


Leading Edge: Intratumor Delivery of Monoclonal Antibodies for the Treatment of Solid Tumors

 , , , ,
, , , ,  , , ,
, , ,
Abstract
1. Therapeutic Antibodies: Beyond Conventional Monoclonal Antibodies (mAbs)
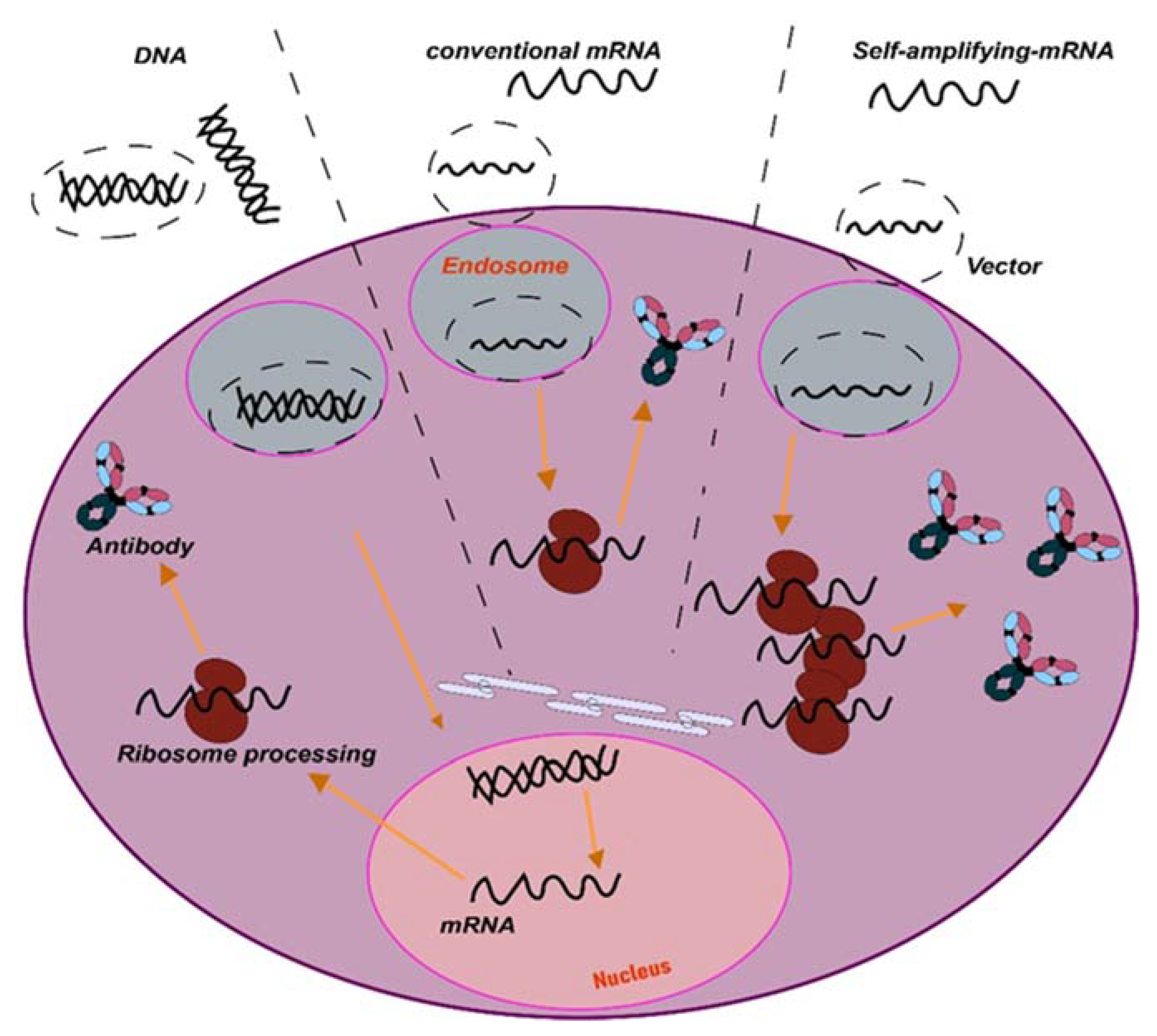
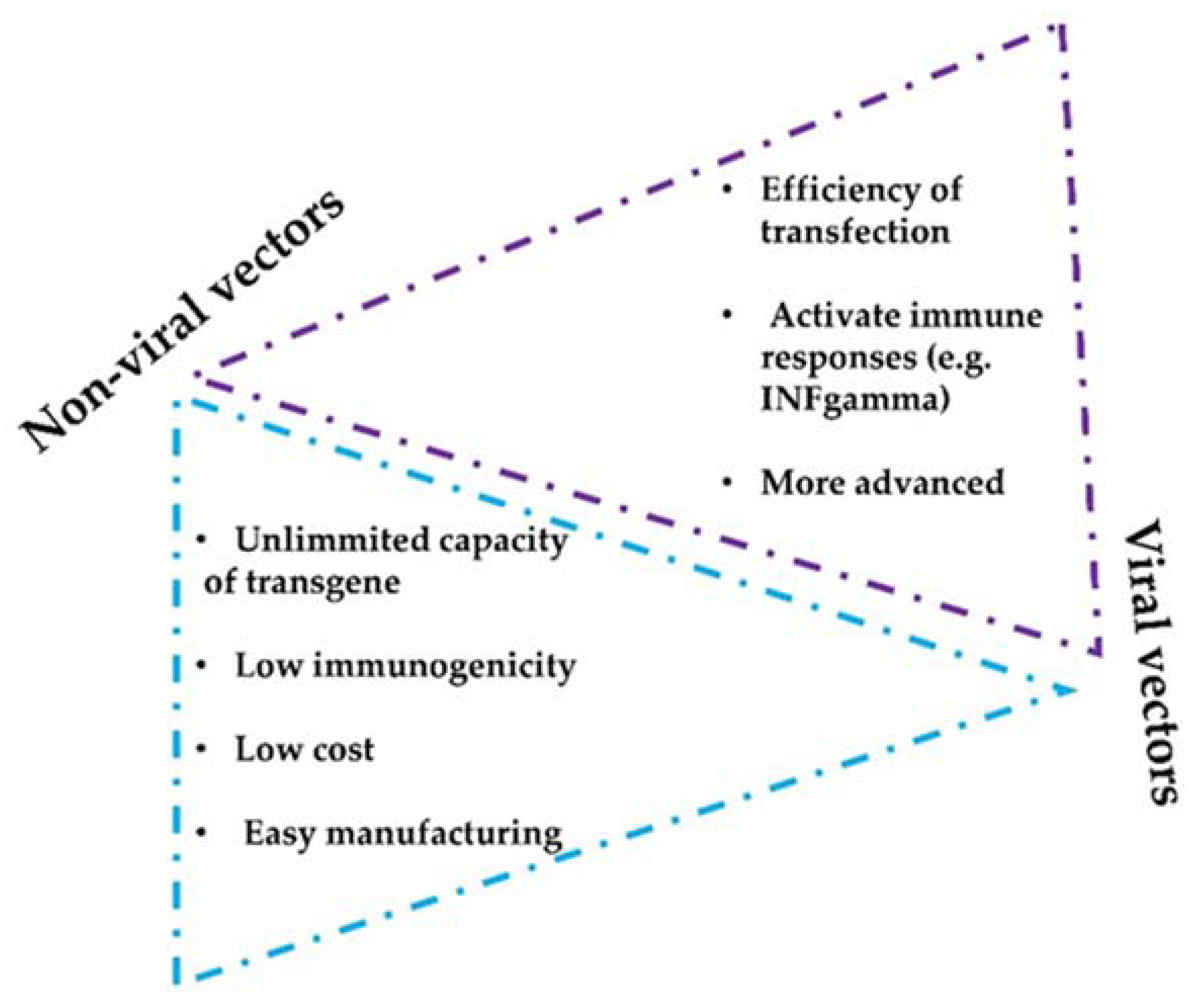
2. In Vivo mAb Gene Delivery Systems
3. Routes of mAbs Administration
4. Intratumor mAbs Delivery in Solid Tumors
5. Preclinical Non-Viral Vectors for mAbs Intratumor Delivery
5.1. Nanoparticles and Lipid Vesicles
5.2. Microneedle Delivery Platforms
5.3. Hydrogels as Delivery Vehicles
5.4. HSC−Platelet−Anti-PD-1 Assembly
5.5. Intratumor Plasmid DNA (pDNA) Electroporation
5.6. Antigen Peptides Conjugated on mAbs
6. Intratumor Delivery of mAbs with Viral Vectors in Preclinical Models
6.1. Oncolytic Viruses for the Treatment of Solid Tumors
6.2. Semliki Forest Virus (SFV)
6.3. Adeno-Associated Viral Vectors
6.4. Adenovirus Vectors
7. Intratumor mAbs Delivery under Clinical Development
8. Selected Clinical Trials According to Therapy Types
8.1. Clinical Trials with Administration of Conventional Monoclonal Antibodies
