
Vascular Endothelial Growth Factor as Molecular Target for Bronchopulmonary Dysplasia Prevention in Very Low Birth Weight Infants
 ,
,  , ,
, ,  ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Lung Development
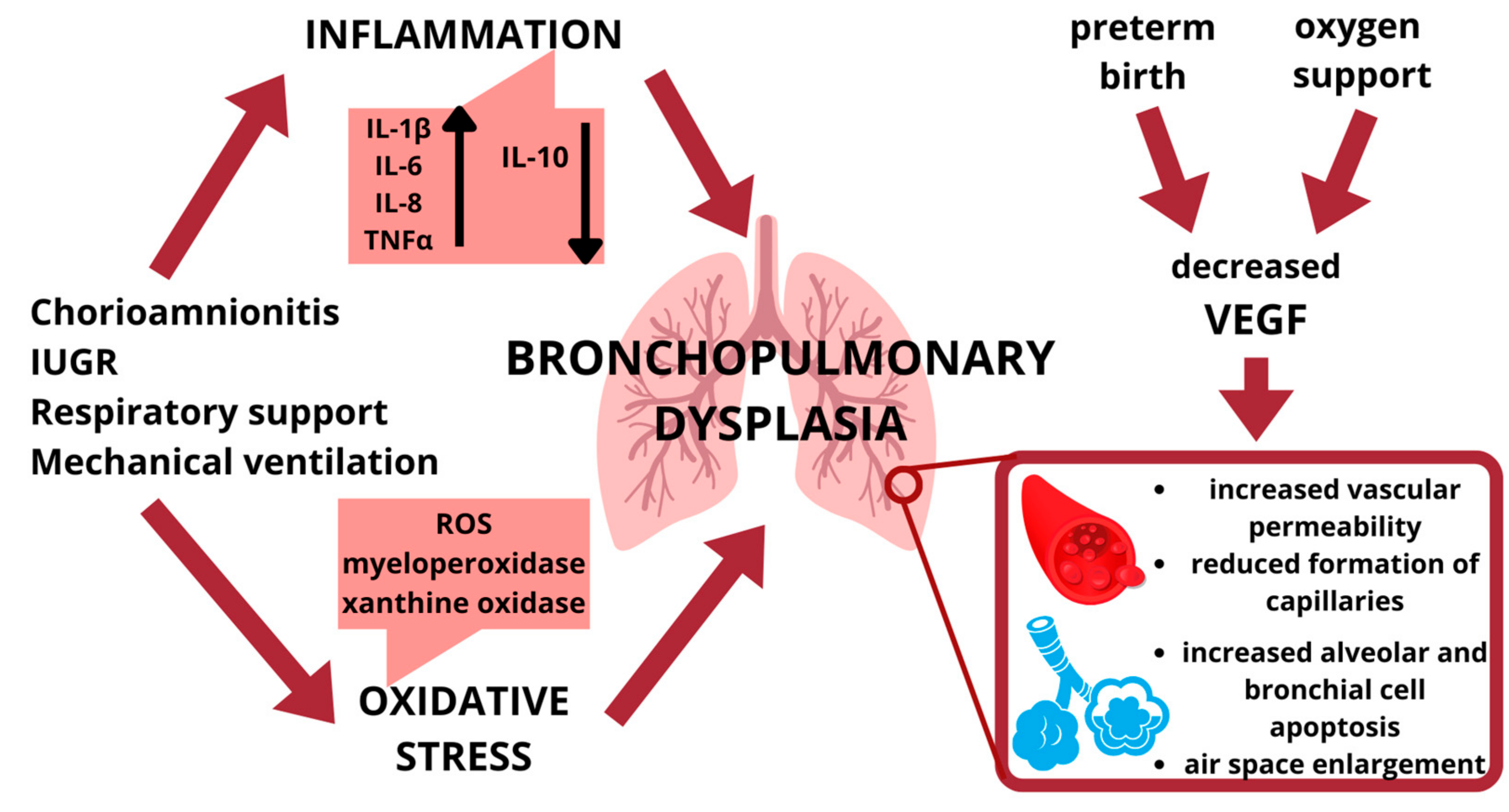
2.1. Lung Inflammation
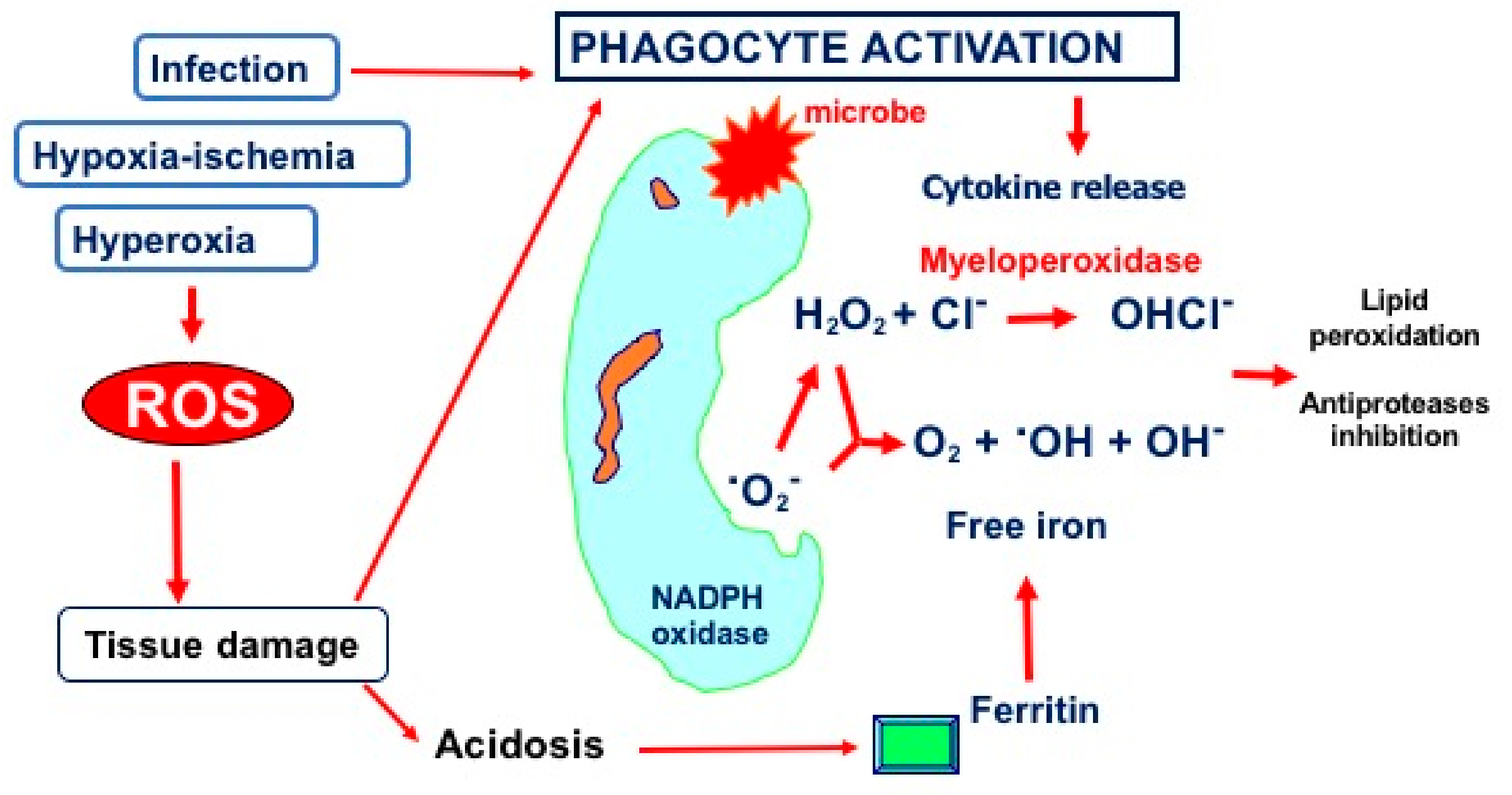
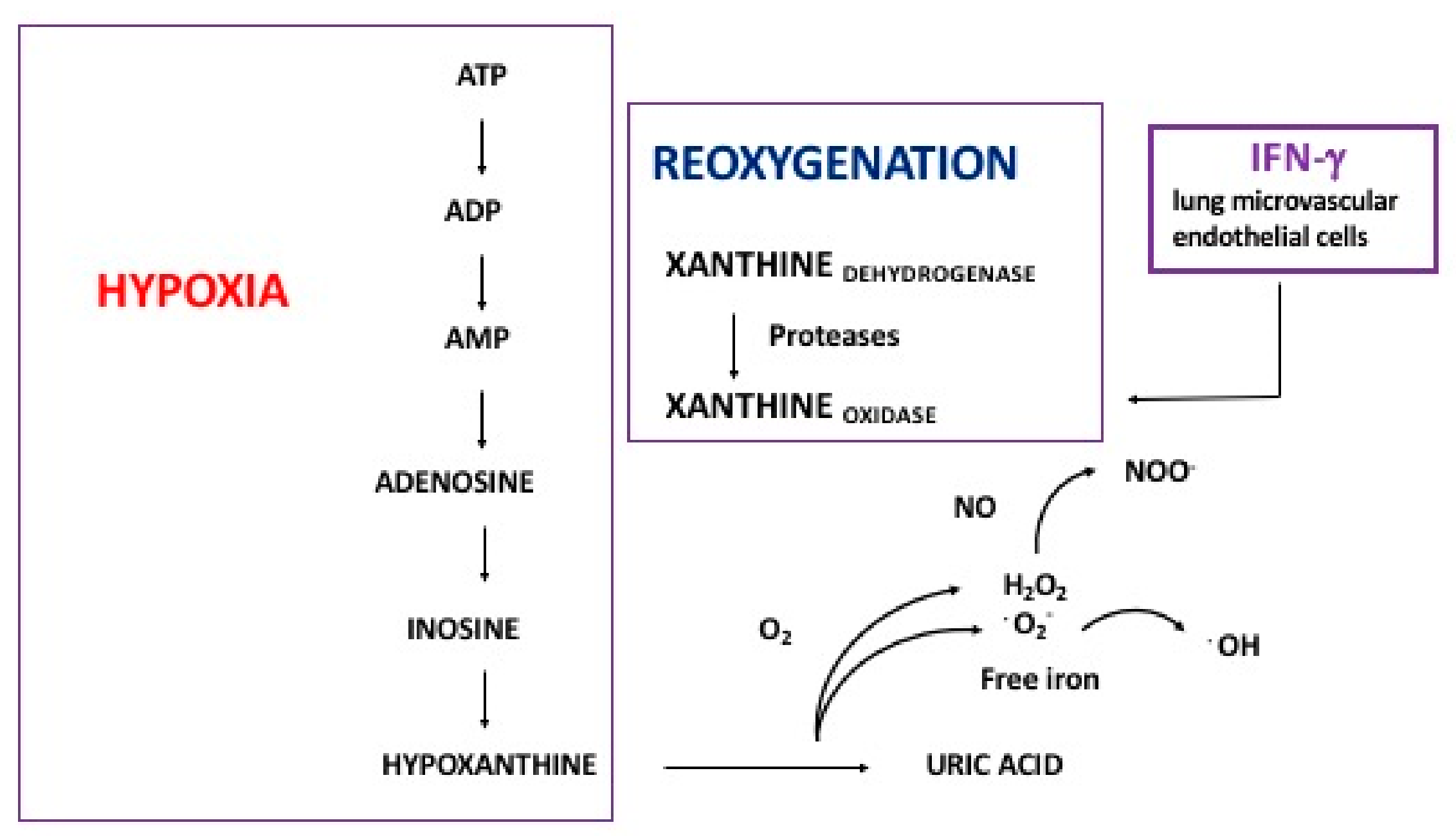
2.2. Hyperoxia and Oxidative Stress
2.3. Vascuologenesis
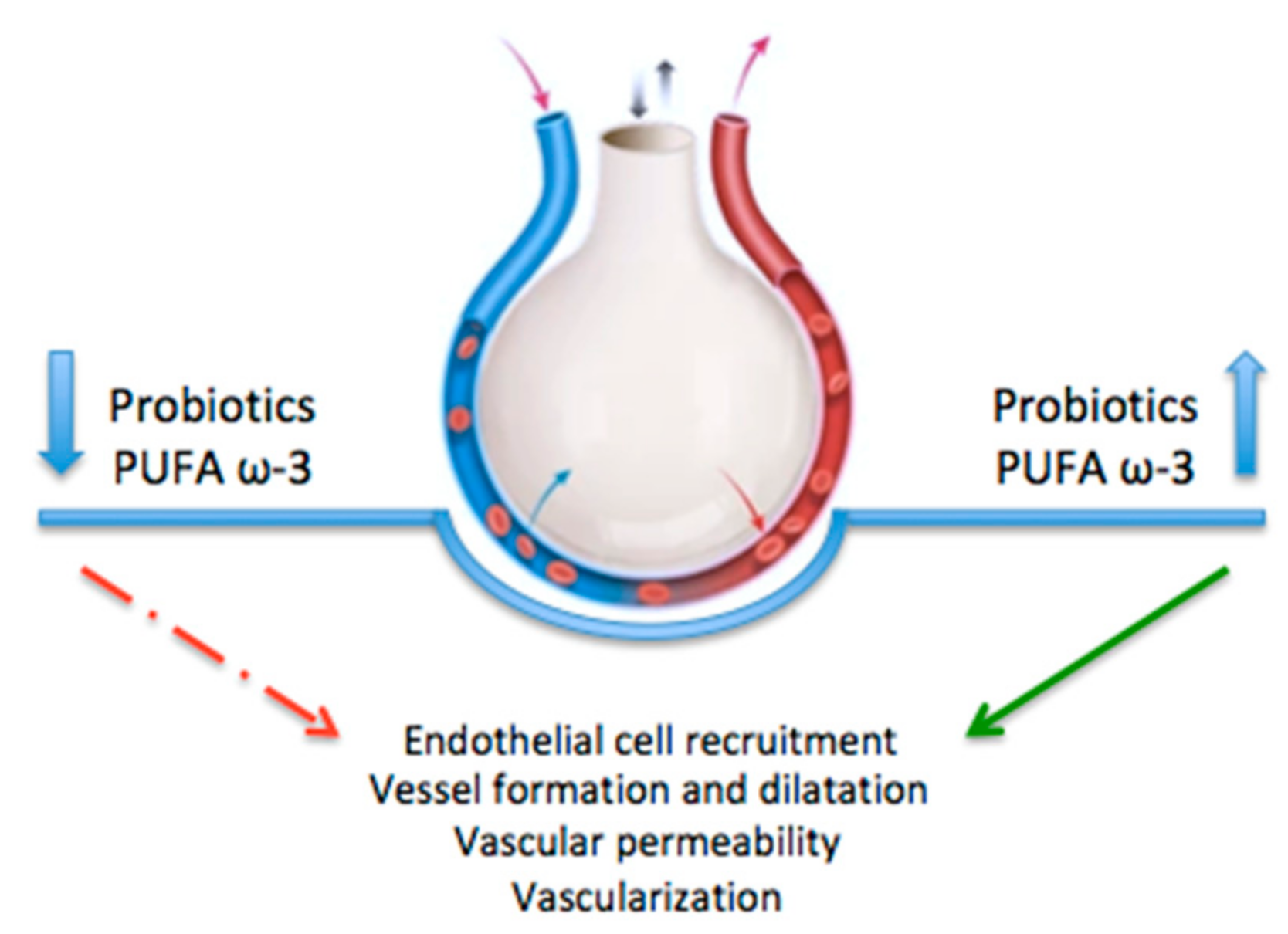
3. Vascular Endothelial Growth Factor
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Naeem, A.; Ahmed, I.; Silveyra, P. Bronchopulmonary Dysplasia: An Update on Experimental Therapeutics. Eur. Med. J. (Chelmsf. Engl.) 2019, 4, 20–29. [Google Scholar] [CrossRef]
- Holzfurtner, L.; Shahzad, T.; Dong, Y.; Rekers, L.; Selting, A.; Staude, B.; Lauer, T.; Schmidt, A.; Rivetti, S.; Zimmer, K.-P.; et al. When Inflammation Meets Lung Development-an Update on the Pathogenesis of Bronchopulmonary Dysplasia. Mol. Cell Pediatr. 2022, 9, 7. [Google Scholar] [CrossRef]
- Gilfillan, M.; Bhandari, A.; Bhandari, V. Diagnosis and Management of Bronchopulmonary Dysplasia. BMJ 2021, 375, n1974. [Google Scholar] [CrossRef]
- Wang, S.-H.; Tsao, P.-N. Phenotypes of Bronchopulmonary Dysplasia. Int. J. Mol. Sci. 2020, 21, E6112. [Google Scholar] [CrossRef]
- Jensen, E.A.; Schmidt, B. Epidemiology of Bronchopulmonary Dysplasia. Birth Defects Res. A Clin. Mol. Teratol. 2014, 100, 145–157. [Google Scholar] [CrossRef]
- Kalikkot Thekkeveedu, R.; Guaman, M.C.; Shivanna, B. Bronchopulmonary Dysplasia: A Review of Pathogenesis and Pathophysiology. Respir. Med. 2017, 132, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Shastri, S.; Sharma, P. Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clin. Med. Insights Pediatr. 2016, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Terrin, G.; Di Chiara, M.; Boscarino, G.; Metrangolo, V.; Faccioli, F.; Onestà, E.; Giancotti, A.; Di Donato, V.; Cardilli, V.; De Curtis, M. Morbidity Associated with Patent Ductus Arteriosus in Preterm Newborns: A Retrospective Case-Control Study. Ital. J. Pediatr. 2021, 47, 9. [Google Scholar] [CrossRef]
- Boscarino, G.; Conti, M.G.; De Luca, F.; Di Chiara, M.; Deli, G.; Bianchi, M.; Favata, P.; Cardilli, V.; Di Nardo, G.; Parisi, P.; et al. Intravenous Lipid Emulsions Affect Respiratory Outcome in Preterm Newborn: A Case-Control Study. Nutrients 2021, 13, 1243. [Google Scholar] [CrossRef]
