

Sensory Phenotype of the Oesophageal Mucosa in Gastro-Oesophageal Reflux Disease


{kind=link}
Abstract
1. Introduction
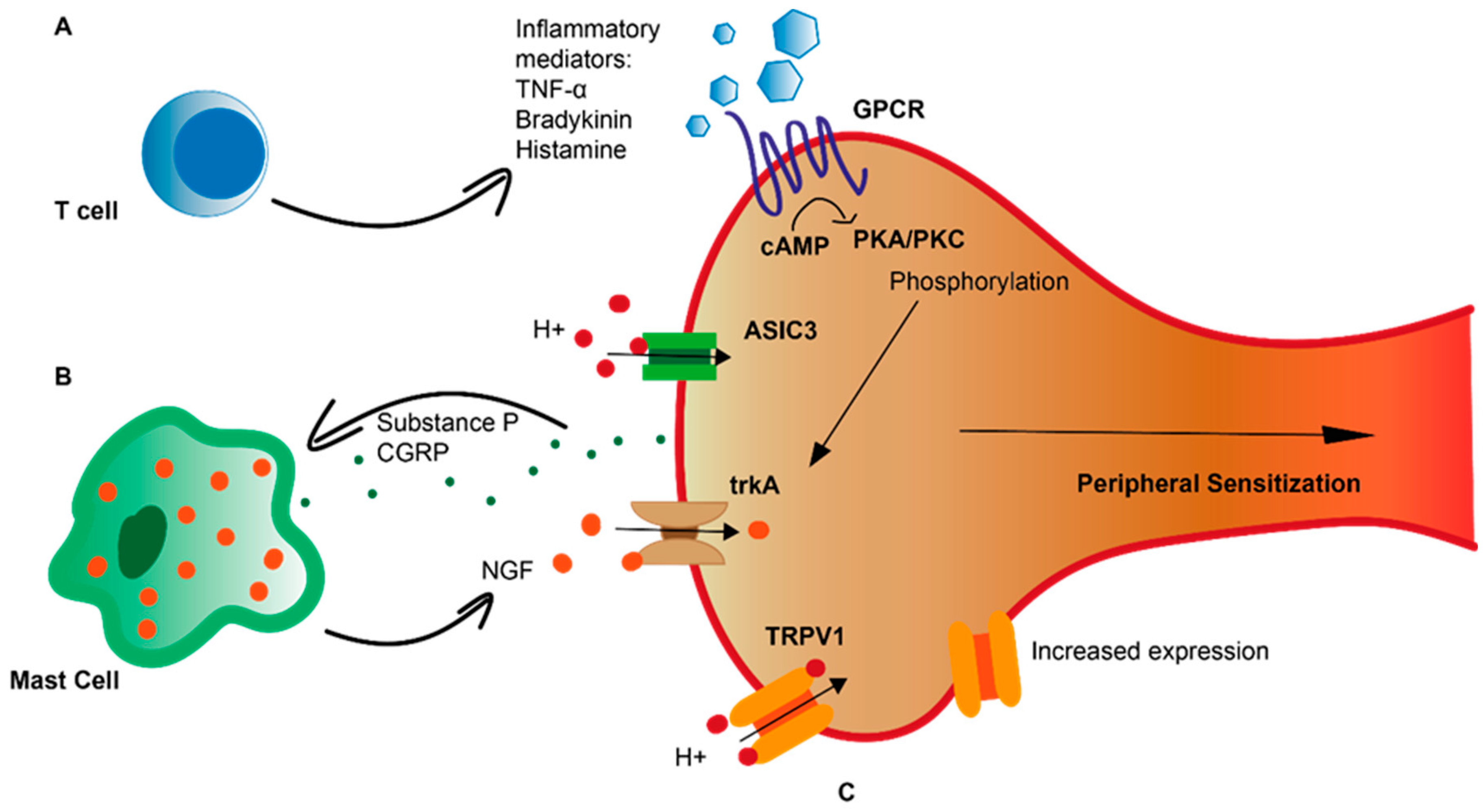
2. Peripheral Sensitization in GORD
3. Inflammation in Heartburn Pathogenesis
4. Microinflammation in GORD
5. Neuro-Immune Interactions in GORD
6. Mucosal Neuro-Anatomy
7. The Epithelial Barrier in GORD
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Lundell, L.R.; Dent, J.; Bennett, J.R.; Blum, A.L.; Armstrong, D.; Galmiche, J.P.; Johnson, F.; Hongo, M.; Richter, J.E.; Spechler, S.J.; et al. Endoscopic Assessment of Oesophagitis: Clinical and Functional Correlates and Further Validation of the Los Angeles Classification. Gut 1999, 45, 172–180. [Google Scholar] [CrossRef]
- Labenz, J.; Labenz, G.; Stephan, D.; Willeke, F. Insufficient Symptom Control under Long-Term Treatment with PPI in GERD—Fact or Fiction? MMW-Fortschr. Med. 2016, 158, 7–11. [Google Scholar] [CrossRef]
- Weijenborg, P.W.; Smout, A.J.P.M.; Verseijden, C.; van Veen, H.A.; Verheij, J.; de Jonge, W.J.; Bredenoord, A.J. Hypersensitivity to Acid Is Associated with Impaired Esophageal Mucosal Integrity in Patients with Gastroesophageal Reflux Disease with and without Esophagitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G323–G329. [Google Scholar] [CrossRef]
- Yang, M.; Li, Z.S.; Chen, D.F.; Zou, D.W.; Xu, X.R.; Fang, D.C.; Xu, G.M.; Stephens, R.L.; Wang, Z.G. Quantitative Assessment and Characterization of Visceral Hyperalgesia Evoked by Esophageal Balloon Distention and Acid Perfusion in Patients with Functional Heartburn, Nonerosive Reflux Disease, and Erosive Esophagitis. Clin. J. Pain 2010, 26, 326–331. [Google Scholar] [CrossRef]
- Nagahara, A.; Miwa, H.; Minoo, T.; Hojo, M.; Kawabe, M.; Osada, T.; Kurosawa, A.; Asaoka, D.; Terai, T.; Ohkusa, T.; et al. Increased Esophageal Sensitivity to Acid and Saline in Patients with Nonerosive Gastro-Esophageal Reflux Disease. J. Clin. Gastroenterol. 2006, 40, 891–895. [Google Scholar] [CrossRef]
- Medda, B.K.; Sengupta, J.N.; Lang, I.M.; Shaker, R. Response Properties of the Brainstem Neurons of the Cat Following Intra-Esophageal Acid-Pepsin Infusion. Neuroscience 2005, 135, 1285–1294. [Google Scholar] [CrossRef]
- Banerjee, B.; Medda, B.K.; Zheng, Y.; Miller, H.; Miranda, A.; Sengupta, J.N.; Shaker, R. Alterations in N-Methyl-D-Aspartate Receptor Subunits in Primary Sensory Neurons Following Acid-Induced Esophagitis in Cats. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G66–G77. [Google Scholar] [CrossRef]