8.2. Clinical Trials with Oncolytic Viruses
8.3. Clinical Trials with Non-Viral Lipid Nanoparticles
9. Key Notes and Conclusions
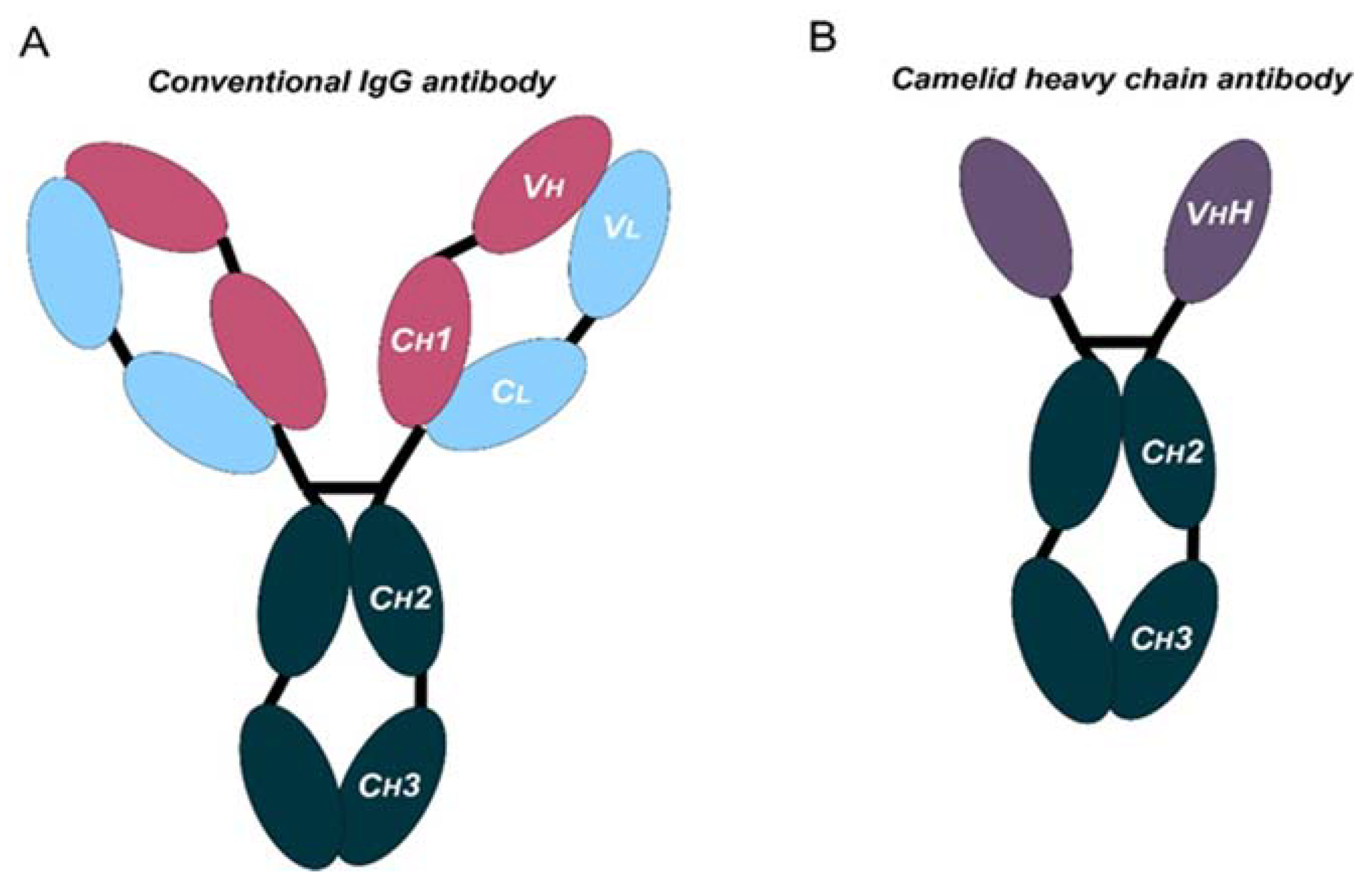
- The use of conventional mAbs is limited by their complex structure. Novel designs such as bispecific antibodies or nanobodies can be good alternatives.
- Direct in vivo delivery of synthetic nucleic acids encoding antibodies such as plasmid DNA or mRNA platforms represent new approaches for in vivo delivery of antibody-like biologics. These platforms have advantages such as rapid product development and simpler manufacturing processes.
- Intratumor delivery increases efficacy, local bioavailability, reduces toxicity, and improves the antitumor immune responses.
- Intratumor delivery can be combined with other systemic strategies and can be implemented with non-viral- and viral-based delivery methods, including nanoparticles or lipid vesicles.
- Virotherapies are promising approaches for cancer treatment and as delivery vehicles of mAbs, but may pose biosafety concerns, especially for the use of oncolytic viruses.
10. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Buss, N.A.; Henderson, S.J.; McFarlane, M.; Shenton, J.M.; de Haan, L. Monoclonal antibody therapeutics: History and future. Curr. Opin. Pharmacol. 2012, 12, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2021. mAbs 2021, 13, 1860476. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Goulet, D.R.; Atkins, W.M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2020, 109, 74–103. [Google Scholar] [CrossRef]
- Bocanegra, A.; Blanco, E.; Fernandez-Hinojal, G.; Arasanz, H.; Chocarro, L.; Zuazo, M.; Morente, P.; Vera, R.; Escors, D.; Kochan, G. PD-L1 in Systemic Immunity: Unraveling Its Contribution to PD-1/PD-L1 Blockade Immunotherapy. Int. J. Mol. Sci. 2020, 21, 5918. [Google Scholar] [CrossRef]
- Deshpande, R.P.; Sharma, S.; Watabe, K. The Confounders of Cancer Immunotherapy: Roles of Lifestyle, Metabolic Disorders and Sociological Factors. Cancers 2020, 12, 2983. [Google Scholar] [CrossRef]
- Edwards, C.J.; Sette, A.; Cox, C.; Di Fiore, B.; Wyre, C.; Sydoruk, D.; Yadin, D.; Hayes, P.; Stelter, S.; Bartlett, P.D.; et al. The multi-specific VH-based Humabody CB213 co-targets PD1 and LAG3 on T cells to promote anti-tumour activity. Br. J. Cancer 2022, 126, 1168–1177. [Google Scholar] [CrossRef]
- Chocarro, L.; Bocanegra, A.; Blanco, E.; Fernandez-Rubio, L.; Arasanz, H.; Echaide, M.; Garnica, M.; Ramos, P.; Pineiro-Hermida, S.; Vera, R.; et al. Cutting-Edge: Preclinical and Clinical Development of the First Approved Lag-3 Inhibitor. Cells 2022, 11, 2351. [Google Scholar] [CrossRef]
- Zuazo, M.; Arasanz, H.; Fernandez-Hinojal, G.; Garcia-Granda, M.J.; Gato, M.; Bocanegra, A.; Martinez, M.; Hernandez, B.; Teijeira, L.; Morilla, I.; et al. Functional systemic CD4 immunity is required for clinical responses to PD-L1/PD-1 blockade therapy. EMBO Mol. Med. 2019, 11, e10293. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- O’Day, S.J.; Maio, M.; Chiarion-Sileni, V.; Gajewski, T.F.; Pehamberger, H.; Bondarenko, I.N.; Queirolo, P.; Lundgren, L.; Mikhailov, S.; Roman, L.; et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: A multicenter single-arm phase II study. Ann. Oncol. 2010, 21, 1712–1717. [Google Scholar] [CrossRef]
- Castelli, M.S.; McGonigle, P.; Hornby, P.J. The pharmacology and therapeutic applications of monoclonal antibodies. Pharmacol. Res. Perspect. 2019, 7, e00535. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef]
- Verhaar, E.R.; Woodham, A.W.; Ploegh, H.L. Nanobodies in cancer. Semin. Immunol. 2021, 52, 101425. [Google Scholar] [CrossRef]
- Yang, E.Y.; Shah, K. Nanobodies: Next Generation of Cancer Diagnostics and Therapeutics. Front. Oncol. 2020, 10, 1182. [Google Scholar] [CrossRef]
- Rissiek, B.; Koch-Nolte, F.; Magnus, T. Nanobodies as modulators of inflammation: Potential applications for acute brain injury. Front. Cell. Neurosci. 2014, 8, 344. [Google Scholar] [CrossRef]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef]
- Panikar, S.S.; Banu, N.; Haramati, J.; Del Toro-Arreola, S.; Riera Leal, A.; Salas, P. Nanobodies as efficient drug-carriers: Progress and trends in chemotherapy. J. Control. Release 2021, 334, 389–412. [Google Scholar] [CrossRef] [PubMed]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Raybould, M.I.J.; Marks, C.; Krawczyk, K.; Taddese, B.; Nowak, J.; Lewis, A.P.; Bujotzek, A.; Shi, J.; Deane, C.M. Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. USA 2019, 116, 4025–4030. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, I.; Bott, S.W.; Patel, A.S.; Wolf, C.G.; Hospodar, A.R.; Sampathkumar, S.; Shrank, W.H. Pricing of monoclonal antibody therapies: Higher if used for cancer? Am. J. Manag. Care 2018, 24, 109–112. [Google Scholar] [PubMed]
- Garnica, M.; Aiello, A.; Ligotti, M.E.; Accardi, G.; Arasanz, H.; Bocanegra, A.; Blanco, E.; Calabro, A.; Chocarro, L.; Echaide, M.; et al. How Can We Improve the Vaccination Response in Older People? Part II: Targeting Immunosenescence of Adaptive Immunity Cells. Int. J. Mol. Sci. 2022, 23, 9797. [Google Scholar] [CrossRef]
- Echaide, M.; Labiano, I.; Delgado, M.; Fernandez de Lascoiti, A.; Ochoa, P.; Garnica, M.; Ramos, P.; Chocarro, L.; Fernandez, L.; Arasanz, H.; et al. Immune Profiling Uncovers Memory T-Cell Responses with a Th17 Signature in Cancer Patients with Previous SARS-CoV-2 Infection Followed by mRNA Vaccination. Cancers 2022, 14, 4464. [Google Scholar] [CrossRef]
- Williams, J.A. Vector Design for Improved DNA Vaccine Efficacy, Safety and Production. Vaccines 2013, 1, 225–249. [Google Scholar] [CrossRef]
- Shah, M.A.; Ali, Z.; Ahmad, R.; Qadri, I.; Fatima, K.; He, N. DNA Mediated Vaccines Delivery Through Nanoparticles. J. Nanosci. Nanotechnol. 2015, 15, 41–53. [Google Scholar] [CrossRef]
- Eygeris, Y.; Gupta, M.; Kim, J.; Sahay, G. Chemistry of Lipid Nanoparticles for RNA Delivery. Acc. Chem. Res. 2022, 55, 2–12. [Google Scholar] [CrossRef]
- Hollevoet, K.; De Vleeschauwer, S.; De Smidt, E.; Vermeire, G.; Geukens, N.; Declerck, P. Bridging the Clinical Gap for DNA-Based Antibody Therapy Through Translational Studies in Sheep. Hum. Gene Ther. 2019, 30, 1431–1443. [Google Scholar] [CrossRef]
- Hollevoet, K.; De Smidt, E.; Geukens, N.; Declerck, P. Prolonged in vivo expression and anti-tumor response of DNA-based anti-HER2 antibodies. Oncotarget 2018, 9, 13623–13636. [Google Scholar] [CrossRef]
- Andrews, C.D.; Luo, Y.; Sun, M.; Yu, J.; Goff, A.J.; Glass, P.J.; Padte, N.N.; Huang, Y.; Ho, D.D. In Vivo Production of Monoclonal Antibodies by Gene Transfer via Electroporation Protects against Lethal Influenza and Ebola Infections. Mol. Ther. Methods Clin. Dev. 2017, 7, 74–82. [Google Scholar] [CrossRef]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef]