- Lapcharoensap, W.; Kan, P.; Powers, R.J.; Shaw, G.M.; Stevenson, D.K.; Gould, J.B.; Wirtschafter, D.D.; Lee, H.C. The Relationship of Nosocomial Infection Reduction to Changes in Neonatal Intensive Care Unit Rates of Bronchopulmonary Dysplasia. J. Pediatr. 2017, 180, 105–109.e1. [Google Scholar] [CrossRef]
- Ryan, S.W.; Nycyk, J.; Shaw, B.N. Prediction of Chronic Neonatal Lung Disease on Day 4 of Life. Eur. J. Pediatr. 1996, 155, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Thébaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary Dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef]
- Laughon, M.; Allred, E.N.; Bose, C.; O’Shea, T.M.; Van Marter, L.J.; Ehrenkranz, R.A.; Leviton, A.; ELGAN Study Investigators. Patterns of Respiratory Disease during the First 2 Postnatal Weeks in Extremely Premature Infants. Pediatrics 2009, 123, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Siffel, C.; Kistler, K.D.; Lewis, J.F.M.; Sarda, S.P. Global Incidence of Bronchopulmonary Dysplasia among Extremely Preterm Infants: A Systematic Literature Review. J. Matern. Fetal Neonatal Med. 2021, 34, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Laughon, M.M.; Langer, J.C.; Bose, C.L.; Smith, P.B.; Ambalavanan, N.; Kennedy, K.A.; Stoll, B.J.; Buchter, S.; Laptook, A.R.; Ehrenkranz, R.A.; et al. Prediction of Bronchopulmonary Dysplasia by Postnatal Age in Extremely Premature Infants. Am. J. Respir. Crit. Care Med. 2011, 183, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Lianou, L.; Petropoulou, C.; Lipsou, N.; Bouza, H. Difference in Mortality and Morbidity Between Extremely and Very Low Birth Weight Neonates. Neonatal Netw. 2022, 41, 257–262. [Google Scholar] [CrossRef]
- Maniscalco, W.M.; Watkins, R.H.; D’Angio, C.T.; Ryan, R.M. Hyperoxic Injury Decreases Alveolar Epithelial Cell Expression of Vascular Endothelial Growth Factor (VEGF) in Neonatal Rabbit Lung. Am. J. Respir. Cell Mol. Biol. 1997, 16, 557–567. [Google Scholar] [CrossRef]
- Marti, H.H.; Risau, W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc. Natl. Acad. Sci. USA 1998, 95, 15809–15814. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Mackman, N.; Moons, L.; Luther, T.; Gressens, P.; Van Vlaenderen, I.; Demunck, H.; Kasper, M.; Breier, G.; Evrard, P.; et al. Role of tissue factor in embryonic blood vessel development. Nature 1996, 383, 73–75. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Vandivier, R.W.; Tuder, R.M. Vascular endothelial growth factor in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L209–L221. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Brown, K.R.; England, K.M.; Goss, K.L.; Snyder, J.M.; Acarregui, M.J. VEGF induces airway epithelial cell proliferation in human fetal lung in vitro. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L1001–L1010. [Google Scholar] [CrossRef]
