
Mitochondrial-Derived Vesicles—Link to Extracellular Vesicles and Implications in Cardiovascular Disease


Abstract
1. Introduction
2. The Formation of Mitochondrial-Derived Vesicles Is a Cellular Stress Response
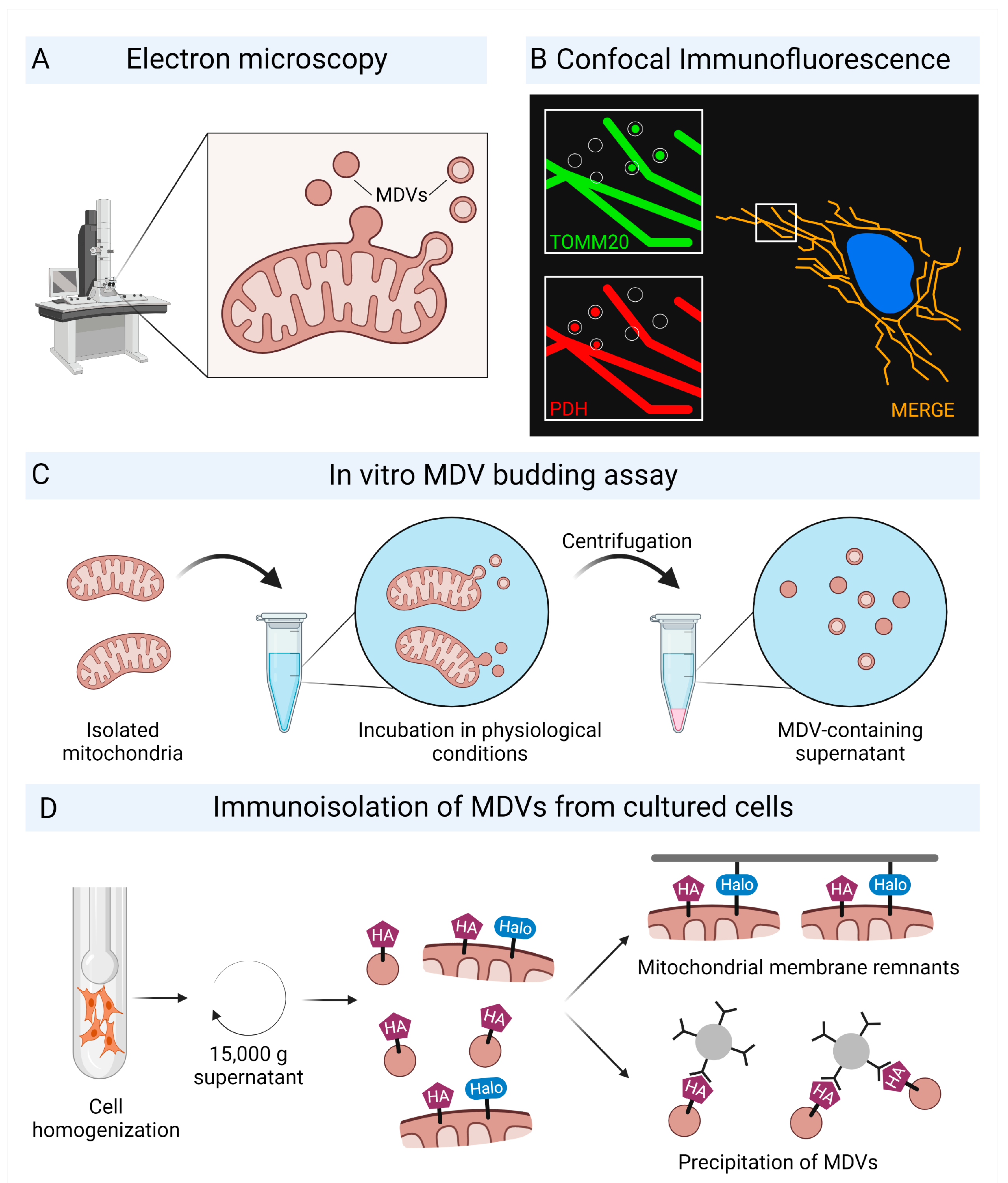
2.1. Characterization of Mitochondrial-Derived Vesicles
2.2. Oxidative Stress Increases the Formation of Mitochondrial-Derived Vesicles
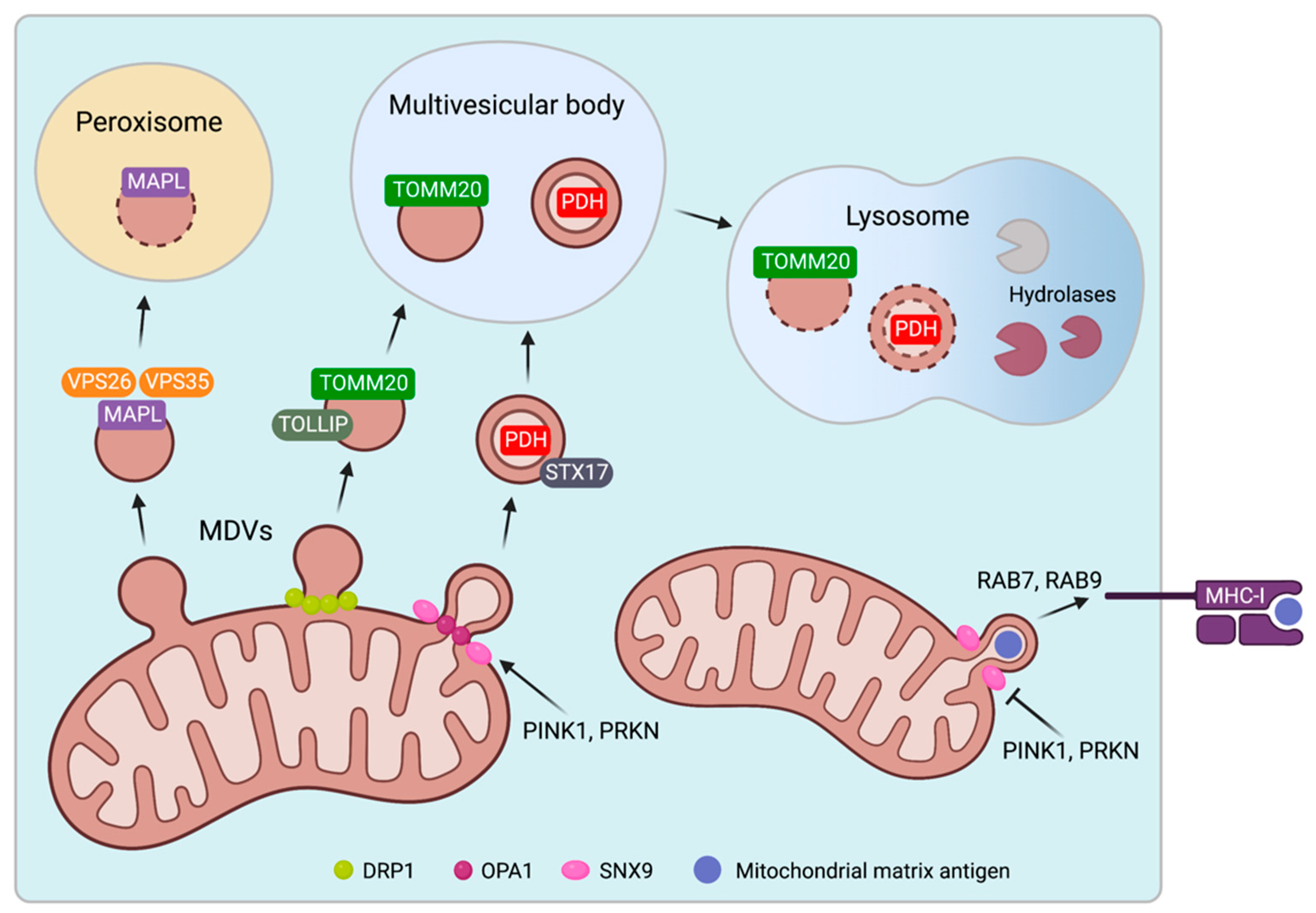
2.3. Biogenesis and Trafficking of Mitochondrial-Derived Vesicles
3. Cellular Release of Extracellular Vesicles with Mitochondrial Cargo
3.1. Mitochondrial Cargo Is Present in Extracellular Vesicles
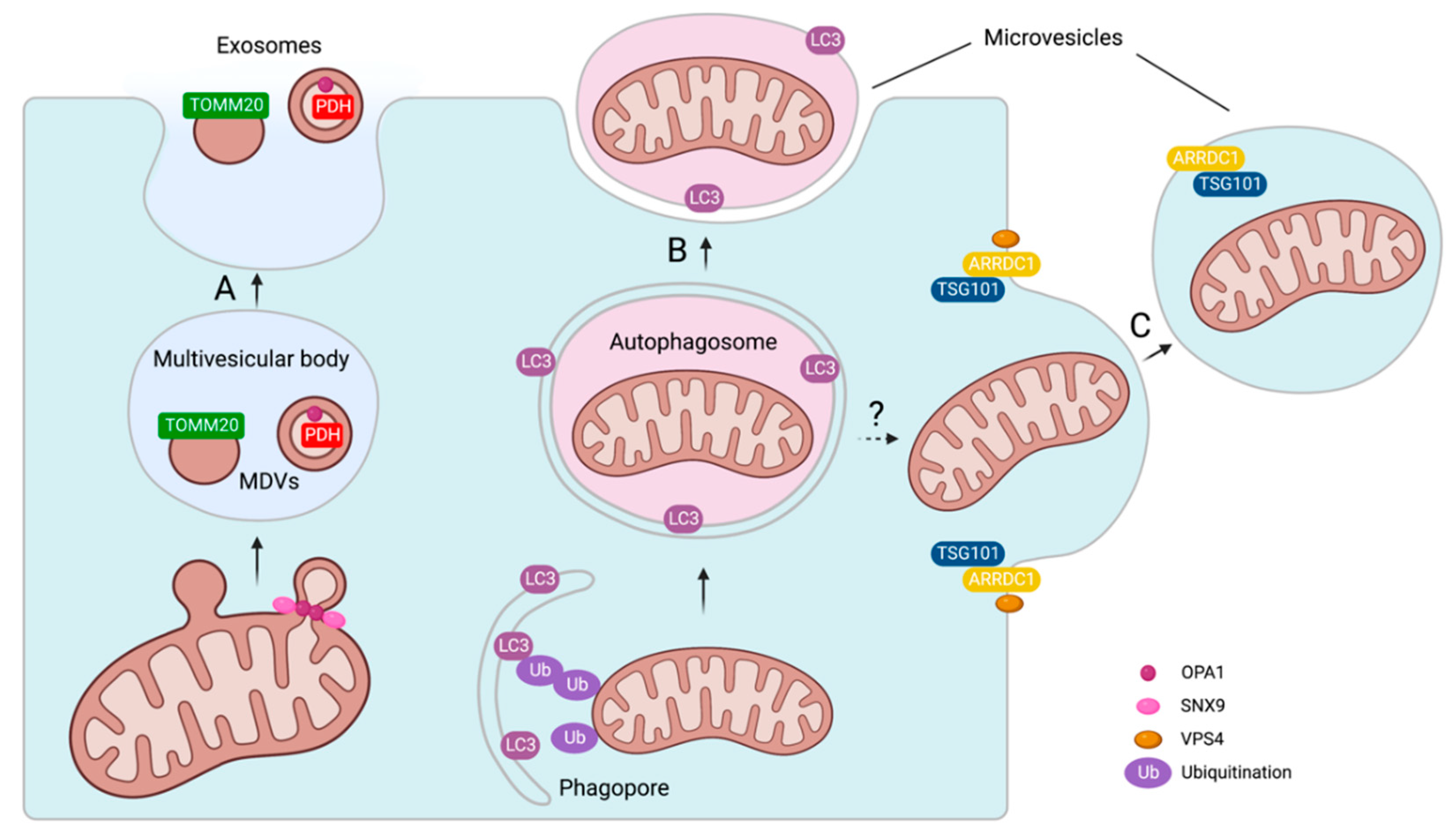
3.2. Mechanisms of Mitochondrial Cargo Loading into Extracellular Vesicles
3.3. Biological Effects of Extracellular Vesicles with Mitochondrial Cargo
4. Therapeutic Applications of Mitochondria in Cardiovascular Disease
5. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neubauer, S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.Y.; Ruiz-Velasco, A.; Bui, T.; Collins, L.; Wang, X.; Liu, W. Mitochondrial function in the heart: The insight into mechanisms and therapeutic potentials. Br. J. Pharmacol. 2019, 176, 4302–4318. [Google Scholar] [CrossRef] [PubMed]
- Schaper, J.; Meiser, E.; Stämmler, G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985, 56, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef]
- Forini, F.; Canale, P.; Nicolini, G.; Iervasi, G. Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine. Pharmaceutics 2020, 12, 1122. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. Embo J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef]
- Cadete, V.J.; Deschênes, S.; Cuillerier, A.; Brisebois, F.; Sugiura, A.; Vincent, A.; Turnbull, D.; Picard, M.; McBride, H.M.; Burelle, Y. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J. Physiol. 2016, 594, 5343–5362. [Google Scholar] [CrossRef]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef] [PubMed]
- Vasam, G.; Nadeau, R.; Cadete, V.J.J.; Lavallée-Adam, M.; Menzies, K.J.; Burelle, Y. Proteomics characterization of mitochondrial-derived vesicles under oxidative stress. FASEB J. 2021, 35, e21278. [Google Scholar] [CrossRef] [PubMed]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Vallabhaneni, K.C.; Haller, H.; Dumler, I. Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev. 2012, 21, 3104–3113. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef]