- Ura, T.; Yamashita, A.; Mizuki, N.; Okuda, K.; Shimada, M. New vaccine production platforms used in developing SARS-CoV-2 vaccine candidates. Vaccine 2021, 39, 197–201. [Google Scholar] [CrossRef]
- Almazan, F.; Dediego, M.L.; Galan, C.; Escors, D.; Alvarez, E.; Ortego, J.; Sola, I.; Zuniga, S.; Alonso, S.; Moreno, J.L.; et al. Construction of a severe acute respiratory syndrome coronavirus infectious cDNA clone and a replicon to study coronavirus RNA synthesis. J. Virol. 2006, 80, 10900–10906. [Google Scholar] [CrossRef]
- Tenholder, M.F. Pulmonary infections in the immunocompromised host. Perspective on procedures. Chest 1988, 94, 676–678. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Arasanz, H.; Bocanegra, A.I.; Morilla, I.; Fernandez-Irigoyen, J.; Martinez-Aguillo, M.; Teijeira, L.; Garnica, M.; Blanco, E.; Chocarro, L.; Ausin, K.; et al. Circulating Low Density Neutrophils Are Associated with Resistance to First Line Anti-PD1/PDL1 Immunotherapy in Non-Small Cell Lung Cancer. Cancers 2022, 14, 3846. [Google Scholar] [CrossRef]
- Khan, Z.; Hammer, C.; Guardino, E.; Chandler, G.S.; Albert, M.L. Mechanisms of immune-related adverse events associated with immune checkpoint blockade: Using germline genetics to develop a personalized approach. Genome Med. 2019, 11, 39. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. New Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Darnell, E.P.; Mooradian, M.J.; Baruch, E.N.; Yilmaz, M.; Reynolds, K.L. Immune-Related Adverse Events (irAEs): Diagnosis, Management, and Clinical Pearls. Curr. Oncol. Rep. 2020, 22, 39. [Google Scholar] [CrossRef] [PubMed]
- Arasanz, H.; Zuazo, M.; Bocanegra, A.; Chocarro, L.; Blanco, E.; Martinez, M.; Morilla, I.; Fernandez, G.; Teijeira, L.; Morente, P.; et al. Hyperprogressive Disease: Main Features and Key Controversies. Int. J. Mol. Sci. 2021, 22, 3736. [Google Scholar] [CrossRef] [PubMed]
- Arasanz, H.; Zuazo, M.; Bocanegra, A.; Gato, M.; Martinez-Aguillo, M.; Morilla, I.; Fernandez, G.; Hernandez, B.; Lopez, P.; Alberdi, N.; et al. Early detection of hyperprogressive disease in non-small cell lung cancer by monitoring of systemic T cell dynamics. Cancers 2020, 12, 344. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Bittner, B.; Richter, W.; Schmidt, J. Subcutaneous Administration of Biotherapeutics: An Overview of Current Challenges and Opportunities. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2018, 32, 425–440. [Google Scholar] [CrossRef]
- Viola, M.; Sequeira, J.; Seica, R.; Veiga, F.; Serra, J.; Santos, A.C.; Ribeiro, A.J. Subcutaneous delivery of monoclonal antibodies: How do we get there? J. Control. Release 2018, 286, 301–314. [Google Scholar] [CrossRef]
- Champiat, S.; Tselikas, L.; Farhane, S.; Raoult, T.; Texier, M.; Lanoy, E.; Massard, C.; Robert, C.; Ammari, S.; De Baere, T.; et al. Intratumoral Immunotherapy: From Trial Design to Clinical Practice. Clin. Cancer Res. 2021, 27, 665–679. [Google Scholar] [CrossRef]
- Goldmacher, G.V.; Khilnani, A.D.; Andtbacka, R.H.I.; Luke, J.J.; Hodi, F.S.; Marabelle, A.; Harrington, K.; Perrone, A.; Tse, A.; Madoff, D.C.; et al. Response Criteria for Intratumoral Immunotherapy in Solid Tumors: ItRECIST. J. Clin. Oncol. 2020, 38, 2667–2676. [Google Scholar] [CrossRef]
- Margolin, K.; Morishima, C.; Velcheti, V.; Miller, J.S.; Lee, S.M.; Silk, A.W.; Holtan, S.G.; Lacroix, A.M.; Fling, S.P.; Kaiser, J.C.; et al. Phase I Trial of ALT-803, A Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 5552–5561. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reynies, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougouin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Sweeney, K.J.; Tetzlaff, M.T.; Vega, F.; Gillenwater, A.; Zuo, Z.; Gross, N.; Nagarajan, P.; Wargo, J.; Nelson, K.; Prieto, V.G.; et al. Tertiary lymphoid structures with overlapping histopathologic features of cutaneous marginal zone lymphoma during neoadjuvant cemiplimab therapy are associated with antitumor response. J. Cutan. Pathol. 2021, 48, 674–679. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Brody, J.D.; Ai, W.Z.; Czerwinski, D.K.; Torchia, J.A.; Levy, M.; Advani, R.H.; Kim, Y.H.; Hoppe, R.T.; Knox, S.J.; Shin, L.K.; et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: A phase I/II study. J. Clin. Oncol. 2010, 28, 4324–4332. [Google Scholar] [CrossRef]
- Hong, W.X.; Haebe, S.; Lee, A.S.; Westphalen, C.B.; Norton, J.A.; Jiang, W.; Levy, R. Intratumoral Immunotherapy for Early-stage Solid Tumors. Clin. Cancer Res. 2020, 26, 3091–3099. [Google Scholar] [CrossRef]
- Lockman, P.R.; Mumper, R.J.; Khan, M.A.; Allen, D.D. Nanoparticle technology for drug delivery across the blood-brain barrier. Drug Dev. Ind. Pharm. 2002, 28, 1–13. [Google Scholar] [CrossRef]
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019, 10, 3850. [Google Scholar] [CrossRef]
- Marabelle, A.; Andtbacka, R.; Harrington, K.; Melero, I.; Leidner, R.; de Baere, T.; Robert, C.; Ascierto, P.A.; Baurain, J.F.; Imperiale, M.; et al. Starting the fight in the tumor: Expert recommendations for the development of human intratumoral immunotherapy (HIT-IT). Ann. Oncol. 2018, 29, 2163–2174. [Google Scholar] [CrossRef]
- Tselikas, L.; de Baere, T.; Isoardo, T.; Susini, S.; Ser-Le Roux, K.; Polrot, M.; Adam, J.; Rouanne, M.; Zitvogel, L.; Moine, L.; et al. Pickering emulsions with ethiodized oil and nanoparticles for slow release of intratumoral anti-CTLA4 immune checkpoint antibodies. J. Immunother. Cancer 2020, 8, e000579. [Google Scholar] [CrossRef]