- Bessou, P.; Perl, E.R. Response of Cutaneous Sensory Units with Unmyelinated Fibers to Noxious Stimuli. J. Neurophysiol. 1969, 32, 1025–1043. [Google Scholar] [CrossRef]
- Knowles, C.H.; Aziz, Q. Visceral Hypersensitivity in Nonerosive Reflux Disease. Gut 2008, 57, 674–683. [Google Scholar] [CrossRef]
- Akbar, A.; Yiangou, Y.; Facer, P.; Walters, J.R.F.; Anand, P.; Ghosh, S. Increased Capsaicin Receptor TRPV1-Expressing Sensory Fibres in Irritable Bowel Syndrome and Their Correlation with Abdominal Pain. Gut 2008, 57, 923. [Google Scholar] [CrossRef]
- Akbar, A.; Yiangou, Y.; Facer, P.; Brydon, W.G.; Walters, J.R.F.; Anand, P.; Ghosh, S. Expression of the TRPV1 Receptor Differs in Quiescent Inflammatory Bowel Disease with or without Abdominal Pain. Gut 2010, 59, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; de La Monte, S.; Ma, J.; Hong, J.; Tong, M.; Cao, W.; Behar, J.; Biancani, P.; Harnett, K.M. HCl-Activated Neural and Epithelial Vanilloid Receptors (TRPV1) in Cat Esophageal Mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G135–G143. [Google Scholar] [CrossRef]
- Guarino, M.P.L.; Cheng, L.; Ma, J.; Harnett, K.; Biancani, P.; Altomare, A.; Panzera, F.; Behar, J.; Cicala, M. Increased TRPV1 Gene Expression in Esophageal Mucosa of Patients with Non-Erosive and Erosive Reflux Disease. Neurogastroenterol. Motil. 2010, 22, 746–751.e219. [Google Scholar] [CrossRef] [PubMed]
- Bhat, Y.M.; Bielefeldt, K. Capsaicin Receptor (TRPV1) and Non-Erosive Reflux Disease. Eur. J. Gastroenterol. Hepatol. 2006, 18, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Matthews, P.J.; Aziz, Q.; Facer, P.; Davis, J.B.; Thompson, D.G.; Anand, P. Increased Capsaicin Receptor TRPV1 Nerve Fibres in the Inflamed Human Oesophagus. Eur. J. Gastroenterol. Hepatol. 2004, 16, 897–902. [Google Scholar] [CrossRef]
- Peles, S.; Medda, B.K.; Zhang, Z.; Banerjee, B.; Lehmann, A.; Shaker, R.; Sengupta, J.N. Differential Effects of Transient Receptor Vanilloid One (TRPV1) Antagonists in Acid-Induced Excitation of Esophageal Vagal Afferent Fibers of Rats. Neuroscience 2009, 161, 515–525. [Google Scholar] [CrossRef]
- Morgan, M.; Nencini, S.; Thai, J.; Ivanusic, J.J. TRPV1 Activation Alters the Function of Aδ and C Fiber Sensory Neurons That Innervate Bone. Bone 2019, 123, 168–175. [Google Scholar] [CrossRef]
- McKemy, D.D.; Neuhausser, W.M.; Julius, D. Identification of a Cold Receptor Reveals a General Role for TRP Channels in Thermosensation. Nature 2002, 416, 52–58. [Google Scholar] [CrossRef]
