
Perinatal Obesity Sensitizes for Premature Kidney Aging Signaling

 , , , ,
, , , ,  ,
,  , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
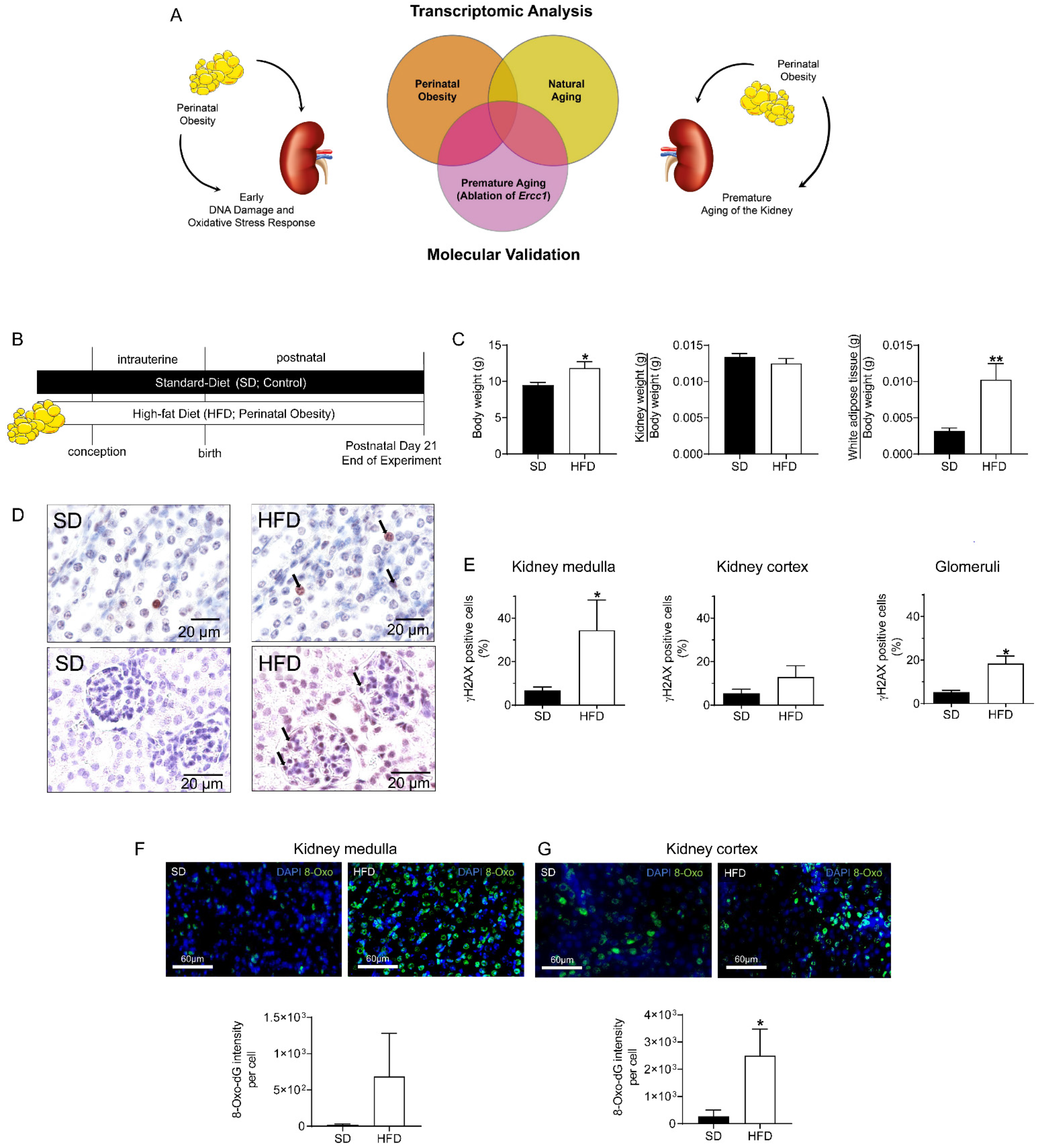
2.1. Maternal HFD Induces DNA Damage Response (DDR) and Oxidative Stress in Male Offspring’s Kidney at P21
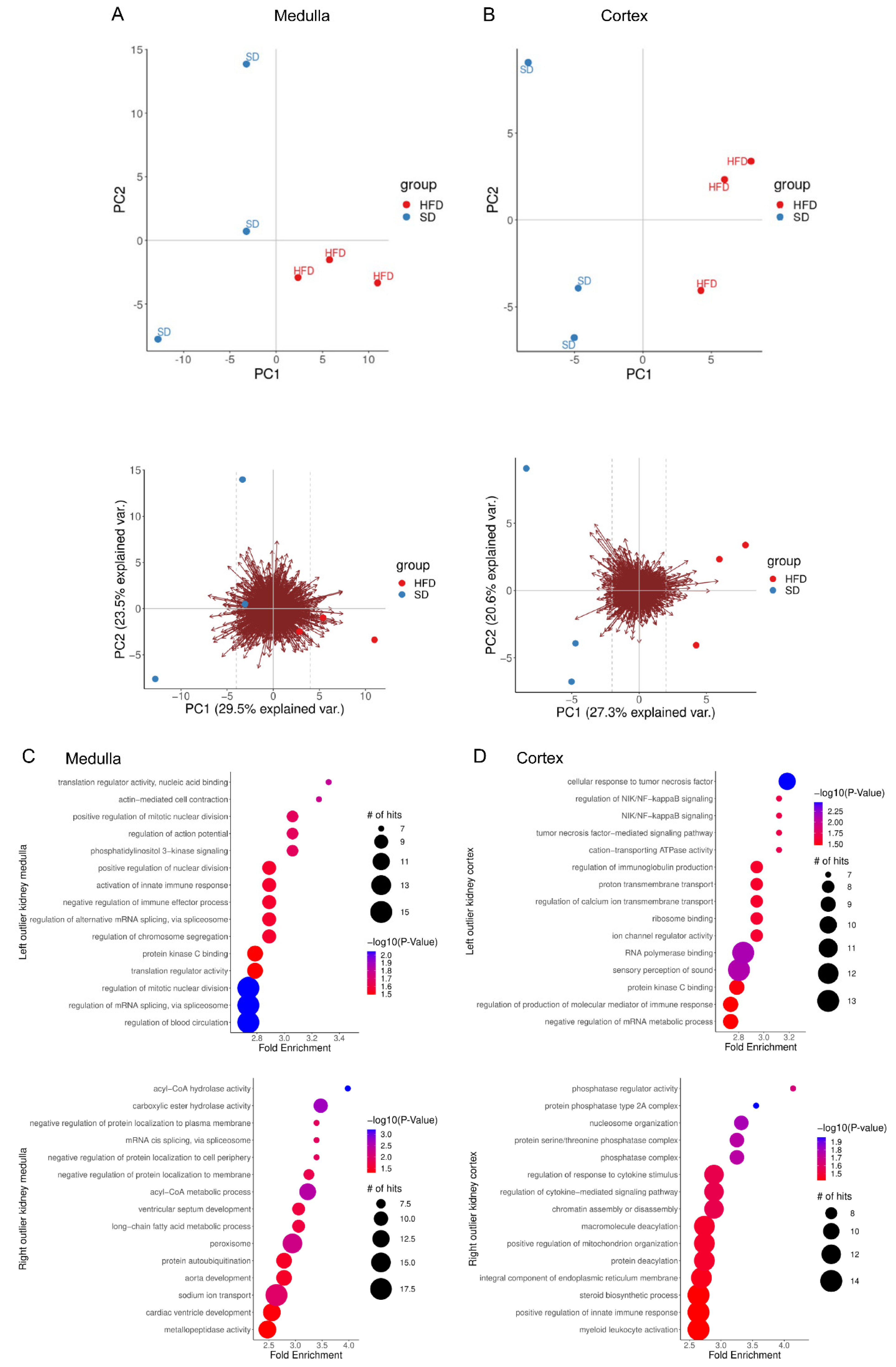
2.2. Perinatal Obesity Induces Aging-Associated Pathways
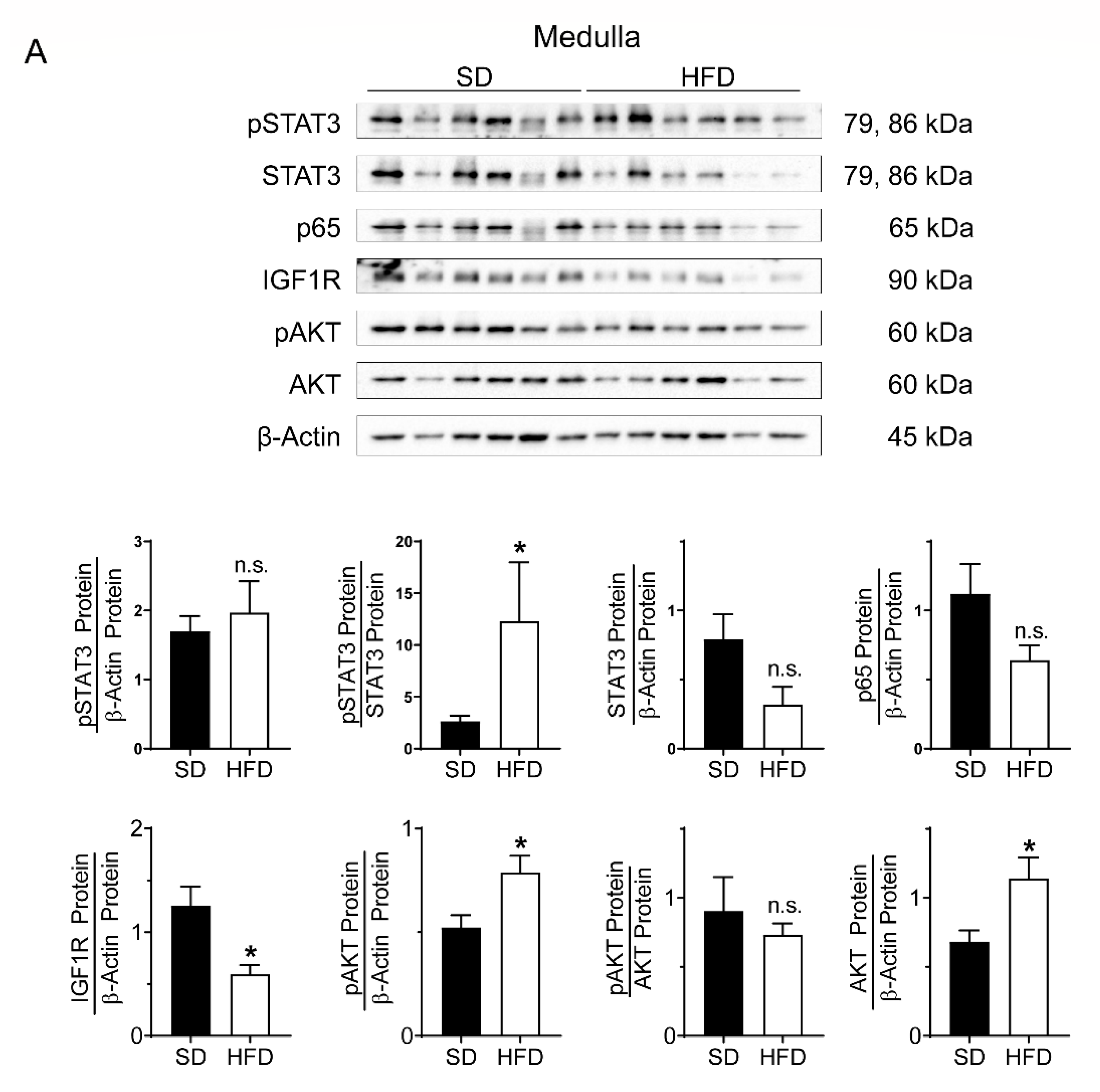
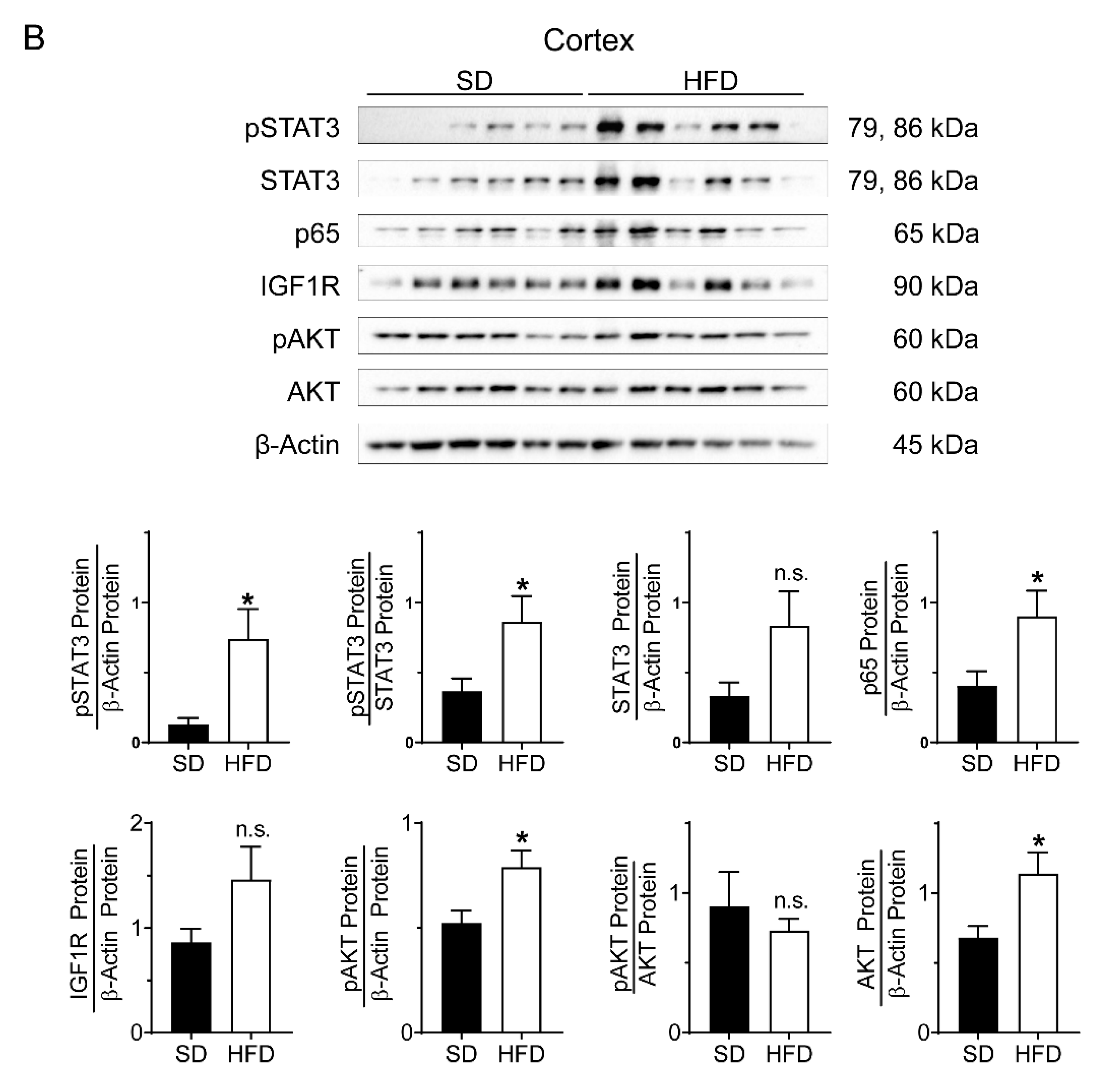
2.3. Perinatal Obesity Induces Compartment-Specific Activation of Inflammatory STAT3 and NFκB Signaling Cascade As Well As A Dysregulation of the Aging-Associated Renal IGF1R/AKT Pathway
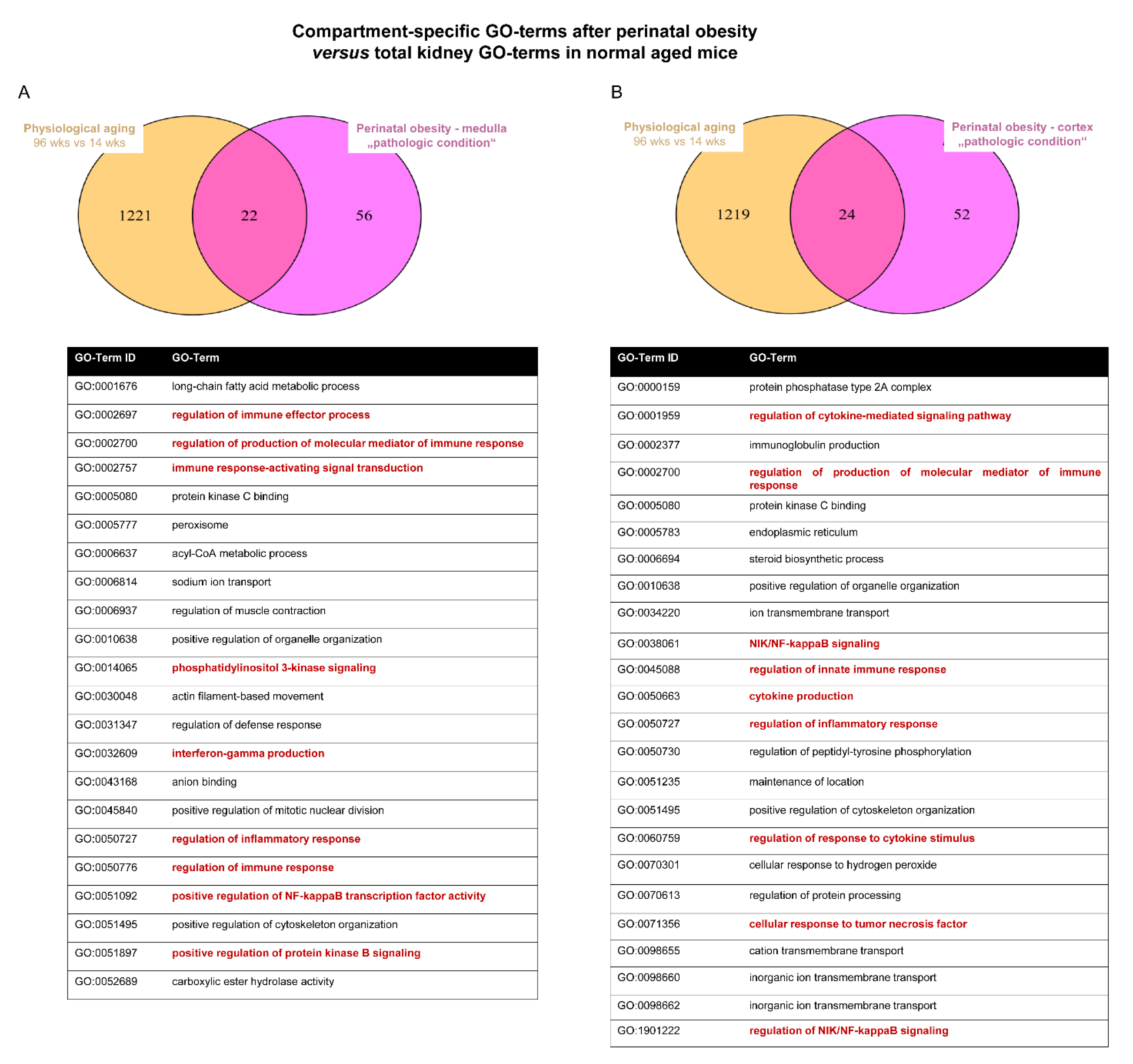
2.4. Converging Aging Signaling in Kidney after Perinatal Obesity
3. Methods
3.1. Animal Procedures
3.2. Physiological Data of Dams and Offspring
3.3. Tissue Preparation
3.4. RNA Sequencing and Transcriptomic Data Analysis
3.5. Immunoblots
3.6. Immunohistochemistry
3.7. Immunofluorescent Staining
3.8. Analysis of Data
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kazancioğlu, R. Risk factors for chronic kidney disease: An update. Kidney Int. Suppl. 2013, 3, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Mattace-Raso, F.; Sijbrands, E.J.; Zoccali, C. The aging kidney revisited: A systematic review. Ageing Res. Rev. 2014, 14, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Mimran, A.; Ribstein, J.; Jover, B. Aging and sodium homeostasis. Kidney Int. Suppl. 1992, 37, S107–S113. [Google Scholar] [PubMed]
- Sands, J.M. Urine Concentrating and Diluting Ability During Aging. J. Gerontol. Ser. A 2012, 67, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Emamian, S.A.; Nielsen, M.B.; Pedersen, J.F.; Ytte, L. Kidney dimensions at sonography: Correlation with age, sex, and habitus in 665 adult volunteers. Am. J. Roentgenol. 1993, 160, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Hoy, W.E.; Douglas-Denton, R.N.; Hughson, M.D.; Cass, A.; Johnson, K.; Bertram, J.F. A stereological study of glomerular number and volume: Preliminary findings in a multiracial study of kidneys at autopsy. Kidney Int. 2003, 63, S31–S37. [Google Scholar] [CrossRef]
