) | 5 mg b.i.d. and titrated to 7.5 mg b.i.d. for 6 months | Randomized, double-blind study | ↓ NYHA class ↑ QoL | [34] |
| Patients with chronic HF (n = 1873) | 5 mg b.i.d. and titrated to 7.5 mg or 2.5 mg b.i.d. for 4 months | Observational and longitudinal study | ↓ NYHA class ↓ decompensation | [35] |
| Children with dilated cardiomyopathy (n = 74) | 0.02 mg/kg b.i.d. (6–12 months old) or 0.05 mg/kg b.i.d. (1–18 years old) or 2.5 mg b.i.d. (>40 kg bw) and titrated for 12 months. | Randomized, double-blind, placebo-controlled, phase II/III clinical trial | ↑ PedQL ↔ NYHA class | [36] |
| Subjects | Dose of Ivabradine | Type of Study | Findings | Reference |
|---|---|---|---|---|
| Hospitalized patients with severe CHF (n = 10) | Infusion at 0.1 mg/kg for 90 min, followed by 0.05–0.075 mg/kg for 90 min | Single-center open-label phase II clinical trial | At 4 h: ↓ HR, ↑ SV ↑ LV systolic work | [40] |
| Hemodynamically stable acute HF patients (n = 63) | Started at 5 mg daily, followed by 10 mg daily for > 90 days | Retrospective cohort | ↓ HR, ↑ LVEF ↔ SBP, ↔ DBP | [18] |
| Patients with chronic HF (n = 1873) | 5 mg b.i.d. and titrated to 7.5 mg or 2.5 mg b.i.d. for 4 months | Observational and longitudinal study | ↑ LVEF | [35] |
| Acute decompensated HFrEF patients (n = 292) | Not given. Follow-up for 1 year after discharge | Retrospective study | ↓ HR ↔ SBP, ↔ LVEF | [28] |
| Moderate-to-severe HF patients with HR < 75 (n = 1188) and >75 bpm (n = 2052) (SHIFT study) | 5 mg b.i.d. titrated to 7.5 mg b.i.d. for a median follow-up of 22.5 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | In HR > 75 bpm group: ↓ HR In HR < 75 bpm group: ↔ HR | [21] |
| Moderate-to-severe HF patients with HR > 70 bpm (n = 298) (SHIFT study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 8 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | ↓ office HR ↓ 24-HR ↓ HR awake ↓ HR asleep | [42] |
| Patients with chronic HF (n = 30) | 5 mg b.i.d. for 4 months | Cross-sectional | ↓ LVEDV, ↓ LVESV ↑ LVEF, ↑ SV, ↑ Ees ↓ VAC | [41] |
| Acute HF patients with inflammatory rheumatic disease (n = 12) | 2.5 mg/d b.i.d. titrated to 5 mg/d b.i.d. for 2 weeks | Retrospective observational study | ↓ HR ↑ LVEF | [23] |
| Moderate-to-severe HF patients with HR > 77 bpm (n = 208) (SHIFT study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 31–35 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | ↓ LVESVI, ↓ LVESV, ↓ LVEDVI, ↓ LVEDV, ↑ LVEF | [19] |
| Patients with HFpEF (n = 84) (EDIFY study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 8 months | Randomized, double-blind, placebo-controlled, multicenter clinical trial | ↓ HR ↔ E/e′, ↔ E, ↔ Ea, ↔ Ees, ↔ Ea/Ees ↔ Total mitral flow duration ↔ Mitral flow integral time velocity ↔ Lateral e′, ↔ Septal e′ ↔ Mean of lateral and septal e′ ↔ LVEDV, ↔ SV, ↔ LAVI | [27] |
| Male patients with chronic HF (n = 22) | 5 mg b.i.d. and titrated to 7.5 mg for 6 months | Longitudinal study | ↓ HR ↔ SBP, ↔ DBP, ↔ LVEF | [43] |
| Patients with systolic chronic HF (n = 98) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 6 months | Open-label, blinded, parallel-group, interventional, prospective-cohort study | ↓ HR | [29] |