- Compernolle, V.; Brusselmans, K.; Acker, T.; Hoet, P.; Tjwa, M.; Beck, H.; Plaisance, S.; Dor, Y.; Keshet, E.; Lupu, F.; et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 2002, 8, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.J.; Pryhuber, G.S.; Huyck, H.; Watkins, R.H.; Metlay, L.A.; Maniscalco, W.M. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 164, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Di Pietro, G.M.; Esposito, S. Bronchopulmonary Dysplasia: Clinical Aspects and Preventive and Therapeutic Strategies. J. Transl. Med. 2018, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Czerska, M.; Mikołajewska, K.; Zieliński, M.; Gromadzińska, J.; Wąsowicz, W. Today’s Oxidative Stress Markers. Med. Pr. 2015, 66, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.H. Mechanisms of Lung Injury and Bronchopulmonary Dysplasia. Am. J. Perinatol. 2016, 33, 1076–1078. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.H.; Romero, R.; Jun, J.K.; Park, K.H.; Park, J.D.; Ghezzi, F.; Kim, B.I. Amniotic Fluid Cytokines (Interleukin-6, Tumor Necrosis Factor-Alpha, Interleukin-1 Beta, and Interleukin-8) and the Risk for the Development of Bronchopulmonary Dysplasia. Am. J. Obstet. Gynecol. 1997, 177, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Rocha, G.; Proença, E.; Guedes, A.; Carvalho, C.; Areias, A.; Ramos, J.P.; Rodrigues, T.; Guimarães, H. Cord Blood Levels of IL-6, IL-8 and IL-10 May Be Early Predictors of Bronchopulmonary Dysplasia in Preterm Newborns Small for Gestational Age. Dis. Markers 2012, 33, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, C.; Köksal, N.; Özkan, H.; Dorum, B.A.; Bağcı, O. Low Serum IGF-1 and Increased Cytokine Levels in Tracheal Aspirate Samples Are Associated with Bronchopulmonary Dysplasia. Turk. J. Pediatr. 2017, 59, 122–129. [Google Scholar] [CrossRef]
- Pierce, M.R.; Bancalari, E. The Role of Inflammation in the Pathogenesis of Bronchopulmonary Dysplasia. Pediatr. Pulmonol. 1995, 19, 371–378. [Google Scholar] [CrossRef]
- Bry, K.; Lappalainen, U. Pathogenesis of Bronchopulmonary Dysplasia: The Role of Interleukin 1beta in the Regulation of Inflammation-Mediated Pulmonary Retinoic Acid Pathways in Transgenic Mice. Semin. Perinatol. 2006, 30, 121–128. [Google Scholar] [CrossRef]