- Roseman, D.A.; Hwang, S.-J.; Oyama-Manabe, N.; Chuang, M.L.; O’Donnell, C.J.; Manning, W.J.; Fox, C.S. Clinical associations of total kidney volume: The Framingham Heart Study. Nephrol. Dial. Transplant. 2016, 32, 1344–1350. [Google Scholar] [CrossRef]
- Wang, X.; Vrtiska, T.J.; Avula, R.T.; Walters, L.R.; Chakkera, H.A.; Kremers, W.K.; Lerman, L.O.; Rule, A.D. Age, kidney function, and risk factors associate differently with cortical and medullary volumes of the kidney. Kidney Int. 2014, 85, 677–685. [Google Scholar] [CrossRef]
- McLachlan, M.; Wasserman, P. Changes in sizes and distensibility of the aging kidney. Br. J. Radiol. 1981, 54, 488–491. [Google Scholar] [CrossRef]
- Denic, A.; Glassock, R.J.; Rule, A.D. Structural and Functional Changes with the Aging Kidney. Adv. Chronic Kidney Dis. 2016, 23, 19–28. [Google Scholar] [CrossRef]
- Denic, A.; Lieske, J.C.; Chakkera, H.A.; Poggio, E.D.; Alexander, M.P.; Singh, P.; Kremers, W.K.; Lerman, L.O.; Rule, A.D. The Substantial Loss of Nephrons in Healthy Human Kidneys with Aging. J. Am. Soc. Nephrol. 2016, 28, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P.; Furth, S.L.; Zoccali, C.; Li, P.K.T.; Garcia-Garcia, G.; Benghanem-Gharbi, M.; Bollaert, R.; Dupuis, S.; Erk, T.; Kalantar-Zadeh, K.; et al. Obesity and kidney disease: Hidden consequences of the epidemic. Kidney Int. 2016, 91, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Song, Y.; Caballero, B.; Cheskin, L. Association between obesity and kidney disease: A systematic review and meta-analysis. Kidney Int. 2008, 73, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.S.; Larson, M.; Leip, E.P.; Culleton, B.; Wilson, P.W.F.; Levy, D. Predictors of New-Onset Kidney Disease in a Community-Based Population. JAMA 2004, 291, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Borrelli, S.; Minutolo, R.; Chiodini, P.; De Nicola, L.; Conte, G. A systematic review and meta-analysis suggests obesity predicts onset of chronic kidney disease in the general population. Kidney Int. 2017, 91, 1224–1235. [Google Scholar] [CrossRef]
- Silverwood, R.J.; Pierce, M.; Thomas, C.; Hardy, R.; Ferro, C.; Sattar, N.; Whincup, P.; Savage, C.; Kuh, D.; Nitsch, D.; et al. Association between Younger Age When First Overweight and Increased Risk for CKD. J. Am. Soc. Nephrol. 2013, 24, 813–821. [Google Scholar] [CrossRef]