- Feng, Y.; Zhu, R.; Shen, J.; Wu, J.; Lu, W.; Zhang, J.; Zhang, J.; Liu, K. Human Bone Marrow Mesenchymal Stem Cells Rescue Endothelial Cells Experiencing Chemotherapy Stress by Mitochondrial Transfer Via Tunneling Nanotubes. Stem Cells Dev. 2019, 28, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Golan, K.; Singh, A.K.; Kollet, O.; Bertagna, M.; Althoff, M.J.; Khatib-Massalha, E.; Petrovich-Kopitman, E.; Wellendorf, A.M.; Massalha, H.; Levin-Zaidman, S.; et al. Bone marrow regeneration requires mitochondrial transfer from donor Cx43-expressing hematopoietic progenitors to stroma. Blood 2020, 136, 2607–2619. [Google Scholar] [CrossRef]
- Guo, Y.; Chi, X.; Wang, Y.; Heng, B.C.; Wei, Y.; Zhang, X.; Zhao, H.; Yin, Y.; Deng, X. Mitochondria transfer enhances proliferation, migration, and osteogenic differentiation of bone marrow mesenchymal stem cell and promotes bone defect healing. Stem Cell Res. Ther. 2020, 11, 245. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.M.; Chai, Y.H.; et al. iPSC-MSCs with High Intrinsic MIRO1 and Sensitivity to TNF-α Yield Efficacious Mitochondrial Transfer to Rescue Anthracycline-Induced Cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef]
- Han, H.; Hu, J.; Yan, Q.; Zhu, J.; Zhu, Z.; Chen, Y.; Sun, J.; Zhang, R. Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol. Med. Rep. 2016, 13, 1517–1524. [Google Scholar] [CrossRef]
- Puhm, F.; Afonyushkin, T.; Resch, U.; Obermayer, G.; Rohde, M.; Penz, T.; Schuster, M.; Wagner, G.; Rendeiro, A.F.; Melki, I.; et al. Mitochondria Are a Subset of Extracellular Vesicles Released by Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial Cells. Circ. Res. 2019, 125, 43–52. [Google Scholar] [CrossRef]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef]
- D’Acunzo, P.; Pérez-González, R.; Kim, Y.; Hargash, T.; Miller, C.; Alldred, M.J.; Erdjument-Bromage, H.; Penikalapati, S.C.; Pawlik, M.; Saito, M.; et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci. Adv. 2021, 7, eabe5085. [Google Scholar] [CrossRef]
- Todkar, K.; Chikhi, L.; Desjardins, V.; El-Mortada, F.; Pépin, G.; Germain, M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 2021, 12, 1971. [Google Scholar] [CrossRef]