- Leroy, S.; Caumette, E.; Waddington, C.; Hébert, A.; Brant, R.; Lavoie, P.M. A Time-Based Analysis of Inflammation in Infants at Risk of Bronchopulmonary Dysplasia. J. Pediatr. 2018, 192, 60–65.e1. [Google Scholar] [CrossRef]
- Oncel, M.Y.; Yurttutan, S.; Alyamac Dizdar, E.; Gokce, I.K.; Gonul, I.I.; Topal, T.; Canpolat, F.E.; Dilmen, U. Beneficial Effect of Etanercept on Hyperoxic Lung Injury Model in Neonatal Rats. J. Investig. Surg. 2016, 29, 1–5. [Google Scholar] [CrossRef]
- Mao, X.; Qiu, J.; Zhao, L.; Xu, J.; Yin, J.; Yang, Y.; Zhang, M.; Cheng, R. Vitamin D and IL-10 Deficiency in Preterm Neonates With Bronchopulmonary Dysplasia. Front. Pediatr. 2018, 6, 246. [Google Scholar] [CrossRef]
- Balany, J.; Bhandari, V. Understanding the Impact of Infection, Inflammation, and Their Persistence in the Pathogenesis of Bronchopulmonary Dysplasia. Front. Med. (Lausanne) 2015, 2, 90. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.H.; Costeloe, K.; Klein, N.J.; Rees, H.; McIntosh, N.; Keeling, J.W.; MacDonald, T.T. Mucosal Tumor Necrosis Factor-α Production and Extensive Disruption of Sulfated Glycosaminoglycans Begin within Hours of Birth in Neonatal Respiratory Distress Syndrome. Pediatr. Res. 1996, 40, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Speer, C.P. Inflammation and Bronchopulmonary Dysplasia: A Continuing Story. Semin. Fetal Neonatal Med. 2006, 11, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Yu, J.; Liu, D.; Tan, Q.; He, Y. The Airway Microbiome and Metabolome in Preterm Infants: Potential Biomarkers of Bronchopulmonary Dysplasia. Front. Pediatr. 2022, 10, 862157. [Google Scholar] [CrossRef]
- Heydarian, M.; Schulz, C.; Stoeger, T.; Hilgendorff, A. Association of Immune Cell Recruitment and BPD Development. Mol. Cell Pediatr. 2022, 9, 16. [Google Scholar] [CrossRef]
- Rogers, L.K.; Cismowski, M.J. Oxidative Stress in the Lung—The Essential Paradox. Curr. Opin. Toxicol. 2018, 7, 37–43. [Google Scholar] [CrossRef]
- Perrone, S.; Negro, S.; Tataranno, M.L.; Buonocore, G. Oxidative Stress and Antioxidant Strategies in Newborns. J. Matern. Fetal Neonatal Med. 2010, 23 (Suppl. 3), 63–65. [Google Scholar] [CrossRef] [PubMed]
- Dumpa, V.; Bhandari, V. Non-Invasive Ventilatory Strategies to Decrease Bronchopulmonary Dysplasia-Where Are We in 2021? Children 2021, 8, 132. [Google Scholar] [CrossRef]