| Patients with systolic HF (n = 43) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 3 months | Longitudinal study | ↓ HR ↔ SBP, DBP ↔ LVEDV, LVESV, LVEF, ↔ E/A, ↓ E/E′ ↓ LA Vmax, ↓ LA Vp ↔ LA Vmin ↔ LA passive emptying volume and fraction ↓ LA active emptying volume and fraction ↓ PA lateral, septum, and tricuspid ↓ PA lateral–PA tricuspid ↔ PA lateral–PA septum ↓ PA septum–PA tricuspid ↓ interatrial conduction delay ↔ left intra-atrial conduction delay ↓ right intra-atrial conduction delay | [44] |
| Moderate-to-severe HF patients (HR > 70 bpm) (n = 143) (SHIFT study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 8 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | ↓ HR, ↔ LVESP, ↑ SV ↔ Pulse pressure, ↔ MAP ↑ Total arterial compliance ↓ Ea, ↔ TPR, ↔ CO, ↔ Ees ↑ LVEF, ↔ LVESV ↔ LVEDV, ↔ Ea/Ees | [45] |
| Patients with cardiomyopathy (n = 33) | 5 mg b.i.d. for 3 months and 7.5 mg b.i.d. for 3 months | Observational study | ↓ HR, ↑ LVEF | [32] |
| Hospitalized patients with acute decompensated systolic heart failure (n = 10) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. until discharged | Observational, open-label, longitudinal, and retrospective study | ↓ HR, ↓ SBP ↔ DBP, ↔ MBP | [33] |
| Moderate-to-severe HF patients (HR > 70 bpm) with left bundle branch block (n = 208) (SHIFT study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 8 months | Randomized, double-blind, placebo-controlled, parallel-group, multicenter clinical trial | ↓ LVESVI, ↓ LVEDVI ↓ LVESV, ↓ LVEDV ↑ LVEF | [46] |
| Patients with HF (n = 10) | 5 mg b.i.d. and titrated to 7.5 mg b.i.d. for 6 months | Randomized, double-blind, double-dummy study | ↑ VO2 | [34] |
| Patients with chronic HF (n = 1873) | 5 mg b.i.d. and titrated to 7.5 mg or 2.5 mg b.i.d. for 4 months | Observational and longitudinal study | ↑ LVEF | [35] |
| Patients with chronic HF (n = 767) (RELIf-CHF study) | 5 mg b.i.d. and titrated to 7.5 mg or 2.5 mg b.i.d. for 12 months | Observational follow-up study | ↓ HR, ↑ LVEF | [20] |
| Patients with stable symptomatic chronic HF (n = 52) | 5 mg b.i.d. and titrated to 7.5 mg 2.5 mg b.i.d. for 12 months | Observational follow-up study | ↓ LVEDV, ↓ LVESV, ↑ LVEF, ↓ DT ↔ TAPSE, ↔ PASP, ↔ RV FAC, ↔ E peak, ↔ A peak, ↔ myocardial performance index ↑ systolic velocity ↑ Early diastolic velocity ↓ Late diastolic velocity ↔ RV IVV, ↔ RV IVA ↑ RV GLS, ↑ RV LS ↑ RV LSRS, ↑ RV LSRE ↑ RV LSRA | [47] |
| Children with dilated cardiomyopathy (n = 74) | 0.02 mg/kg b.i.d. (6–12 months old) or 0.05 mg/kg b.i.d. (1–18 years old) or 2.5 mg b.i.d. (>40 kg bw) and titrated for 12 months. | Randomized, double-blind, placebo-controlled, phase II/III clinical trial | ↓ HR, ↑ LVEF | [36] |
| Models | Dose and Duration of Ivabradine | Findings | Reference |
|---|---|---|---|
| Surface ECG recordings and transesophageal electrophysiological study in female C57BL/10 mice | Single dose of 10 mg/kg (i.p.) | ↓ HR ↑ QRS duration ↔ QR duration ↑ QT1 intervals ↑ QT2-P intervals ↑ S2Q2 intervals | [50] |
| Chronic-hypertension-induced cardiac hypertrophy in pigs | 1 mg/kg/d infusion for 28 days | ↓ HR, ↑ SV, ↑ LVEDP ↑ LV twist, ↔ LV twisting rate ↑ LV untwisting rate ↑ LV untwisting velocity at MVO ↔ LV apical rotation ↑ LV basal rotation ↑ untwist during isovolumic relaxation time | [51] |