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health ex-amination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Gallus, S.; Lugo, A.; Murisic, B.; Bosetti, C.; Boffetta, P.; La Vecchia, C. Overweight and obesity in 16 European countries. Eur. J. Nutr. 2014, 54, 679–689. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J.P. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef]
- Plagemann, A. Perinatal programming and functional teratogenesis: Impact on body weight regulation and obesity. Physiol. Behav. 2005, 86, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Dötsch, J.; Alcazar, M.A.A.; Janoschek, R.; Nüsken, E.; Weber, L.T.; Nüsken, K.D. Perinatal programming of renal function. Curr. Opin. Pediatr. 2016, 28, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.E.; Yoo, K.H. Obesity and chronic kidney disease: Prevalence, mechanism, and management. Clin. Exp. Pediatr. 2021, 64, 511–518. [Google Scholar] [CrossRef]
- Voggel, J.; Mohr, J.; Nusken, K.D.; Dotsch, J.; Nusken, E.; Alejandre Alcazar, M.A. Translational insights into mechanisms and pre-ventive strategies after renal injury in neonates. Semin. Fetal Neonatal Med. 2022, 27, 101245. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, T.; Huber, E.K.; Dinger, K.; Wilke, R.; Vohlen, C.; Selle, J.; Kadah, M.; Persigehl, T.; Heneweer, C.; Dotsch, J.; et al. Maternal high-fat diet induces long-term obesity with sex-dependent metabolic programming of adipocyte differentiation, hypertrophy and dysfunction in the offspring. Clin. Sci. 2020, 134, 921–939. [Google Scholar] [CrossRef]
- Kasper, P.; Vohlen, C.; Dinger, K.; Mohr, J.; Hucklenbruch-Rother, E.; Janoschek, R.; Koth, J.; Matthes, J.; Appel, S.; Dotsch, J.; et al. Renal Metabolic Programming Is Linked to the Dynamic Regulation of a Leptin-Klf15 Axis and Akt/AMPKalpha Signaling in Male Offspring of Obese Dams. Endocrinology 2017, 158, 3399–3415. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.; Simão, S.; Silva, E.; Pinto, V.; Amaral, J.S.; Afonso, J.; Serrão, M.P.; Pinho, M.J.; Soares-Da-Silva, P. Aging increases oxidative stress and renal expression of oxidant and antioxidant enzymes that are associated with an increased trend in systolic blood pressure. Oxid. Med. Cell. Longev. 2009, 2, 138–145. [Google Scholar] [CrossRef]