- Escors, D.; Breckpot, K. Lentiviral Vectors in Gene Therapy: Their Current Status and Future Potential. Arch. Immunol. Ther. Exp. 2010, 58, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Breckpot, K.; Escors, D.; Arce, F.; Lopes, L.; Karwacz, K.; Van Lint, S.; Keyaerts, M.; Collins, M. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J. Virol. 2010, 84, 5627–5636. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Allawadhi, P.; Khurana, A.; Singh, V.; Navik, U.; Pasumarthi, S.K.; Khurana, I.; Banothu, A.K.; Weiskirchen, R.; Bharani, K.K. Gene therapy: Comprehensive overview and therapeutic applications. Life Sci. 2022, 294, 120375. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.J.; Li, L.; Hwang, B.; Cadena, M.; Theus, A.S.; Finamore, T.A.; Bauser-Heaton, H.; Mahmoudi, M.; Roeder, R.K.; Serpooshan, V. Tissue engineered drug delivery vehicles: Methods to monitor and regulate the release behavior. J. Control. Release 2022, 349, 143–155. [Google Scholar] [CrossRef]
- Zhao, Y.; Bilal, M.; Qindeel, M.; Khan, M.I.; Dhama, K.; Iqbal, H.M.N. Nanotechnology-based immunotherapies to combat cancer metastasis. Mol. Biol. Rep. 2021, 48, 6563–6580. [Google Scholar] [CrossRef]
- Guevara, M.L.; Persano, S.; Persano, F. Lipid-Based Vectors for Therapeutic mRNA-Based Anti-Cancer Vaccines. Curr. Pharm. Des. 2019, 25, 1443–1454. [Google Scholar] [CrossRef]
- Karwacz, K.; Bricogne, C.; Macdonald, D.; Arce, F.; Bennett, C.L.; Collins, M.; Escors, D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8(+) T cells. EMBO Mol. Med. 2011, 3, 581–592. [Google Scholar] [CrossRef]
- Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Ibanez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, Y.; Cao, W.; Xia, F.; Liu, B.; Niu, J.; Alfranca, G.; Sun, X.; Ma, L.; de la Fuente, J.M.; et al. A tumor microenvironment responsive biodegradable CaCO(3)/MnO(2)- based nanoplatform for the enhanced photodynamic therapy and improved PD-L1 immunotherapy. Theranostics 2019, 9, 6867–6884. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, C.; Zhang, X.; Chen, G.; Hu, Q.; Li, H.; Wang, J.; Wen, D.; Zhang, Y.; Lu, Y.; et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol. 2019, 14, 89–97. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Wang, J.; Hu, Q.; Langworthy, B.; Ye, Y.; Sun, W.; Lin, J.; Wang, T.; Fine, J.; et al. PD-1 Blockade Cellular Vesicles for Cancer Immunotherapy. Adv. Mater. 2018, 30, e1707112. [Google Scholar] [CrossRef]
- Yu, Z.L.; Liu, J.Y.; Chen, G. Small extracellular vesicle PD-L1 in cancer: The knowns and unknowns. NPJ Precis. Oncol. 2022, 6, 42. [Google Scholar] [CrossRef]
- Aznar, M.A.; Planelles, L.; Perez-Olivares, M.; Molina, C.; Garasa, S.; Etxeberría, I.; Perez, G.; Rodriguez, I.; Bolaños, E.; Lopez-Casas, P.; et al. Immunotherapeutic effects of intratumoral nanoplexed poly I:C. J. Immunother. Cancer. 2019, 7, 116. [Google Scholar] [CrossRef]
- Lee, K.; Goudie, M.J.; Tebon, P.; Sun, W.; Luo, Z.; Lee, J.; Zhang, S.; Fetah, K.; Kim, H.J.; Xue, Y.; et al. Non-transdermal microneedles for advanced drug delivery. Adv. Drug Deliv. Rev. 2020, 165–166, 41–59. [Google Scholar] [CrossRef]
- Wang, C.; Ye, Y.; Hochu, G.M.; Sadeghifar, H.; Gu, Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett. 2016, 16, 2334–2340. [Google Scholar] [CrossRef]
- Ye, Y.; Wang, J.; Hu, Q.; Hochu, G.M.; Xin, H.; Wang, C.; Gu, Z. Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses through Delivery of Checkpoint Inhibitors. ACS Nano 2016, 10, 8956–8963. [Google Scholar] [CrossRef]
- Yan, D.; Sherman, J.H.; Keidar, M. Cold atmospheric plasma, a novel promising anti-cancer treatment modality. Oncotarget 2017, 8, 15977–15995. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, Y.; Li, Q.; Yu, C.; Chu, W. Natural Polymer-based Stimuli-responsive Hydrogels. Curr. Med. Chem. 2020, 27, 2631–2657. [Google Scholar] [CrossRef]
- Kahn, J.S.; Hu, Y.; Willner, I. Stimuli-Responsive DNA-Based Hydrogels: From Basic Principles to Applications. Acc. Chem. Res. 2017, 50, 680–690. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, Y.; Hedenqvist, M.S.; Chen, C.; Cai, C.; Li, H.; Liu, H.; Fu, J. Multifunctional conductive hydrogels and their applications as smart wearable devices. J. Mater. Chem. B 2021, 9, 2561–2583. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Yang, P.; Huang, P.; Zhang, C.; Kong, D.; Wang, W. Injectable polypeptide hydrogel-based co-delivery of vaccine and immune checkpoint inhibitors improves tumor immunotherapy. Theranostics 2019, 9, 2299–2314. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fang, M.; Zhang, J.; Wang, J.; Song, Y.; Shi, J.; Li, W.; Wu, G.; Ren, J.; Wang, Z.; et al. Hydrogel dual delivered celecoxib and anti-PD-1 synergistically improve antitumor immunity. Oncoimmunology 2016, 5, e1074374. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, H.; Zhou, D.; Chen, Z.; Gu, Z. Local and Targeted Delivery of Immune Checkpoint Blockade Therapeutics. Acc. Chem. Res. 2020, 53, 2521–2533. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, L.; Yshii, L.; Junius, S.; Geukens, N.; Liston, A.; Hollevoet, K.; Declerck, P. Intratumoral DNA-based delivery of checkpoint-inhibiting antibodies and interleukin 12 triggers T cell infiltration and anti-tumor response. Cancer Gene Ther. 2022, 29, 984–992. [Google Scholar] [CrossRef]