- Amunts, A.; Brown, A.; Toots, J.; Scheres, S.H.W.; Ramakrishnan, V. The structure of the human mitochondrial ribosome. Science 2015, 348, 95–98. [Google Scholar] [CrossRef]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef]
- Toyofuku, M.; Nomura, N.; Eberl, L. Types and origins of bacterial membrane vesicles. Nat. Rev. Microbiol. 2019, 17, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Fujimoto, M.; Katayama, K.; Yamaoka, S.; Tsutsumi, N.; Arimura, S. Formation of Mitochondrial Outer Membrane Derived Protrusions and Vesicles in Arabidopsis thaliana. PLoS ONE 2016, 11, e0146717. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Hughes, C.E.; Henderson, K.A.; Yazvenko, N.; Gottschling, D.E. Selective sorting and destruction of mitochondrial membrane proteins in aged yeast. eLife 2016, 5, e13943. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- McLelland, G.L.; Lee, S.A.; McBride, H.M.; Fon, E.A. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J. Cell Biol. 2016, 214, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.A.; Phillips, E.O.; Collier, C.L.; Jb Robinson, A.; Routledge, D.; Wood, R.E.; Assar, E.A.; Tumbarello, D.A. Tollip coordinates Parkin-dependent trafficking of mitochondrial-derived vesicles. EMBO J. 2020, 39, e102539. [Google Scholar] [CrossRef]
- König, T.; Nolte, H.; Aaltonen, M.J.; Tatsuta, T.; Krols, M.; Stroh, T.; Langer, T.; McBride, H.M. MIROs and DRP1 drive mitochondrial-derived vesicle biogenesis and promote quality control. Nat. Cell Biol. 2021, 23, 1271–1286. [Google Scholar] [CrossRef]
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine dehydrogenase/xanthine oxidase and oxidative stress. Age 1997, 20, 127–140. [Google Scholar] [CrossRef]
- Rieske, J.S.; Lipton, S.H.; Baum, H.; Silman, H.I. Factors affecting the binding of antimycin A to complex 3 of the mitochondrial respiratory chain. J. Biol. Chem. 1967, 242, 4888–4896. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, H.; Wu, Y.; Zhu, Y.; Zhang, J.; Yang, G.; Yan, Q.; Li, J.; Li, T.; Liu, L. Mitochondrial-Derived Vesicles Protect Cardiomyocytes Against Hypoxic Damage. Front. Cell Dev. Biol. 2020, 8, 214. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Roberts, R.F.; Bayne, A.N.; Goiran, T.; Lévesque, D.; Boisvert, F.M.; Trempe, J.F.; Fon, E.A. Proteomic Profiling of Mitochondrial-Derived Vesicles in Brain Reveals Enrichment of Respiratory Complex Sub-assemblies and Small TIM Chaperones. J. Proteome Res. 2021, 20, 506–517. [Google Scholar] [CrossRef]