- Romano, A.D.; Serviddio, G.; de Matthaeis, A.; Bellanti, F.; Vendemiale, G. Oxidative stress and aging. J. Nephrol. 2010, 23, S29–S36. [Google Scholar]
- Vlassara, H.; Torreggiani, M.; Post, J.B.; Zheng, F.; Uribarri, J.; Striker, G.E. Role of oxidants/inflammation in declining renal function in chronic kidney disease and normal aging. Kidney Int. 2009, 76, S3–S11. [Google Scholar] [CrossRef]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell Commun. Signal. 2017, 12, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Spoto, B.; Pisano, A.; Zoccali, C. Insulin resistance in chronic kidney disease: A systematic review. Am. J. Physiol. Physiol. 2016, 311, F1087–F1108. [Google Scholar] [CrossRef]
- Thomas, S.S.; Zhang, L.; Mitch, W.E. Molecular mechanisms of insulin resistance in chronic kidney disease. Kidney Int. 2015, 88, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yuan, H.; Cao, W.; Wang, T.; Yu, H.; Fu, Y.; Jiang, B.; Zhou, H.; Guo, H.; Zhao, X. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics 2019, 9, 3980–3991. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Debès, C.; Grönke, S.; Karalay, Ö.; Tain, L.; Nakamura, S.; Hahn, O.; Weigelt, C.; Zirkel, A.; Sofiadis, K.; Brant, L.; et al. Aging-associated changes in tran-scriptional elongation influence metazoan longevity. bioRxiv 2019. [Google Scholar]
- Schermer, B.; Bartels, V.; Frommolt, P.; Habermann, B.; Braun, F.; Schultze, J.L.; Roodbergen, M.; Hoeijmakers, J.H.; Schumacher, B.; Nurnberg, P.; et al. Transcriptional profiling reveals progeroid Ercc1(-/Delta) mice as a model system for glomerular aging. BMC Genom. 2013, 14, 559. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F. Sex differences in metabolic homeostasis, diabetes, and obesity. Biol. Sex Differ. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Picó, C.; Palou, M.; Priego, T.; Sánchez, J.; Palou, A. Metabolic programming of obesity by energy restriction during the perinatal period: Different outcomes depending on gender and period, type and severity of restriction. Front. Physiol. 2012, 3, 436. [Google Scholar] [CrossRef]