- Tang, X. Interleukin-33 (IL-33) Increases Hyperoxia-Induced Bronchopulmonary Dysplasia in Newborn Mice by Regulation of Inflammatory Mediators. Med. Sci. Monit. 2018, 24, 6717–6728. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dong, W. Oxidative Stress and Bronchopulmonary Dysplasia. Gene 2018, 678, 177–183. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Bose, C.L.; Dammann, C.E.L.; Laughon, M.M. Bronchopulmonary Dysplasia and Inflammatory Biomarkers in the Premature Neonate. Arch. Dis. Child. Fetal Neonatal Ed. 2008, 93, F455–F461. [Google Scholar] [CrossRef]
- Park, H.S.; Kim, S.R.; Lee, Y.C. Impact of Oxidative Stress on Lung Diseases. Respirology 2009, 14, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-W.; Byzova, T.V. Oxidative Stress in Angiogenesis and Vascular Disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef]
- Rossino, M.G.; Lulli, M.; Amato, R.; Cammalleri, M.; Monte, M.D.; Casini, G. Oxidative Stress Induces a VEGF Autocrine Loop in the Retina: Relevance for Diabetic Retinopathy. Cells 2020, 9, 1452. [Google Scholar] [CrossRef]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular Endothelial Growth Factor (VEGF) and Its Receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef]
- Stevens, A.; Soden, J.; Brenchley, P.E.; Ralph, S.; Ray, D.W. Haplotype Analysis of the Polymorphic Human Vascular Endothelial Growth Factor Gene Promoter. Cancer Res. 2003, 63, 812–816. [Google Scholar] [PubMed]
- Narayanan, M.; Owers-Bradley, J.; Beardsmore, C.S.; Mada, M.; Ball, I.; Garipov, R.; Panesar, K.S.; Kuehni, C.E.; Spycher, B.D.; Williams, S.E.; et al. Alveolarization continues during childhood and adolescence: New evidence from helium-3 magnetic resonance. Am. J. Respir. Crit. Care Med. 2012, 185, 186–191. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, Y.; Zhang, X.; Li, X.; Liu, J. Association of VEGF Gene Polymorphisms with Susceptibility to Diabetic Retinopathy: A Systematic Review and Meta-Analysis. Horm. Metab. Res. 2020, 52, 264–279. [Google Scholar] [CrossRef]
- Galambos, C.; Sims-Lucas, S.; Abman, S.H. Three-dimensional reconstruction identifies misaligned pulmonary veins as intrapulmonary shunt vessels in alveolar capillary dysplasia. J. Pediatr. 2014, 164, 192–195. [Google Scholar] [CrossRef]