| Experimental chronic- hypertension-induced cardiac remodeling in pigs | 1 mg/kg (i.v. bolus, single) | ↓ HR, ↔ CO ↔ dp/dtmax, ↔ LV pressure ↑ LV end-diastole internal diameter ↑ LV end-systole internal diameter ↑ LV relaxation filling ↑ LV early filling ↑ LV peak early filling rate | [52] |
| Experimental hypertension- induced cardiac remodeling in SHR | 10 mg/kg/d in drinking water for 6 weeks | ↓ HR, ↔ SBP, ↑ LVEF ↑ LVFS, ↓ E/A, ↓ E/Em | [53] |
| Isoproterenol-induced heart failure in rats | 10 mg/kg/d (p.o.) for 6 weeks | ↓ HR | [54] |
| Isoproterenol-induced heart failure in rats | 10 mg/kg/d (p.o.) for 14 days | ↓ HR | [55] |
| Diastolic-dysfunction-induced heart failure in diabetic mice | 20 mg/kg/d in drinking water for 4 weeks | ↓ HR, ↑ E/A, ↓ EDT ↑ −dp/dtmin, ↓ Tau, ↓ IVRT | [56] |
| Diabetic cardiomyopathy in mice | 20 mg/kg/d (p.o.) for 12 weeks | ↓ HR, ↑ LVEF | [13] |
| Myocardial I/R-induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 28 days | ↓ HR, ↑ LVFS ↑ LVEF, ↑ delta LVEF | [57] |
| Experimental HFpEF in mice | 10 mg/kg/d (low) and 20 mg/kg/d (high) (p.o.) for 4 weeks | High dose: ↓ HR, ↓ LVEDP, ↔ LVEF ↓ LV −dp/dtmax, ↔ LV +dp/dtmax, ↓ EDT, ↔ LVFS, ↓ IVRT Low dose: ↓ HR | [58] |
| Experimental HFrEF in mice | 10 mg/kg/d and 20 mg/kg/d (p.o.) for 8 weeks | High dose: ↓ HR, ↓ LVEDP, ↓ IVRT ↓ LV −dp/dtmax ↑ LV +dp/dtmax ↓ EDT, ↑ LVEF, ↑ LVFS Low dose: ↓ HR | [58] |
| Post-MI-induced heart failure in rats | 10 mg/kg/min (via osmotic pump) for 2 weeks | ↓ HR, ↑ CO, ↑ SV, ↔ LVEF ↔ LV +dp/dt ↔ LV −dp/dt ↔ LVEDP | [59] |
| Myocardial I/R-induced cardiac remodeling in pigs | 0.3 mg/kg (i.v.) | ↓ HR, ↑ SV, ↓ CO, ↑ CVP ↔ MAP ↔ systemic arterial pressure ↔ pulmonary arterial pressure | [60] |
| Hypertension-induced heart failure in rats | 10 mg/kg/d in drinking water for 10 weeks | ↓ HR, ↔ SBP, ↓ E/A, ↓ E/E′ ↑ LVFS, ↑ LVEF | [11] |
| MI-induced cardiac remodeling in rats | 10 mg/kg/d in drinking water for 8 weeks | ↓ HR, ↑ LVEF, ↓ LVEDP ↑ LVDP, ↑ LV +dp/dt ↑ LV −dp/dt ↓ LV diastolic wall stress | [61] |
| Experimental hypertension- induced cardiac remodeling in rats | 10 mg/kg/d in drinking water for 4 weeks | ↓ HR, ↓ SBP, ↑ LVEF ↑ LVFS | [62] |
| Severe post-MI chronic HF in rats | 10 mg/kg/d in drinking water for 3 months | ↓ HR, ↑ LVEF, ↓ LVEDP ↓ LVEDV, ↓ LVESV ↑ SV, ↔ CO | [63] |
| Abdominal-aorta- constriction-induced chronic heart failure in rats | 10 mg/kg/d (p.o.) for 12 weeks | ↓ LVEDP, ↑ LV +dp/dt ↓ L V −dp/dt | [12] |
| Open chest with LV post- ischemia dysfunction in pigs | Bolus infusion of 0.5 mg/kg | ↓ HR, ↑ SV, ↔ CO ↑ diastolic filling time ↔ MAP, cardiac efficiency | [64] |
| Chronic ischemic heart failure in diabetic rats | 10 mg/kg/d (i.p.) for 7 weeks | ↓ HR, ↑ LVFS, ↓ LVEDP | [65] |
| LAD coronary-artery- ligated-induced cardiac remodeling in rats | 10 mg/kg/d in drinking water for 90 days | ↓ HR, ↑ LVEF, ↔ LVEDV ↔ LVESV | [14] |
| LAD coronary-artery- ligated-induced cardiac remodeling in rats | 6–8 mg/kg/d (i.p.) for 4 weeks | ↓ HR, ↑ SV, ↔ LVEDV ↔ LVESV, ↓ LVEDV/LV mass ↑ LVEF, ↓ LVEDP ↑ LV coronary reserve ↔ coronary conductance | [66] |