- Jacobs, L.; De Smidt, E.; Geukens, N.; Declerck, P.; Hollevoet, K. DNA-Based Delivery of Checkpoint Inhibitors in Muscle and Tumor Enables Long-Term Responses with Distinct Exposure. Mol. Ther. 2020, 28, 1068–1077. [Google Scholar] [CrossRef]
- Lee, E.J.; Jang, G.Y.; Lee, S.E.; Lee, J.W.; Han, H.D.; Park, Y.M.; Kang, T.H. A novel form of immunotherapy using antigen peptides conjugated on PD-L1 antibody. Immunol. Lett. 2021, 240, 137–148. [Google Scholar] [CrossRef]
- Moon, Y.; Shim, M.K.; Choi, J.; Yang, S.; Kim, J.; Yun, W.S.; Cho, H.; Park, J.Y.; Kim, Y.; Seong, J.K.; et al. Anti-PD-L1 peptide-conjugated prodrug nanoparticles for targeted cancer immunotherapy combining PD-L1 blockade with immunogenic cell death. Theranostics 2022, 12, 1999–2014. [Google Scholar] [CrossRef]
- Hollevoet, K.; Declerck, P.J. State of play and clinical prospects of antibody gene transfer. J. Transl. Med. 2017, 15, 131. [Google Scholar] [CrossRef]
- Collins, M.; Thrasher, A. Gene therapy: Progress and predictions. Proc. Biol. Sci. 2015, 282, 20143003. [Google Scholar] [CrossRef]
- Liechtenstein, T.; Perez-Janices, N.; Blanco-Luquin, I.; Schwarze, J.; Dufait, I.; Lanna, A.; De Ridder, M.; Guerrero-Setas, D.; Breckpot, K.; Escors, D. Anti-melanoma vaccines engineered to simultaneously modulate cytokine priming and silence PD-L1 characterized using ex vivo myeloid-derived suppressor cells as a readout of therapeutic efficacy. Oncoimmunology 2014, 3, e29178. [Google Scholar] [CrossRef]
- Alemany, R. Oncolytic Adenoviruses in Cancer Treatment. Biomedicines 2014, 2, 36–49. [Google Scholar] [CrossRef]
- Raja, J.; Ludwig, J.M.; Gettinger, S.N.; Schalper, K.A.; Kim, H.S. Oncolytic virus immunotherapy: Future prospects for oncology. J. Immunother. Cancer 2018, 6, 140. [Google Scholar] [CrossRef]
- de Leeuw, O.; Peeters, B. Complete nucleotide sequence of Newcastle disease virus: Evidence for the existence of a new genus within the subfamily Paramyxovirinae. J. Gen. Virol. 1999, 80 Pt 1, 131–136. [Google Scholar] [CrossRef]
- Schirrmacher, V.; van Gool, S.; Stuecker, W. Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy. Biomedicines 2019, 7, 66. [Google Scholar] [CrossRef]
- Vijayakumar, G.; McCroskery, S.; Palese, P. Engineering Newcastle Disease Virus as an Oncolytic Vector for Intratumoral Delivery of Immune Checkpoint Inhibitors and Immunocytokines. J. Virol. 2020, 94, e01677-19. [Google Scholar] [CrossRef]
- Zuo, S.; Wei, M.; Xu, T.; Kong, L.; He, B.; Wang, S.; Wang, S.; Wu, J.; Dong, J.; Wei, J. An engineered oncolytic vaccinia virus encoding a single-chain variable fragment against TIGIT induces effective antitumor immunity and synergizes with PD-1 or LAG-3 blockade. J. Immunother. Cancer 2021, 9, e002843. [Google Scholar] [CrossRef]
- Lin, C.; Ren, W.; Luo, Y.; Li, S.; Chang, Y.; Li, L.; Xiong, D.; Huang, X.; Xu, Z.; Yu, Z.; et al. Intratumoral Delivery of a PD-1-Blocking scFv Encoded in Oncolytic HSV-1 Promotes Antitumor Immunity and Synergizes with TIGIT Blockade. Cancer Immunol. Res. 2020, 8, 632–647. [Google Scholar] [CrossRef]
- Quetglas, J.I.; Ruiz-Guillen, M.; Aranda, A.; Casales, E.; Bezunartea, J.; Smerdou, C. Alphavirus vectors for cancer therapy. Virus Res. 2010, 153, 179–196. [Google Scholar] [CrossRef]
- Rodriguez-Madoz, J.R.; Prieto, J.; Smerdou, C. Semliki forest virus vectors engineered to express higher IL-12 levels induce efficient elimination of murine colon adenocarcinomas. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 12, 153–163. [Google Scholar] [CrossRef]
- Guan, M.; Rodriguez-Madoz, J.R.; Alzuguren, P.; Gomar, C.; Kramer, M.G.; Kochanek, S.; Prieto, J.; Smerdou, C.; Qian, C. Increased efficacy and safety in the treatment of experimental liver cancer with a novel adenovirus-alphavirus hybrid vector. Cancer Res. 2006, 66, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Briones, M.C.; Martisova, E.; Casales, E.; Silva-Pilipich, N.; Bunuales, M.; Galindo, J.; Mancheno, U.; Gorraiz, M.; Lasarte, J.J.; Kochan, G.; et al. Short-Term Local Expression of a PD-L1 Blocking Antibody from a Self-Replicating RNA Vector Induces Potent Antitumor Responses. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1892–1905. [Google Scholar] [CrossRef]
- Chikkanna-Gowda, C.P.; Sheahan, B.J.; Fleeton, M.N.; Atkins, G.J. Regression of mouse tumours and inhibition of metastases following administration of a Semliki Forest virus vector with enhanced expression of IL-12. Gene Ther. 2005, 12, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Quetglas, J.I.; Labiano, S.; Aznar, M.A.; Bolanos, E.; Azpilikueta, A.; Rodriguez, I.; Casales, E.; Sanchez-Paulete, A.R.; Segura, V.; Smerdou, C.; et al. Virotherapy with a Semliki Forest Virus-Based Vector Encoding IL12 Synergizes with PD-1/PD-L1 Blockade. Cancer Immunol. Res. 2015, 3, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Silva-Pilipich, N.; Lasarte-Cia, A.; Lozano, T.; Martin-Otal, C.; Lasarte, J.J.; Smerdou, C. Intratumoral electroporation of a self-amplifying RNA expressing IL-12 induces antitumor effects in mouse models of cancer. Mol. Ther. Nucleic Acids 2022, 29, 387–399. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Archer, J.; Fuerte-Stone, J.; Khandhar, A.P.; Voigt, E.; Granger, B.; Bombardi, R.G.; Govero, J.; Tan, Q.; Durnell, L.A.; et al. Intramuscular Delivery of Replicon RNA Encoding ZIKV-117 Human Monoclonal Antibody Protects against Zika Virus Infection. Mol. Ther. Methods Clin. Dev. 2020, 18, 402–414. [Google Scholar] [CrossRef]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Keeler, A.M.; Flotte, T.R. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu. Rev. Virol. 2019, 6, 601–621. [Google Scholar] [CrossRef]