- Alcazar, M.A.; Dinger, K.; Rother, E.; Ostreicher, I.; Vohlen, C.; Plank, C.; Dotsch, J. Prevention of early postnatal hyperalimentation protects against activation of transforming growth factor-beta/bone morphogenetic protein and interleukin-6 signaling in rat lungs after intrauterine growth restriction. J. Nutr. 2014, 144, 1943–1951. [Google Scholar] [CrossRef]
- Alejandre Alcazar, M.A.; Ostreicher, I.; Appel, S.; Rother, E.; Vohlen, C.; Plank, C.; Dotsch, J. Developmental regulation of inflam-matory cytokine-mediated Stat3 signaling: The missing link between intrauterine growth restriction and pulmonary dysfunc-tion? J. Mol. Med. 2012, 90, 945–957. [Google Scholar] [CrossRef]
- Dinger, K.; Kasper, P.; Hucklenbruch-Rother, E.; Vohlen, C.; Jobst, E.; Janoschek, R.; Bae-Gartz, I.; Van Koningsbruggen-Rietschel, S.; Plank, C.; Dötsch, J.; et al. Early-onset obesity dysregulates pulmonary adipocytokine/insulin signaling and induces asthma-like disease in mice. Sci. Rep. 2016, 6, 24168. [Google Scholar] [CrossRef]
- Selle, J.; Dinger, K.; Jentgen, V.; Zanetti, D.; Will, J.; Georgomanolis, T.; Vohlen, C.; Wilke, R.; Kojonazarov, B.; Klymenko, O.; et al. Maternal and perinatal obesity induce bronchial obstruction and pulmonary hypertension via IL-6-FoxO1-axis in later life. Nat. Commun. 2022, 13, 4352. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome. Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Stasi, A.; Cosola, C.; Caggiano, G.; Cimmarusti, M.T.; Palieri, R.; Acquaviva, P.M.; Rana, G.; Gesualdo, L. Obesity-Related Chronic Kidney Disease: Principal Mechanisms and New Approaches in Nutritional Management. Front. Nutr. 2022, 9, 925619. [Google Scholar] [CrossRef]
- Barbieri, D.; Goicoechea, M.; Sanchez-Niño, M.D.; Ortiz, A.; Verde, E.; Verdalles, U.; De José, A.P.; Delgado, A.; Hurtado, E.; Sánchez-Cámara, L.; et al. Obesity and chronic kidney disease progression—The role of a new adipocytokine: C1q/tumour necrosis factor-related protein-1. Clin. Kidney J. 2018, 12, 420–426. [Google Scholar] [CrossRef]
- Crosby, L.; Davis, B.; Joshi, S.; Jardine, M.; Paul, J.; Neola, M.; Barnard, N.D. Ketogenic Diets and Chronic Disease: Weighing the Benefits against the Risks. Front. Nutr. 2021, 8, 702802. [Google Scholar] [CrossRef]