| LAD coronary-artery- ligated-induced cardiac remodeling in rats | 10 mg/kg/d (i.g.) for 7 days | ↑ LVSP, ↓ LVEDP ↑ +dp/dtmax, ↓ −dp/dtmax | [67] |
| Doxorubicin-induced LV dysfunction in rats | 10 mg/kg (i.p.), alternate days for 2 weeks | ↓ HR, ↔ MAP, ↑ +dp/dtmax ↑ Tau, ↑ SDNN, ↓ LF ↔ HF, ↓ LF/HF, ↑ RMSSD ↑ Total power | [68] |
| Pulmonary-arterial- hypertension-induced heart failure in rats | 10 mg/kg/d (p.o.) for 3 weeks | ↔ HR, ↑ RV S′, ↑ LV E’ ↓ RV fractional area ↓ RV IVCT, ↓ LV IVCT ↓ Time to mitral valve opening ↓ Time to RV peak radial motion ↓ Time to maximum LVSB ↓ Time to maximum TAPSE ↓ Time to tricuspid valve opening ↓ RV Tau (τ) | [69] |
| Hypertension-induced cardiac remodeling in SHR | 1 mg/kg/d (i.p.) for 14 days | ↓ HR, ↓ SBP, ↓ DBP, ↓ MAP | [70] |
| Transverse-aortic- constriction-induced cardiac hypertrophy in mice | 10, 20, 40, and 80 mg/kg/d (i.g.) for 4 weeks | All doses: ↓ HR, ↓ LV Vols, ↑ LVEF ↑ LVFS 10 and 20 mg/kg/d: ↓ LV Vold | [15] |
| Myocardial I/R-induced cardiac remodeling in pigs | 0.3 mg/kg for 7 days | ↑ LVEF | [71] |
| Pulmonary-hypertension- induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 3 weeks | ↓ HR, ↓ RV longitudinal ↑ RV S′, ↓ RV S:D ratio ↓ RV TDI-MPI, ↓ TDI IVRT ↓ RDI IVRT/R-R, ↑ SV, ↑ CO ↑ RV +dp/dt, ↓ RV −dp/dt ↓ RV Tau | [72] |
| RV pressure-loaded-induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 3 weeks | ↓ HR, ↑ FAC, ↑ TAPSE ↓ RV MPI, ↓ RV S:D ratio ↓ RV longitudinal ↓ RV TDI-MPI, ↓ TDI IVRT ↓ RDI IVRT/R-R, ↑ SV, ↑ CO ↓ RV EDP, ↑ RV +dp/dt ↓ RV −dp/dt, ↓ RV Ees ↓ RV Tau | [72] |
| SU5416+Hypoxia-induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 3 weeks | ↓ HR, ↑ FAC, ↑ TAPSE ↓ RV MPI, ↓ RV TDI-MPI ↓ TDI IVCT, ↓ TDI IVRT ↓ RDI IVRT/R-R, ↑ SV, ↑ CO ↓ RV EDP, ↓ RV Ees, ↓ RV EDPVR, ↓ RV Tau | [72] |
| Hyperthyroid cardiomyopathy in rats | 10 mg/kg/d (p.o.) for 28 days | ↓ HR, ↓ EDT, ↑ Ea, ↓ E/Ea ↓ Scirc, ↓ SRcirc, ↓ Slong ↑ SRlong, ↑ Srad, ↑ SRrad | [73] |
| Cardiogenic-shock-induced cardiac remodeling in pigs | 0.3 mg/kg (i.v. bolus) | ↓ HR, ↑ SV, ↑ LVEF | [74] |
| Models | Dose and Duration of Ivabradine | Findings | Reference |
|---|---|---|---|
| Hypertension-induced HF in rats | 10 mg/kg/d in drinking water for 10 weeks | ↔ LV HCN2 gene ↔ LV HCN4 gene ↔ LA HCN2 gene ↔ LA HCN4 gene ↓ RA HCN2 gene ↔ RA HCN4 gene ↓ LV NE, ↓ RA NE, ↓ LA NE ↓ urine NE ↓ urine normetanephrine ↑ RA Ach, ↔ LA Ach ↓ serum NE ↓ serum epinephrine ↔ serum dopamine ↑ LV tyrosine hydroxylase protein ↑ LA tyrosine hydroxylase protein ↑ RA tyrosine hydroxylase protein ↓ LV ACE gene ↔ LV ET-1 gene ↓ LV AVP gene ↓ LV β1 adrenoceptor gene ↓ LV NGF gene | [11] |
| Post-MI-induced HF in rats | 10 mg/kg/min (via osmotic pump) for 2 weeks | ↓ HCN4 expression | [59] |
| Hypertension-induced cardiac remodeling in SHR | 1 mg/kg/d (i.p.) for 14 days | ↓ LV HCN4 mRNA | [70] |
| Chronic ischemic heart failure in diabetic rats | 10 mg/kg/d (i.p.) for 7 weeks | ↓ plasma NE ↑ NE uptake-1 in stellate ganglion tissues | [65] |
| Severe post-MI chronic HF in rats | 10 mg/kg/d in drinking water for 3 months | ↓ LV ACE mRNA ↓ LV AT1R mRNA ↓ LV ACE protein ↓ LV. AT1R protein | [63] |