- Shao, W.; Earley, L.F.; Chai, Z.; Chen, X.; Sun, J.; He, T.; Deng, M.; Hirsch, M.L.; Ting, J.; Samulski, R.J.; et al. Double-stranded RNA innate immune response activation from long-term adeno-associated virus vector transduction. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Reul, J.; Frisch, J.; Engeland, C.E.; Thalheimer, F.B.; Hartmann, J.; Ungerechts, G.; Buchholz, C.J. Tumor-Specific Delivery of Immune Checkpoint Inhibitors by Engineered AAV Vectors. Front. Oncol. 2019, 9, 52. [Google Scholar] [CrossRef]
- Silva-Pilipich, N.; Martisova, E.; Ballesteros-Briones, M.C.; Hervas-Stubbs, S.; Casares, N.; Gonzalez-Sapienza, G.; Smerdou, C.; Vanrell, L. Long-Term Systemic Expression of a Novel PD-1 Blocking Nanobody from an AAV Vector Provides Antitumor Activity without Toxicity. Biomedicines 2020, 8, 562. [Google Scholar] [CrossRef]
- Del Rosario, J.M.M.; Smith, M.; Zaki, K.; Risley, P.; Temperton, N.; Engelhardt, O.G.; Collins, M.; Takeuchi, Y.; Hufton, S.E. Protection From Influenza by Intramuscular Gene Vector Delivery of a Broadly Neutralizing Nanobody Does Not Depend on Antibody Dependent Cellular Cytotoxicity. Front. Immunol. 2020, 11, 627. [Google Scholar] [CrossRef]
- Bunuales, M.; Ballesteros-Briones, M.C.; Gonzalez-Aparicio, M.; Hervas-Stubbs, S.; Martisova, E.; Mancheno, U.; Ricobaraza, A.; Lumbreras, S.; Smerdou, C.; Hernandez-Alcoceba, R. Adenovirus-Mediated Inducible Expression of a PD-L1 Blocking Antibody in Combination with Macrophage Depletion Improves Survival in a Mouse Model of Peritoneal Carcinomatosis. Int. J. Mol. Sci. 2021, 22, 4176. [Google Scholar] [CrossRef]
- Chowdhury, F.; Johnson, P.W.; Glennie, M.J.; Williams, A.P. Ex vivo assays of dendritic cell activation and cytokine profiles as predictors of in vivo effects in an anti-human CD40 monoclonal antibody ChiLob 7/4 phase I trial. Cancer Immunol. Res. 2014, 2, 229–240. [Google Scholar] [CrossRef]
- Irenaeus, S.M.M.; Nielsen, D.; Ellmark, P.; Yachnin, J.; Deronic, A.; Nilsson, A.; Norlen, P.; Veitonmaki, N.; Wennersten, C.S.; Ullenhag, G.J. First-in-human study with intratumoral administration of a CD40 agonistic antibody, ADC-1013, in advanced solid malignancies. Int. J. Cancer 2019, 145, 1189–1199. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug Name (s) | Format | Target (s) | Type of Cancer | Current Status | ClinicalTrials.gov Identifier | Nº Participants | FDA Approval Status |
|---|---|---|---|---|---|---|---|
| KN046 | Tetravalent, bispecific, Fc-fusion protein | CTLA-4, PD-L1 | Advanced solid tumors and lymphoma | Phase II/III | NCT03872791 | 52 | No |
| NCT04474119 | 482 | No | |||||
| NCT04925947 | 29 | FDA-regulated Drug Product | |||||
| Envolimab KN035 | Monospecific, Fc-fusion protein | PD-L1 | Advanced solid tumors, multiple primary neoplasm | Phase II | NCT03667170 | 200 | No |
| NCT04182789 | 20 | No | |||||
| NCT04891198 | 200 | No | |||||
| αPD1-MSLN-CAR T cells | single-chain variable fragments (scFv) | αPD1-MSLN-CAR T | MSLN-positive Advanced Solid Tumors | Early phase I | NCT05373147 | 21 | No |
| INBRX-109 | Tetravalent, monospecific, | Death receptor 5 | Advanced solid tumors, conventional chondrosarcoma | Phase I/II | NCT03715933 | 240 | FDA-regulated Drug Product |
| Fc-fusion protein | NCT04950075 | 201 | FDA-regulated Drug Product | ||||
| KN044 | Monospecific, Fc-fusion protein | CTLA-4 | Advanced solid tumors | Phase I | NCT04126590 | 39 | FDA-regulated Drug Product |
| Target (s) | Delivery | Type of Cancer | Current Status | ClinicalTrials.gov Identifier | Nº Participants | FDA Approval Status |
|---|---|---|---|---|---|---|
| PD-1 | Intravenous infusion | Advanced gastric adenocarcinoma | Phase II | NCT03704246 | 123 | No |
| Intravenous injection | Gastric cancer | Phase I | NCT03713905 | 400 | No | |
| Intravenous injection | Colorectal cancer. | Phase I/ II | NCT03711058 | 54 | FDA-regulated Drug Product | |
| Intravenous infusion | Advanced Solid Tumors | Phase I/ II | NCT04775680 | 60 | No | |
| Intravenous injection | Gastric cancer | Phase II | NCT03704246 | 123 | No | |
| Intravenous injection | Advanced solid tumors | Phase I | NCT04478461 | 21 | No | |
| Intravenous injection | Advanced refractory solid tumors. | Phase I | NCT02791334 | 215 | FDA-regulated Drug Product | |
| PD-1/TIM-3 Bispecific Antibody | Intravenous injection | Advanced and/or metastatic solid tumors | Phase I | NCT03708328 | 134 | FDA-regulated Drug Product |
| Anti-CD47/PD-1 Bifunctional Antibody | Intravenous injection | Advanced solid tumors | Phase II | NCT04886271 | 210 | No |
| PD-1/VEGF Bispecific Antibody | Intravenous infusion | Solid tumors | Phase I/ II | NCT04597541 | 59 | No |
| PD-1/CTLA-4 Bispecific Antibody | Intravenous injection | Advanced or metastatic solid tumors | Phase I/ II | NCT03852251 | 338 | No |
| PD-1 formulated with MK-5180 | Subcutaneous Injection | Advanced Solid Tumors | Phase I | NCT05017012 | 72 | No |
| CTLA-4 | Intravenous injection | Advanced Solid Tumors | Phase I | NCT03849469 | 242 | FDA-regulated Drug Product |
| CD39 | Intravenous infusion | Locally advanced or metastatic solid tumors | Phase I | NCT05075564 | 60 | FDA-regulated Drug Product |
| OX40 | Intratumoral or intravenous injection | Advanced solid tumors | Phase I | NCT03831295 | 12 | FDA-regulated Drug Product |
| LAG3 | Intravenous injection | Advanced solid tumors | Phase I/ II | NCT01968109 | 1499 | FDA-regulated Drug Product |
| CCR5 | Subcutaneous Injection | Locally advanced or metastatic solid tumors | Phase II | NCT04504942 | 30 | FDA-regulated Drug Product |