- Rodrigues, P.; Furriol, J.; Tormo, E.; Ballester, S.; Lluch, A.; Eroles, P. The Single-Nucleotide Polymorphisms +936 C/T VEGF and −710 C/T VEGFR1 Are Associated with Breast Cancer Protection in a Spanish Population. Breast Cancer Res. Treat. 2012, 133, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Northway, W.H., Jr.; Rosan, R.C.; Porter, D.Y. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N. Engl. J. Med. 1967, 276, 357–368. [Google Scholar] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Estes, M.L.; Mund, J.A.; Ingram, D.A.; Case, J. Identification of endothelial cells and progenitor cell subsets in human peripheral blood. Curr. Protoc. Cytom. 2010, 52, 9–33. [Google Scholar] [CrossRef]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular Endothelial Growth Factor Receptor-2: Structure, Function, Intracellular Signalling and Therapeutic Inhibition. Cell Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Popova, A.P.; Bozyk, P.D.; Bentley, J.K.; Linn, M.J.; Goldsmith, A.M.; Schumacher, R.E.; Weiner, G.M.; Filbrun, A.G.; Hershenson, M.B. Isolation of tracheal aspirate mesenchymal stromal cells predicts bronchopulmonary dysplasia. Pediatrics 2010, 126, e1127–e1133. [Google Scholar] [CrossRef] [PubMed]
- Myint, M.Z.Z.; Jia, J.; Adlat, S.; Oo, Z.M.; Htoo, H.; Hayel, F.; Chen, Y.; Bah, F.B.; Sah, R.K.; Bahadar, N.; et al. Effect of Low VEGF on Lung Development and Function. Transgenic Res. 2021, 30, 35–50. [Google Scholar] [CrossRef]
- Popova, A.P.; Bozyk, P.D.; Goldsmith, A.M.; Linn, M.J.; Lei, J.; Bentley, J.K.; Hershenson, M.B. Autocrine production of TGF-beta1 promotes myofibroblastic differentiation of neonatal lung mesenchymal stem cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L735–L743. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Rossiter, H.B.; Wagner, P.D.; Breen, E.C. Lung-Targeted VEGF Inactivation Leads to an Emphysema Phenotype in Mice. J. Appl. Physiol. (1985) 2004, 97, 1559–1566; discussion 1549. [Google Scholar] [CrossRef]
- Wang, H.; St Julien, K.R.; Stevenson, D.K.; Hoffmann, T.J.; Witte, J.S.; Lazzeroni, L.C.; Krasnow, M.A.; Quaintance, C.C.; Oehlert, J.W.; Jelliffe-Pawlowski, L.L.; et al. A genome-wide association study (gwas) for bronchopulmonary dysplasia. Pediatrics 2013, 132, 290–297. [Google Scholar] [CrossRef]
- Lassus, P.; Ristimäki, A.; Ylikorkala, O.; Viinikka, L.; Andersson, S. Vascular Endothelial Growth Factor in Human Preterm Lung. Am. J. Respir. Crit. Care Med. 1999, 159, 1429–1433. [Google Scholar] [CrossRef]
- Mariduena, J.; Ramagopal, M.; Hiatt, M.; Chandra, S.; Laumbach, R.; Hegyi, T. Vascular Endothelial Growth Factor Levels and Bronchopulmonary Dysplasia in Preterm Infants. J. Matern. Fetal Neonatal Med. 2022, 35, 1517–1522. [Google Scholar] [CrossRef]