| Experimental hypertension-induced cardiac remodeling in SHR | 10 mg/kg/d in drinking water for 6 weeks | ↔ serum Ang 1–10 (Ang I), Ang 1–8 (Ang II), Ang 2–8 (Ang III), Ang 3–8 (Ang IV), Ang 1–7, Ang 1–5 ↓ (Ang 1–5)/(Ang 1–7) ↔ serum renin ↔ serum ACE ↔ serum aldosterone ↔ serum aldosterone/Ang II ratio | [53] |
| LAD coronary-artery-ligated-induced cardiac remodeling in rats | 6–8 mg/kg/d (i.p.) for 4 weeks | ↓ plasma Ang II ↓ LV AT1R protein ↔ LV bradykinin protein | [66] |
| Experimental hypertension-induced cardiac remodeling in rats | 10 mg/kg/d in drinking water for 4 weeks | ↔ serum aldosterone ↔ serum renin ↔ serum Ang 1–10 (Ang I), Ang 1–8 (Ang II), Ang 2–8 (Ang III), Ang 3–8 (Ang IV), Ang 1–7, Ang 1–5 | [62] |
| Models | Dose and Duration of Ivabradine | Findings | Reference |
|---|---|---|---|
| Experimental HFpEF in mice | 10 mg/kg/d (low) and 20 mg/kg/d (high) (p.o.) for 4 weeks | High dose: ↓ LV fibrotic area | [58] |
| Experimental HFrEF in mice | 10 mg/kg/d and 20 mg/kg/d (p.o.) for 8 weeks | High dose: ↓ LV fibrotic area ↓ LV α-SMA protein ↓ LV CTGF protein ↓ LV Col-1 and Col-3 protein ↓ LV TGF-β1 protein ↓ LV TGFR2 protein ↓ LV pSmad2/3 protein Low dose: ↓ LV fibrotic area ↓ LV α-SMA protein ↓ LV CTGF protein ↓ LV Col-1 and Col-3 protein ↓ LV TGF-β1 protein ↓ LV TGFR2 protein ↓ LV pSmad2/3 protein | [58] |
| Ang II-induced primary ventricular fibroblast proliferation | 3 and 10 µM | Both concentrations: ↓ fibroblast proliferation rate ↓ α-SMA protein ↓ CTGF protein ↓ Col-1 and Col-3 protein ↓ TGF-β1 protein ↓ TGFR2 protein ↓ pSmad2/3 protein | [58] |
| Cardiogenic-shock-induced cardiac remodeling in pigs | 0.3 mg/kg (i.v. bolus) | ↓ MMP-9 protein ↔ EMMPRIN mRNA ↓ EMMPRIN protein ↑ EMMPRIN+Cav3 colonization | [74] |
| Isoproterenol-induced HF in rats | 10 mg/kg/d (p.o.) for 14 days | ↓ serum MMP-9 | [55] |
| Hyperthyroid cardiomyopathy in rats | 10 mg/kg/d (p.o.) for 28 days | ↔ cardiac fibrosis | [73] |
| Myocardial I/R-induced cardiac remodeling in pigs | 0.3 mg/kg for 7 days | ↓ MMP-9 protein | [71] |
| Diastolic-dysfunction-induced HF in diabetic mice | 20 mg/kg/d in drinking water for 4 weeks | ↓ α-SMA protein ↓ Collagen 1 protein ↓ Collagen 3 protein ↓ TIMP2 protein ↑ MMP2 protein | [56] |
| High-glucose-treated rat primary ventricular cardiac fibroblasts | 10–40 µM | All concentrations: ↓ Collagen 1 protein ↓ Collagen 3 protein ↓ α-SMA protein ↓ TIMP2 protein ↓ MMP2 protein | [56] |
| Diabetic cardiomyopathy in mice | 20 and 40 mg/kg/d in drinking for 12 weeks | Both doses: ↓ collagen | [106] |
| Transverse-aortic-constriction- induced cardiac hypertrophy in mice | 10, 20, 40, and 80 mg/kg/d (i.g.) for 4 weeks | All doses: ↓ Col 1 mRNA ↓ Col 3 mRNA ↓ PI3K protein ↓ mTORC1 ↔ mTORC2 ↓ Akt ↓ p-Akt ↓ p-p70S6K1 | [15] |
| Diabetic cardiomyopathy in mice | 20 mg/kg/d (p.o.) for 12 weeks | ↓ Col 1 protein ↓ Col 3 protein | [13] |
| SU5416+Hypoxia-induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 3 weeks | ↓ RV collagen area ↓ RV collagen I/III protein ratio ↓ RVTGF-β1 protein ↓ RV pSMAD2/Smad2,3 protein ↓ RV pSMAD3/Smad2,3 protein ↓ RV CTGF protein | [72] |
| RV pressure-loaded-induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 3 weeks | ↓ RV collagen area ↓ RV collagen I/III protein ratio ↓ RVTGF-β1 protein ↓ RV pSMAD2/Smad2,3 protein ↓ RV pSMAD3/Smad2,3 protein ↓ RV CTGF protein | [72] |