- Hoyer-Allo, K.J.R.; Späth, M.R.; Brodesser, S.; Zhu, Y.; Binz-Lotter, J.; Höhne, M.; Brönneke, H.; Bohl, K.; Johnsen, M.; Kubacki, T.; et al. Caloric restriction reduces the pro-inflammatory eicosanoid 20-hydroxyeicosatetraenoic acid to protect from acute kidney injury. Kidney Int. 2022, 102, 560–576. [Google Scholar] [CrossRef] [PubMed]
- Braun, F.; Rinschen, M.M.; Bartels, V.; Frommolt, P.; Habermann, B.; Hoeijmakers, J.H.J.; Schumacher, B.; Dollé, M.E.T.; Müller, R.-U.; Benzing, T.; et al. Altered lipid metabolism in the aging kidney identified by three layered omic analysis. Aging 2016, 8, 441–454. [Google Scholar] [CrossRef]
- Geylis, M.; Coreanu, T.; Novack, V.; Landau, D. Risk factors for childhood chronic kidney disease: A population-based study. Pediatr. Nephrol. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Tran, S.; Chen, Y.-W.; Chenier, I.; Chan, J.S.D.; Quaggin, S.; Hébert, M.-J.; Ingelfinger, J.R.; Zhang, S.-L. Maternal Diabetes Modulates Renal Morphogenesis in Offspring. J. Am. Soc. Nephrol. 2008, 19, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Chenier, I.; Tran, S.; Scotcher, M.; Chang, S.-Y.; Zhang, S.-L. Maternal diabetes programs hypertension and kidney injury in offspring. Pediatr. Nephrol. 2010, 25, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Alcazar, M.A.A.; Boehler, E.; Rother, E.; Amann, K.; Vohlen, C.; von Hörsten, S.; Plank, C.; Dötsch, J. Early Postnatal Hyperalimentation Impairs Renal Function via SOCS-3 Mediated Renal Postreceptor Leptin Resistance. Endocrinology 2012, 153, 1397–1410. [Google Scholar] [CrossRef]
- Guo, W.; Guan, X.; Pan, X.; Sun, X.; Wang, F.; Ji, Y.; Huang, P.; Deng, Y.; Zhang, Q.; Han, Q.; et al. Post-Natal Inhibition of NF-kappaB Activation Prevents Renal Damage Caused by Prenatal LPS Exposure. PLoS ONE 2016, 11, e0153434. [Google Scholar]
- Nüsken, E.; Fink, G.; Lechner, F.; Voggel, J.; Wohlfarth, M.; Sprenger, L.; Mehdiani, N.; Weber, L.T.; Liebau, M.C.; Brachvogel, B.; et al. Altered molecular signatures during kidney development after intrauterine growth restriction of different origins. J. Mol. Med. 2020, 98, 395–407. [Google Scholar] [CrossRef]