- Mygind, K.J.; Störiko, T.; Freiberg, M.L.; Samsøe-Petersen, J.; Schwarz, J.; Andersen, O.M.; Kveiborg, M. Sorting nexin 9 (SNX9) regulates levels of the transmembrane ADAM9 at the cell surface. J. Biol. Chem. 2018, 293, 8077–8088. [Google Scholar] [CrossRef]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Braschi, E.; Goyon, V.; Zunino, R.; Mohanty, A.; Xu, L.; McBride, H.M. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 2010, 20, 1310–1315. [Google Scholar] [CrossRef]
- Priya, A.; Kalaidzidis, I.V.; Kalaidzidis, Y.; Lambright, D.; Datta, S. Molecular insights into Rab7-mediated endosomal recruitment of core retromer: Deciphering the role of Vps26 and Vps35. Traffic 2015, 16, 68–84. [Google Scholar] [CrossRef]
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Cocozza, F.; Grisard, E.; Martin-Jaular, L.; Mathieu, M.; Théry, C. SnapShot: Extracellular Vesicles. Cell 2020, 182, 262–262.e1. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.L.; Sahai, E.A.; Yeo, M.; Bosch, M.; Dewar, A.; Olson, M.F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 2001, 3, 339–345. [Google Scholar] [CrossRef]
- Atkin-Smith, G.K.; Tixeira, R.; Paone, S.; Mathivanan, S.; Collins, C.; Liem, M.; Goodall, K.J.; Ravichandran, K.S.; Hulett, M.D.; Poon, I.K. A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat. Commun. 2015, 6, 7439. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Zhang, X.; Hubal, M.J.; Kraus, V.B. Immune cell extracellular vesicles and their mitochondrial content decline with ageing. Immun. Ageing 2020, 17, 1. [Google Scholar] [CrossRef]
- Song, W.; Bossy, B.; Martin, O.J.; Hicks, A.; Lubitz, S.; Knott, A.B.; Bossy-Wetzel, E. Assessing mitochondrial morphology and dynamics using fluorescence wide-field microscopy and 3D image processing. Methods 2008, 46, 295–303. [Google Scholar] [CrossRef]
- Wiemerslage, L.; Lee, D. Quantification of mitochondrial morphology in neurites of dopaminergic neurons using multiple parameters. J. Neurosci. Methods 2016, 262, 56–65. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef]