| 4-1BB | Intravenous infusion | Advanced Solid Malignancies | Phase I | NCT04144842 | 50 | No |
| PD-L1xCD27 Bispecific Antibody | Intravenous infusion | Advanced Solid Malignancies | Phase I | NCT04440943 | 27 | FDA-regulated Drug Product |
| PD-L1 | Intravenous infusion | Advanced Solid Malignancies | Phase I | NCT03590054 | 35 | FDA-regulated Drug Product |
| Anti-PD-L1/Anti-CTLA4 | Intravenous injection | Advanced Solid Malignancies | Phase I/II | NCT03518606 | 150 | No |
| Target | Non i.t /co-i.t Therapy | Type of Cancer | ClinicalTrials.gov Identifier | Nº Participants | FDA Approval Status | |
|---|---|---|---|---|---|---|
| CTLA-4 | Injection of ipilimumab during a biopsy procedure. | Head and neck Cancer | NCT02812524 | 18 | FDA-regulated Drug Product | |
| Combination with intravenous nivolumab | Melanoma | NCT02857569 | 90 | Not provided | ||
| Combination with intravenous nivolumab | Glioblastoma | NCT03233152 | 6 | No | ||
| Intratumoral Tilsotolimod combination with intratumoral ipilimumab and intravenous nivolumab. | Advanced cancers | NCT04270864 | 72 | No | ||
| PD-1 | mRNA-2752, a lipid nanoparticle encapsulating mRNAs encoding human OX40L, IL-23, and IL-36γ. | Ductal Carcinoma in Situ (DCIS) | NCT02872025 | 48 | FDA-regulated Drug Product | |
| Intra-lesional nivolumab therapy | Cutaneous Kaposi Sarcoma | NCT03316274 | 12 | FDA-regulated Drug Product | ||
| Pre-operative cemiplimab administered intralesionally | Cutaneous Squamous Cell Carcinoma | NCT03889912 | 61 | FDA-regulated Drug Product | ||
| Combination of PD-1 and CTLA4 | Metastatic Prostatic Adenocarcinoma | NCT04090775 | 12 | FDA-regulated Drug Product | ||
| mAbs delivery and | CD40 | Alone intratumorally or intravenously administered ADC-1013 | Advanced Solid Tumors | NCT02379741 | 24 | Not provided |
| non-viral theraphy | APX005M in Combination with systemic prembrolizumab | Metastatic Melanoma | NCT02706353 | 41 | FDA-regulated Drug Product | |
| ABBV-927 and ABBV-181 | Advanced solid tumors | NCT02988960; NCT03818542 | 163;3 | FDA-regulated Drug Product | ||
| Intratumoral Selicrelumab with atezolizumab | Relapsed B Cell Lymphoma | NCT03892525 | 4 | No | ||
| Fc-engineered anti-CD40 agonist | Lesions to the Skin | NCT04059588 | 28 | FDA-regulated Drug Product | ||
| SL-172154: fusion protein SIRPα-Fc-CD40L | Squamous Cell Carcinoma: Head and Neck or Skin | NCT04502888 | 5 | FDA-regulated Drug Product | ||
| D2C7-IT in Combination With 2141-V11 | Malignant Glioma | NCT04547777 | 30 | FDA-regulated Drug Product | ||
| Intratumoral TriMix Injections (CD40 and CD27) | Breast Cancer Patients | NCT03788083 | 36 | No | ||
| OX40 | mRNA 2416 alone or in combination with durvalumab | Advanced Malignancies | NCT03323398 | 79 | FDA-regulated Drug Product | |
| Combinaiton with TLR9 agonist SD-101 and radiation | Low-Grade B-Cell Non-Hodgkin Lymphomas | NCT03410901 | 15 | FDA-regulated Drug Product | ||
| CD137 | Urelumab combined with nivolumab | Solid Tumors | NCT03792724 | 32 | Product Manufactured in and Exported from the U.S | |
| CD40 | MEM-288:oncolytic adenovirus vector encoding transgenes for human IFNβ and a recombinant chimeric form of CD40-ligand | Solid tumors | NCT05076760 | 18 | FDA-regulated Drug Product | |
| CD40 | AdCD40L is a replication-deficient virus carrying the gene for CD40 ligand | Melanoma | NCT01455259 | 30 | Not provided | |
| Viral therapy | PD-1 | MVR-C5252: oncolytic vectors expressing IL-12 and anti-PD-1 antibody | Recurrent or progressive glioblastoma | NCT05095441 | 51 | FDA-regulated Drug Product |
| PD-1 | ONCOS-102: Oncolytic Adenovirus Expressing GMCSF and combined with prembrolizumab | Melanoma progressing after (PD1) Blockade | NCT03003676 | 21 | Not provided | |
| CTLA-4 | ISI-JX: Pexa-Vecan oncolytic virus genetically modified to express GM-CSF with intratumoural administration of ipilimumab | Advanced /solid tumors | NCT02977156 | 22 | Not provided | |
| PD-1 and CTLA-4 | ONCR-177 (which encodes CCL4, IL-12, Flt3L, anti-CTLA-4, anti-PD1alone or combined with pembrolizumab | Advanced/metastatic solid tumors | NCT04348916 | 132 | FDA-regulated Drug Product |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco, E.; Chocarro, L.; Fernández-Rubio, L.; Bocanegra, A.; Arasanz, H.; Echaide, M.; Garnica, M.; Piñeiro-Hermida, S.; Kochan, G.; Escors, D. Leading Edge: Intratumor Delivery of Monoclonal Antibodies for the Treatment of Solid Tumors. Int. J. Mol. Sci. 2023, 24, 2676. https://doi.org/10.3390/ijms24032676
Blanco E, Chocarro L, Fernández-Rubio L, Bocanegra A, Arasanz H, Echaide M, Garnica M, Piñeiro-Hermida S, Kochan G, Escors D. Leading Edge: Intratumor Delivery of Monoclonal Antibodies for the Treatment of Solid Tumors. International Journal of Molecular Sciences. 2023; 24(3):2676. https://doi.org/10.3390/ijms24032676
Chicago/Turabian StyleBlanco, Ester, Luisa Chocarro, Leticia Fernández-Rubio, Ana Bocanegra, Hugo Arasanz, Miriam Echaide, Maider Garnica, Sergio Piñeiro-Hermida, Grazyna Kochan, and David Escors. 2023. "Leading Edge: Intratumor Delivery of Monoclonal Antibodies for the Treatment of Solid Tumors" International Journal of Molecular Sciences 24, no. 3: 2676. https://doi.org/10.3390/ijms24032676
APA StyleBlanco, E., Chocarro, L., Fernández-Rubio, L., Bocanegra, A., Arasanz, H., Echaide, M., Garnica, M., Piñeiro-Hermida, S., Kochan, G., & Escors, D. (2023). Leading Edge: Intratumor Delivery of Monoclonal Antibodies for the Treatment of Solid Tumors. International Journal of Molecular Sciences, 24(3), 2676. https://doi.org/10.3390/ijms24032676