- Kasper, P.; Selle, J.; Vohlen, C.; Wilke, R.; Kuiper-Makris, C.; Klymenko, O.; Bae-Gartz, I.; Schomig, C.; Quaas, A.; Schumacher, B.; et al. Perinatal Obesity Induces Hepatic Growth Re-striction with Increased DNA Damage Response, Senescence, and Dysregulated Igf-1-Akt-Foxo1 Signaling in Male Offspring of Obese Mice. Int. J. Mol. Sci. 2022, 23, 5609. [Google Scholar] [CrossRef]
- Ahima, R.S. Connecting obesity, aging and diabetes. Nat. Med. 2009, 15, 996–997. [Google Scholar] [CrossRef]
- Tzanetakou, I.P.; Katsilambros, N.L.; Benetos, A.; Mikhailidis, D.P.; Perrea, D.N. “Is obesity linked to aging?”: Adipose tissue and the role of telomeres. Ageing Res. Rev. 2012, 11, 220–229. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Perez, L.M.; Pareja-Galeano, H.; Sanchis-Gomar, F.; Emanuele, E.; Lucia, A.; Galvez, B.G. ‘Adipaging’: Ageing and obesity share bio-logical hallmarks related to a dysfunctional adipose tissue. J. Physiol. 2016, 594, 3187–3207. [Google Scholar] [CrossRef]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Sun, Y.; Ge, X.; Li, X.; He, J.; Wei, X.; Du, J.; Sun, J.; Li, X.; Xun, Z.; Liu, W.; et al. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis. 2020, 11, 914. [Google Scholar] [CrossRef]
- Bae-Gartz, I.; Janoschek, R.; Kloppe, C.-S.; Vohlen, C.; Roels, F.; Oberthür, A.; Alcazar, M.A.A.; Lippach, G.; Muether, P.S.; Dinger, K.; et al. Running Exercise in Obese Pregnancies Prevents IL-6 Trans-signaling in Male Offspring. Med. Sci. Sports Exerc. 2016, 48, 829–838. [Google Scholar] [CrossRef]
- Janoschek, R.; Bae-Gartz, I.; Vohlen, C.; Alcazar, M.A.; Dinger, K.; Appel, S.; Dotsch, J.; Hucklenbruch-Rother, E. Dietary intervention in obese dams protects male offspring from WAT induction of TRPV4, adiposity, and hyperinsulinemia. Obesity 2016, 24, 1266–1273. [Google Scholar] [CrossRef]
- Bartke, A.; List, E.O.; Kopchick, J.J. The somatotropic axis and aging: Benefits of endocrine defects. Growth Horm. IGF Res. 2016, 27, 41–45. [Google Scholar] [CrossRef]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Mao, K.; Quipildor, G.F.; Tabrizian, T.; Novaj, A.; Guan, F.; Walters, R.O.; Delahaye, F.; Hubbard, G.B.; Ikeno, Y.; Ejima, K.; et al. Late-life targeting of the IGF-1 receptor improves healthspan and lifespan in female mice. Nat. Commun. 2018, 9, 2394. [Google Scholar] [CrossRef]