- Jang, S.C.; Crescitelli, R.; Cvjetkovic, A.; Belgrano, V.; Olofsson Bagge, R.; Sundfeldt, K.; Ochiya, T.; Kalluri, R.; Lötvall, J. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J. Extracell Vesicles 2019, 8, 1635420. [Google Scholar] [CrossRef]
- D’Acunzo, P.; Kim, Y.; Ungania, J.M.; Pérez-González, R.; Goulbourne, C.N.; Levy, E. Isolation of mitochondria-derived mitovesicles and subpopulations of microvesicles and exosomes from brain tissues. Nat. Protoc. 2022, 17, 2517–2549. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Lazo, S.; Noren Hooten, N.; Green, J.; Eitan, E.; Mode, N.A.; Liu, Q.R.; Zonderman, A.B.; Ezike, N.; Mattson, M.P.; Ghosh, P.; et al. Mitochondrial DNA in extracellular vesicles declines with age. Aging Cell 2021, 20, e13283. [Google Scholar] [CrossRef]
- Futter, C.E.; Pearse, A.; Hewlett, L.J.; Hopkins, C.R. Multivesicular endosomes containing internalized EGF-EGF receptor complexes mature and then fuse directly with lysosomes. J. Cell Biol. 1996, 132, 1011–1023. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef]
- Hartwig, J.H. Mechanisms of actin rearrangements mediating platelet activation. J. Cell Biol. 1992, 118, 1421–1442. [Google Scholar] [CrossRef]
- Keerthikumar, S.; Chisanga, D.; Ariyaratne, D.; Al Saffar, H.; Anand, S.; Zhao, K.; Samuel, M.; Pathan, M.; Jois, M.; Chilamkurti, N.; et al. ExoCarta: A Web-Based Compendium of Exosomal Cargo. J. Mol. Biol. 2016, 428, 688–692. [Google Scholar] [CrossRef]
- Crewe, C.; Funcke, J.B.; Li, S.; Joffin, N.; Gliniak, C.M.; Ghaben, A.L.; An, Y.A.; Sadek, H.A.; Gordillo, R.; Akgul, Y.; et al. Extracellular vesicle-based interorgan transport of mitochondria from energetically stressed adipocytes. Cell Metab. 2021, 33, 1853–1868.e11. [Google Scholar] [CrossRef]
- D’Souza, A.; Burch, A.; Dave, K.M.; Sreeram, A.; Reynolds, M.J.; Dobbins, D.X.; Kamte, Y.S.; Zhao, W.; Sabatelle, C.; Joy, G.M.; et al. Microvesicles transfer mitochondria and increase mitochondrial function in brain endothelial cells. J. Control. Release 2021, 338, 505–526. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Willis, C.M.; Manferrari, G.; Rogall, R.; Fernandez-Vizarra, E.; Williamson, J.C.; Braga, A.; van den Bosch, A.; Leonardi, T.; et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 2021, 19, e3001166. [Google Scholar] [CrossRef]