- Grubor, B.; Meyerholz, D.K.; Lazic, T.; DeMacedo, M.M.; Derscheid, R.J.; Hostetter, J.M.; Gallup, J.M.; DeMartini, J.C.; Ackermann, M.R. Regulation of surfactant protein and defensin mRNA expression in cultured ovine type II pneumocytes by all-trans retinoic acid and VEGF. Int. J. Exp. Pathol. 2006, 87, 393–403. [Google Scholar] [CrossRef]
- Le Cras, T.D.; Markham, N.E.; Tuder, R.M.; Voelkel, N.F.; Abman, S.H. Treatment of Newborn Rats with a VEGF Receptor Inhibitor Causes Pulmonary Hypertension and Abnormal Lung Structure. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L555–L562. [Google Scholar] [CrossRef]
- Tang, J.-R.; Markham, N.E.; Lin, Y.-J.; McMurtry, I.F.; Maxey, A.; Kinsella, J.P.; Abman, S.H. Inhaled Nitric Oxide Attenuates Pulmonary Hypertension and Improves Lung Growth in Infant Rats after Neonatal Treatment with a VEGF Receptor Inhibitor. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L344–L351. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-R.; Seedorf, G.; Balasubramaniam, V.; Maxey, A.; Markham, N.; Abman, S.H. Early Inhaled Nitric Oxide Treatment Decreases Apoptosis of Endothelial Cells in Neonatal Rat Lungs after Vascular Endothelial Growth Factor Inhibition. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1271–L1280. [Google Scholar] [CrossRef]
- Michael, Z.; Spyropoulos, F.; Ghanta, S.; Christou, H. Bronchopulmonary Dysplasia: An Update of Current Pharmacologic Therapies and New Approaches. Clin. Med. Insights Pediatr. 2018, 12, 1179556518817322. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.-H.; Li, J.; Snyder, M.; Shaw, G.M.; O’Brodovich, H.M. The Genetic Predisposition to Bronchopulmonary Dysplasia. Curr. Opin. Pediatr. 2016, 28, 318–323. [Google Scholar] [CrossRef]
- Kwinta, P.; Bik-Multanowski, M.; Mitkowska, Z.; Tomasik, T.; Legutko, M.; Pietrzyk, J.J. Genetic Risk Factors of Bronchopulmonary Dysplasia. Pediatr. Res. 2008, 64, 682–688. [Google Scholar] [CrossRef]
- Filonzi, L.; Perrone, S.; Tataranno, M.L.; Magnani, C.; Dadomo, H.; Bottoni, A.; Vaghi, M.; Nonnis Marzano, F. Molecular Polymorphisms of Vascular Endothelial Growth Factor Gene and Bronchopulmonary Dysplasia in Very Low Birth Weight Infants. Dis. Markers 2022, 2022, 2793846. [Google Scholar] [CrossRef]
- Poggi, C.; Giusti, B.; Gozzini, E.; Sereni, A.; Romagnuolo, I.; Kura, A.; Pasquini, E.; Abbate, R.; Dani, C. Genetic Contributions to the Development of Complications in Preterm Newborns. PLoS ONE 2015, 10, e0131741. [Google Scholar] [CrossRef]