| Pulmonary-hypertension-induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 3 weeks | ↓ RV collagen area ↓ RV collagen I/III protein ratio ↓ RVTGF-β1 protein ↓ RV pSMAD2/Smad2,3 protein ↓ RV pSMAD3/Smad2,3 protein ↓ RV CTGF protein | [72] |
| Experimental hypertension-induced cardiac remodeling in SHR | 10 mg/kg/d in drinking water for 6 weeks | ↓ LV collagen ↓ LV hydroxyproline | [53] |
| Experimental hypertension-induced cardiac remodeling in rats | 10 mg/kg/d in drinking water for 4 weeks | ↔ LV hydroxyproline | [62] |
| Isoproterenol-induced HF in rats | 10 mg/kg/d (p.o.) for 6 weeks | ↓ LV hydroxyproline ↓ LV collagen | [54] |
| LAD coronary-artery-ligated- induced cardiac remodeling in rats | 6–8 mg/kg/d (i.p.) for 4 weeks | ↓ LV collagen ↓ LV TGF-β protein ↔ LV VEGF-A protein ↔ LV bradykinin protein | [66] |
| Severe post-MI chronic HF in rats | 10 mg/kg/d in drinking water for 3 months | ↓ collagen volume fraction | [63] |
| Hypertension-induced HF in rats | 10 mg/kg/d in drinking water for 10 weeks | ↔ LV Col 1a1 gene ↔ RA Col 1a1 gene ↔ LA Col 1a1 gene | [11] |
| Abdominal-aorta-constriction- induced chronic heart failure in rats | 10 mg/kg/d (p.o.) for 12 weeks | ↓ CTGF protein ↓ TGF-β1 gene ↓ COL-1 gene | [12] |
| Models | Dose and Duration of Ivabradine | Findings | Reference |
|---|---|---|---|
| LAD coronary-artery-ligated- induced cardiac remodeling in rats | 10 mg/kg/d in drinking water for 90 days | ↑ energy charge, ↑ creatine phosphate ↓ ADP, ↔ ATP, ↔ AMP | [14] |
| LAD coronary-artery-ligated- induced cardiac remodeling in rats | 10 mg/kg/d (i.g.) for 7 days | ↑ LC3II/I protein ↓ p62 protein ↑ beclin 1 protein ↑ ATG5 protein, ↑ ATG7 protein ↓ p-PI3K, ↓ p-AKT ↓ p-mTOR, ↓ p-p70S6K | [67] |
| Transverse-aortic-constriction- induced cardiac hypertrophy in mice | 10, 20, 40, and 80 mg/kg/d (i.g.) for 4 weeks | All doses: ↓ cleaved caspase-3 protein ↑ caspase-3 protein ↓ PI3K protein ↓ Akt, ↓ p-Akt, ↓ p-p70S6K1 | [15] |
| Diabetic cardiomyopathy in mice | 20 and 40 mg/kg/d in drinking for 12 weeks | Both doses: ↓ TUNEL | [107] |
| Diabetic cardiomyopathy in mice | 20 mg/kg/d (p.o.) for 12 weeks | ↓ cleaved caspase-3 protein ↓ TUNEL | [13] |
| Subjects | Dose of Ivabradine | Findings | Reference |
|---|---|---|---|
| Patients with cardiomyopathy (n = 33) | 5 mg b.i.d. for 3 months and 7.5 mg b.i.d. for 3 months | ↓ TNFα, ↔ IL-6 ↔ hsCRP | [32] |
| Patients with cardiomyopathy (n = 33) | 5 mg b.i.d. for 3 months and 7.5 mg b.i.d. for 3 months | ↔ sST2, ↓ GDF-15 ↓ H-FABP, ↔ suPAR | [124] |
| Patients with HF (n = 10) | 5 mg b.i.d. and titrated to 7.5 mg b.i.d. for 6 months | ↓ IL-6, ↓ TNFα | [34] |
| Diabetic cardiomyopathy in mice | 20 mg/kg/d (p.o.) for 12 weeks | ↓ Heart TNF-α mRNA and protein ↓ Heart IL-1β mRNA and protein ↓ Heart IL-6 mRNA and protein ↓ p-JNK, ↓ p-38 | [13] |
| High-glucose-treated rat primary ventricular cardiac fibroblasts | 10–40 µM | All concentrations: ↓ p-JNK protein ↓ p-p38 protein ↓ cell proliferation | [56] |
| Diastolic-dysfunction-induced HF in diabetic mice | 20 mg/kg/d in drinking water for 4 weeks | ↓ p-JNK protein ↓ p-p38 protein | [56] |