- Ikeda, G.; Santoso, M.R.; Tada, Y.; Li, A.M.; Vaskova, E.; Jung, J.H.; O’Brien, C.; Egan, E.; Ye, J.; Yang, P.C. Mitochondria-Rich Extracellular Vesicles From Autologous Stem Cell-Derived Cardiomyocytes Restore Energetics of Ischemic Myocardium. J. Am. Coll Cardiol. 2021, 77, 1073–1088. [Google Scholar] [CrossRef]
- O’Brien, C.G.; Ozen, M.O.; Ikeda, G.; Vaskova, E.; Jung, J.H.; Bayardo, N.; Santoso, M.R.; Shi, L.; Wahlquist, C.; Jiang, Z.; et al. Mitochondria-Rich Extracellular Vesicles Rescue Patient-Specific Cardiomyocytes From Doxorubicin Injury: Insights Into the SENECA Trial. JACC CardioOncol 2021, 3, 428–440. [Google Scholar] [CrossRef]
- Rosina, M.; Ceci, V.; Turchi, R.; Chuan, L.; Borcherding, N.; Sciarretta, F.; Sánchez-Díaz, M.; Tortolici, F.; Karlinsey, K.; Chiurchiù, V.; et al. Ejection of damaged mitochondria and their removal by macrophages ensure efficient thermogenesis in brown adipose tissue. Cell Metab. 2022, 34, 533–548.e12. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Hazeldine, J.; Hampson, P.; Opoku, F.A.; Foster, M.; Lord, J.M. N-Formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury 2015, 46, 975–984. [Google Scholar] [CrossRef]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Cowan, D.B.; Yao, R.; Akurathi, V.; Snay, E.R.; Thedsanamoorthy, J.K.; Zurakowski, D.; Ericsson, M.; Friehs, I.; Wu, Y.; Levitsky, S.; et al. Intracoronary Delivery of Mitochondria to the Ischemic Heart for Cardioprotection. PLoS ONE 2016, 11, e0160889. [Google Scholar] [CrossRef]
- Shin, B.; Saeed, M.Y.; Esch, J.J.; Guariento, A.; Blitzer, D.; Moskowitzova, K.; Ramirez-Barbieri, G.; Orfany, A.; Thedsanamoorthy, J.K.; Cowan, D.B.; et al. A Novel Biological Strategy for Myocardial Protection by Intracoronary Delivery of Mitochondria: Safety and Efficacy. JACC Basic Transl. Sci. 2019, 4, 871–888. [Google Scholar] [CrossRef]
- Blitzer, D.; Guariento, A.; Doulamis, I.P.; Shin, B.; Moskowitzova, K.; Barbieri, G.R.; Orfany, A.; Del Nido, P.J.; McCully, J.D. Delayed Transplantation of Autologous Mitochondria for Cardioprotection in a Porcine Model. Ann. Thorac. Surg. 2020, 109, 711–719. [Google Scholar] [CrossRef]