- Pan, J.; Zhan, C.; Yuan, T.; Wang, W.; Shen, Y.; Sun, Y.; Wu, T.; Gu, W.; Chen, L.; Yu, H. Effects and molecular mechanisms of intrauterine infection/inflammation on lung development. Respir. Res. 2018, 19, 93. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Cai, C.L.; Kumar, D.; Cai, F.; D’Souza, A.; Fordjour, L.; Ahmad, T.; Valencia, G.B.; Aranda, J.V.; Beharry, K.D. Benefits of pre-, pro- and Syn-biotics for lung angiogenesis in malnutritional rats exposed to intermittent hypoxia. Am. J. Transl. Res. 2014, 6, 59–470. [Google Scholar]
- Zhong, Y.; Catheline, D.; Houeijeh, A.; Sharma, D.; Du, L.; Besengez, C.; Deruelle, P.; Legrand, P.; Storme, L. Maternal omega-3 PUFA supplementation prevents hyperoxia-induced pulmonary hypertension in the offspring. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L116–L132. [Google Scholar] [CrossRef]
- Manti, S.; Galdo, F.; Parisi, G.F.; Napolitano, M.; Decimo, F.; Leonardi, S.; Miraglia Del Giudice, M. Long-term effects of bronchopulmonary dysplasia on lung function: A pilot study in preschool children’s cohort. J. Asthma. 2021, 58, 1186–1193. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrone, S.; Manti, S.; Buttarelli, L.; Petrolini, C.; Boscarino, G.; Filonzi, L.; Gitto, E.; Esposito, S.M.R.; Nonnis Marzano, F. Vascular Endothelial Growth Factor as Molecular Target for Bronchopulmonary Dysplasia Prevention in Very Low Birth Weight Infants. Int. J. Mol. Sci. 2023, 24, 2729. https://doi.org/10.3390/ijms24032729
Perrone S, Manti S, Buttarelli L, Petrolini C, Boscarino G, Filonzi L, Gitto E, Esposito SMR, Nonnis Marzano F. Vascular Endothelial Growth Factor as Molecular Target for Bronchopulmonary Dysplasia Prevention in Very Low Birth Weight Infants. International Journal of Molecular Sciences. 2023; 24(3):2729. https://doi.org/10.3390/ijms24032729
Chicago/Turabian StylePerrone, Serafina, Sara Manti, Luca Buttarelli, Chiara Petrolini, Giovanni Boscarino, Laura Filonzi, Eloisa Gitto, Susanna Maria Roberta Esposito, and Francesco Nonnis Marzano. 2023. "Vascular Endothelial Growth Factor as Molecular Target for Bronchopulmonary Dysplasia Prevention in Very Low Birth Weight Infants" International Journal of Molecular Sciences 24, no. 3: 2729. https://doi.org/10.3390/ijms24032729
APA StylePerrone, S., Manti, S., Buttarelli, L., Petrolini, C., Boscarino, G., Filonzi, L., Gitto, E., Esposito, S. M. R., & Nonnis Marzano, F. (2023). Vascular Endothelial Growth Factor as Molecular Target for Bronchopulmonary Dysplasia Prevention in Very Low Birth Weight Infants. International Journal of Molecular Sciences, 24(3), 2729. https://doi.org/10.3390/ijms24032729