| LAD coronary-artery-ligated- induced cardiac remodeling in rats | 10 mg/kg/d (i.g.) for 7 days | ↓ TNFα ↓ IL-1β ↓ IL-6 | [67] |
| Hypertension-induced cardiac remodeling in SHR | 1 mg/kg/d (i.p.) for 14 days | ↓ Number of inflammatory nuclei | [70] |
| Myocardial I/R-induced cardiac remodeling in pigs | 0.3 mg/kg for 7 days | ↑ de-expression of heart CyPA ↓ plasma CyPA protein ↑ cardiac CyPA protein ↔ heart CyPA mRNA ↑ CyPA-LG-EMMPRIN protein ↔ CyPA degradation | [71] |
| Hyperthyroid cardiomyopathy in rats | 10 mg/kg/d (p.o.) for 28 days | ↔ cardiac inflammation | [73] |
| High-glucose-induced apoptosis in cardiomyocytes | 1–40 μM (pretreatment) | 5–40 μM: ↓ p-IKKα/β protein ↔ IKKβ protein ↓ p-IκBα protein ↑ t-IκBα protein ↑ cyto NF-κB protein ↓ nuclear NF-κB protein | [107] |
| Diabetic cardiomyopathy in mice | 20 and 40 mg/kg in drinking water for 12 weeks | Both doses: ↓ p-IKKα/β protein ↔ IKKβ protein ↓ p-IκBα protein ↑ t-IκBα protein ↑ cyto NF-κB protein ↓ nuclear NF-κB protein | [107] |
| Abdominal-aorta- constriction-induced chronic heart failure in rats | 10 mg/kg/d (p.o.) for 12 weeks | ↑ SOD protein | [12] |
| Subjects | Dose of Ivabradine | Findings | Reference |
|---|---|---|---|
| Patients with chronic HF (n = 30) | 5 mg b.i.d. for 4 months | ↓ LVESD ↔ LVEDD | [36] |
| Acute HF patients with inflammatory rheumatic disease (n = 12) | 2.5 mg/d b.i.d. titrated to 5 mg/d b.i.d. for 2 weeks | ↓ Plasma NT-proBNP | [23] |
| Patients with HFpEF (n = 84) (EDIFY study) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 8 months | ↔ NT-proBNP ↔ LV mass index | [27] |
| Patients with systolic chronic HF (n = 98) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. for 6 months | ↓ Cystatin-C, ↓ CA-125 ↓ NT-proBNP | [29] |
| Patients with systolic HF (n = 43) | NA (3 months) | ↔ PW, IVS, ↔ LA diameter | [44] |
| Patients with cardiomyopathy (n = 33) | 5 mg b.i.d. for 3 months and 7.5 mg b.i.d. for 3 months | ↔ LVEDD ↔ serum BNP | [32] |
| Hospitalized patients with acute decompensated systolic heart failure (n = 10) | Started at 5 mg b.i.d. and titrated to 7.5 mg b.i.d. or 2.5 mg b.i.d. until discharged | ↓ NT-ProBNP | [33] |
| Patients with HF (n = 10) | 5 mg b.i.d. and titrated to 7.5 mg b.i.d. for 6 months | ↓ NT-ProBNP | [34] |
| Patients with chronic HF (n = 1873) | 5 mg b.i.d. and titrated to 7.5 mg or 2.5 mg b.i.d. for 4 months | ↓ BNP | [35] |
| Patients with chronic HF (n = 767) (RELIf-CHF study) | 5 mg b.i.d. and titrated to 7.5 mg or 2.5 mg b.i.d. for 12 months | ↓ BNP | [20] |
| Transverse-aortic-constriction-induced cardiac hypertrophy in mice | 10, 20, 40, and 80 mg/kg/d (i.g.) for 4 weeks | All doses: ↓ IVSd, ↓ LVIDs, ↓ LVPWd ↓ LV mass, ↓ HW/BW 10 and 20 mg/kg/d: ↓ IVSs, ↓ LVIDd | [15] |
| Open chest with LV post-ischemia dysfunction in pigs | Bolus infusion of 0.5 mg/kg | ↑ LVED dimension | [64] |
| Chronic ischemic heart failure in diabetic rats | 10 mg/kg/d (i.p.) for 7 weeks | ↓ LVDD, ↓ LVSD, ↓ BNP | [65] |
| LAD coronary-artery-ligated-induced cardiac remodeling in rats | 10 mg/kg/d in drinking water for 90 days | ↓ HW, ↓ ANP | [14] |
| LAD coronary-artery-ligated-induced cardiac remodeling in rats | 6–8 mg/kg/d (i.p.) for 4 weeks | ↑ LVW/BW ↑ LV coronary reserve ↓ LV arteriolar length | [66] |