- Guariento, A.; Blitzer, D.; Doulamis, I.; Shin, B.; Moskowitzova, K.; Orfany, A.; Ramirez-Barbieri, G.; Staffa, S.J.; Zurakowski, D.; Del Nido, P.J.; et al. Preischemic autologous mitochondrial transplantation by intracoronary injection for myocardial protection. J. Thorac. Cardiovasc. Surg. 2020, 160, e15–e29. [Google Scholar] [CrossRef]
- Esmaeili, A.; Alini, M.; Baghaban Eslaminejad, M.; Hosseini, S. Engineering strategies for customizing extracellular vesicle uptake in a therapeutic context. Stem Cell Res. Ther. 2022, 13, 129. [Google Scholar] [CrossRef]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef]
- Dickhout, A.; Koenen, R.R. Extracellular Vesicles as Biomarkers in Cardiovascular Disease; Chances and Risks. Front. Cardiovasc. Med. 2018, 5, 113. [Google Scholar] [CrossRef]
- Chen, Y.; Li, G.; Liu, M.L. Microvesicles as Emerging Biomarkers and Therapeutic Targets in Cardiometabolic Diseases. Genom. Proteom. Bioinform. 2018, 16, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Li, Q. Exosomes as Diagnostic Biomarkers in Cardiovascular Diseases. Adv. Exp. Med. Biol. 2017, 998, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.Y.; Lee, C.K.; Huang, C.; Ou, Y.H.; Charles, C.J.; Richards, A.M.; Neupane, Y.R.; Pavon, M.V.; Zharkova, O.; Pastorin, G.; et al. Extracellular Vesicles in Cardiovascular Diseases: Alternative Biomarker Sources, Therapeutic Agents, and Drug Delivery Carriers. Int. J. Mol. Sci. 2019, 20, 3272. [Google Scholar] [CrossRef] [PubMed]
- Klyachko, N.L.; Arzt, C.J.; Li, S.M.; Gololobova, O.A.; Batrakova, E.V. Extracellular Vesicle-Based Therapeutics: Preclinical and Clinical Investigations. Pharmaceutics 2020, 12, 1171. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Single-Membrane MDVs | Double-Membrane MDVs | |
|---|---|---|
 |  | |
| Origin | Budding from the outer mitochondrial membrane | Budding from the inner and outer mitochondrial membrane |
| Cargo | Outer mitochondrial membrane proteins | Inner mitochondrial membrane and matrix proteins |
| Protein marker | Translocase of outer mitochondrial membrane 20 (TOMM20), Mitochondrial-anchored protein ligase (MAPL) | Pyruvate dehydrogenase subunits E2/E3bp (PDH), NADH:ubiquinone oxidoreductase subunit A9 (NDUFA9), mitochondrial stress 70 protein (mtHSP70) |
| Type of Vesicles | Size | Origin/Mechanism of Secretion | Cargo |