- Garinis, G.A.; Uittenboogaard, L.M.; Stachelscheid, H.; Fousteri, M.; van Ijcken, W.; Breit, T.M.; Van Steeg, H.; Mullenders, L.H.F.; van der Horst, G.; Brüning, J.C.; et al. Persistent transcription-blocking DNA lesions trigger somatic growth attenuation associated with longevity. Nature 2009, 11, 604–615. [Google Scholar] [CrossRef]
- Chow, H.-M.; Shi, M.; Cheng, A.; Gao, Y.; Chen, G.; Song, X.; So, R.W.L.; Zhang, J.; Herrup, K. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat. Neurosci. 2019, 22, 1806–1819. [Google Scholar] [CrossRef]
- Cheng, H.-T.; Huang, J.-W.; Chiang, C.-K.; Yen, C.-J.; Hung, K.-Y.; Wu, K.-D. Metabolic Syndrome and Insulin Resistance as Risk Factors for Development of Chronic Kidney Disease and Rapid Decline in Renal Function in Elderly. J. Clin. Endocrinol. Metab. 2012, 97, 1268–1276. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selle, J.; Bohl, K.; Höpker, K.; Wilke, R.; Dinger, K.; Kasper, P.; Abend, B.; Schermer, B.; Müller, R.-U.; Kurschat, C.; et al. Perinatal Obesity Sensitizes for Premature Kidney Aging Signaling. Int. J. Mol. Sci. 2023, 24, 2508. https://doi.org/10.3390/ijms24032508
Selle J, Bohl K, Höpker K, Wilke R, Dinger K, Kasper P, Abend B, Schermer B, Müller R-U, Kurschat C, et al. Perinatal Obesity Sensitizes for Premature Kidney Aging Signaling. International Journal of Molecular Sciences. 2023; 24(3):2508. https://doi.org/10.3390/ijms24032508
Chicago/Turabian StyleSelle, Jaco, Katrin Bohl, Katja Höpker, Rebecca Wilke, Katharina Dinger, Philipp Kasper, Bastian Abend, Bernhard Schermer, Roman-Ulrich Müller, Christine Kurschat, and et al. 2023. "Perinatal Obesity Sensitizes for Premature Kidney Aging Signaling" International Journal of Molecular Sciences 24, no. 3: 2508. https://doi.org/10.3390/ijms24032508
APA StyleSelle, J., Bohl, K., Höpker, K., Wilke, R., Dinger, K., Kasper, P., Abend, B., Schermer, B., Müller, R.-U., Kurschat, C., Nüsken, K.-D., Nüsken, E., Meyer, D., Savai Pullamsetti, S., Schumacher, B., Dötsch, J., & Alcazar, M. A. A. (2023). Perinatal Obesity Sensitizes for Premature Kidney Aging Signaling. International Journal of Molecular Sciences, 24(3), 2508. https://doi.org/10.3390/ijms24032508