| Hypertension-induced cardiac remodeling in SHR | 1 mg/kg/d (i.p.) for 14 days | ↔ LVW/BW ↓ cardiomyocyte diameter | [70] |
| Myocardial I/R-induced cardiac remodeling in pigs | 0.3 mg/kg for 7 days | ↓ heart necrosis | [71] |
| Pulmonary-hypertension-induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 3 weeks | ↔ HW/BW ↓ cardiomyocyte diameter | [72] |
| RV pressure-loaded-induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 3 weeks | ↔ HW/BW, ↓ RVEDD ↓ cardiomyocyte diameter | [72] |
| SU5416+Hypoxia-induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 3 weeks | ↓ HW/BW, ↓ RVEDD ↓ cardiomyocyte diameter | [72] |
| Hypertension-induced HF in rats | 10 mg/kg/d in drinking water for 10 weeks | ↓ LV IVST, ↓ LVDD ↓ LVSD, ↓ LVSWS ↓ LV mass, ↓ LA dimension ↓ LV/BW, ↓ LA/BW ↓ LV ANP gene ↓ LA ANP gene ↔ RA ANP gene ↔ LV β-MHC gene | [11] |
| Hyperthyroid cardiomyopathy in rats | 10 mg/kg/d (p.o.) for 28 days | ↓ HW/BW ↔ cardiomyocyte width ↓ LVESD, ↔ LVEDD ↓ LA dimension, ↔ IVSd | [73] |
| Myocardial I/R-induced cardiac remodeling in rats | 10 mg/kg/d (p.o.) for 28 days | ↔ LVIDd, ↔ LVIDs | [57] |
| MI-induced cardiac remodeling in rats | 10 mg/kg/d in drinking water for 8 weeks | ↑ HW/BW, ↑ LV/BW ↑ posterior wall-end diastolic thickness | [61] |
| Severe post-MI chronic HF in rats | 10 mg/kg/d in drinking water for 3 months | ↓ LA dimension, ↓ VW/BW | [63] |
| Isoproterenol-induced HF in rats | 10 mg/kg/d (p.o.) for 14 days | ↓ serum NT-proBNP | [55] |
| Experimental HFpEF in mice | 10 mg/kg/d (low) and 20 mg/kg/d (high) (p.o.) for 4 weeks | High dose: ↓ cardiomyocyte size ↓ HW/TL, ↓ IVSd, ↓ LVPWd | [58] |
| Experimental HFrEF in mice | 10 mg/kg/d and 20 mg/kg/d (p.o.) for 8 weeks | High dose: ↓ cardiomyocyte size, ↓ HW/TL | [58] |
| Experimental hypertension-induced cardiac remodeling in rats | 10 mg/kg/d in drinking water for 4 weeks | ↔ VW/BW | [62] |
| Isoproterenol-induced HF in rats | 10 mg/kg/d (p.o.) for 6 weeks | ↓ LVW/BW, ↔ RVW/BW | [54] |
| Experimental hypertension-induced cardiac remodeling in SHR | 10 mg/kg/d in drinking water for 6 weeks | ↔ LVW/BW | [53] |
| Cardiogenic-shock-induced cardiac remodeling in pigs | 0.3 mg/kg (i.v. bolus) | ↓ c-Tn-1 | [74] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamisah, Y.; Che Hassan, H.H. Therapeutic Use and Molecular Aspects of Ivabradine in Cardiac Remodeling: A Review. Int. J. Mol. Sci. 2023, 24, 2801. https://doi.org/10.3390/ijms24032801
Kamisah Y, Che Hassan HH. Therapeutic Use and Molecular Aspects of Ivabradine in Cardiac Remodeling: A Review. International Journal of Molecular Sciences. 2023; 24(3):2801. https://doi.org/10.3390/ijms24032801
Chicago/Turabian StyleKamisah, Yusof, and Hamat H. Che Hassan. 2023. "Therapeutic Use and Molecular Aspects of Ivabradine in Cardiac Remodeling: A Review" International Journal of Molecular Sciences 24, no. 3: 2801. https://doi.org/10.3390/ijms24032801
APA StyleKamisah, Y., & Che Hassan, H. H. (2023). Therapeutic Use and Molecular Aspects of Ivabradine in Cardiac Remodeling: A Review. International Journal of Molecular Sciences, 24(3), 2801. https://doi.org/10.3390/ijms24032801