|---|---|---|---|
| MDVs | 70–100 nm | Budding from mitochondrial surface/ fusion of multivesicular bodies with plasma membrane? | Mitochondrial proteins |
| Exosomes | 30–150 nm | Endolysosomal system/ fusion of multivesicular bodies with plasma membrane | Proteins, lipids, nucleic acids |
| Microvesicles | 100–1000 nm | Outward budding of the plasma membrane | Proteins, lipids, nucleic acids, organelles |
| Apoptotic bodies | >1000 nm | Cell shrinkage and fragmentation | Organelles, nuclear fragments |
| Condition/ Disease | Mitochondrial Administration | Origin of Mitochondria | Status | Trial Identifier |
|---|---|---|---|---|
| Infertility | Injection into oocytes during in vitro fertilization | Autologous (ovarian stem cells) | Completed | NCT02586298 |
| Extracorporeal membrane oxygenation complication | Intramyocardial injection during surgery/intracoronary infusion with catheter | Autologous (skeletal muscle) | Recruiting | NCT02851758 |
| Cerebral ischemia | Endovascular infusion with catheter | Autologous (skeletal muscle) | Recruiting | NCT04998357 |
| Polymyositis or dermatomyositis | Intravenous infusion | Allogeneic (umbilical cord-derived MSCs) | Recruiting | NCT04976140 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heyn, J.; Heuschkel, M.A.; Goettsch, C. Mitochondrial-Derived Vesicles—Link to Extracellular Vesicles and Implications in Cardiovascular Disease. Int. J. Mol. Sci. 2023, 24, 2637. https://doi.org/10.3390/ijms24032637
Heyn J, Heuschkel MA, Goettsch C. Mitochondrial-Derived Vesicles—Link to Extracellular Vesicles and Implications in Cardiovascular Disease. International Journal of Molecular Sciences. 2023; 24(3):2637. https://doi.org/10.3390/ijms24032637
Chicago/Turabian StyleHeyn, Jonas, Marina Augusto Heuschkel, and Claudia Goettsch. 2023. "Mitochondrial-Derived Vesicles—Link to Extracellular Vesicles and Implications in Cardiovascular Disease" International Journal of Molecular Sciences 24, no. 3: 2637. https://doi.org/10.3390/ijms24032637
APA StyleHeyn, J., Heuschkel, M. A., & Goettsch, C. (2023). Mitochondrial-Derived Vesicles—Link to Extracellular Vesicles and Implications in Cardiovascular Disease. International Journal of Molecular Sciences, 24(3), 2637. https://doi.org/10.3390/ijms24032637