- Banovcin, P.; Duricek, M.; Zatko, T.; Liptak, P.; Hyrdel, R.; Kollarik, M. The Infusion of Menthol into the Esophagus Evokes Cold Sensations in Healthy Subjects but Induces Heartburn in Patients with Gastroesophageal Reflux Disease (GERD). Dis. Esophagus 2019, 32, doz038. [Google Scholar] [CrossRef]
- Yu, X.; Hu, Y.; Ru, F.; Kollarik, M.; Undem, B.J.; Yu, S. TRPM8 Function and Expression in Vagal Sensory Neurons and Afferent Nerves Innervating Guinea Pig Esophagus. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G489–G496. [Google Scholar] [CrossRef]
- Kress, M.; Waldmann, R. Chapter 8 Acid Sensing Ionic Channels. Curr. Top. Membr. 2006, 57, 241–276. [Google Scholar] [CrossRef]
- Gu, Q.; Lee, L.Y. Acid-Sensing Ion Channels and Pain. Pharmaceuticals 2010, 3, 1411–1425. [Google Scholar] [CrossRef]
- Price, M.P.; McIlwrath, S.L.; Xie, J.; Cheng, C.; Qiao, J.; Tarr, D.E.; Sluka, K.A.; Brennan, T.J.; Lewin, G.R.; Welsh, M.J. The DRASIC Cation Channel Contributes to the Detection of Cutaneous Touch and Acid Stimuli in Mice. Neuron 2001, 32, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Ru, F.; Banovcin, P.; Kollarik, M. Acid Sensitivity of the Spinal Dorsal Root Ganglia C-Fiber Nociceptors Innervating the Guinea Pig Esophagus. Neurogastroenterol. Motil. 2015, 27, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Akiba, Y.; Mizumori, M.; Kuo, M.; Ham, M.; Guth, P.H.; Engel, E.; Kaunitz, J.D. CO2 Chemosensing in Rat Oesophagus. Gut 2008, 57, 1654–1664. [Google Scholar] [CrossRef]
- Ustaoglu, A.; Sawada, A.; Lee, C.; Lei, W.-Y.; Chen, C.-L.; Hackett, R.; Sifrim, D.; Peiris, M.; Woodland, P. Heartburn Sensation in Non-Erosive Reflux Disease: Pattern of Superficial Sensory Nerves Expressing TRPV1 and Epithelial Cells Expressing ASIC3 Receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G804–G815. [Google Scholar] [CrossRef]
- Schuligoi, R.; Jocic, M.; Heinemann, A.; Schoninkle, E.; Pabst, M.A.; Holzer, P. Gastric Acid-Evoked c-Los Messenger RNA Expression in Rat Brainstem Is Signaled by Capsaicin-Resistant Vagal Afferents. Gastroenterology 1998, 115, 649–660. [Google Scholar] [CrossRef]
- Bielefeldt, K.; Davis, B.M. Differential Effects of ASIC3 and TRPV1 Deletion on Gastroesophageal Sensation in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G130–G138. [Google Scholar] [CrossRef]
- Wu, L.; Oshima, T.; Shan, J.; Sei, H.; Tomita, T.; Ohda, Y.; Fukui, H.; Watari, J.; Miwa, H. PAR-2 Activation Enhances Weak Acid-Induced ATP Release through TRPV1 and ASIC Sensitization in Human Esophageal Epithelial Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G695–G702. [Google Scholar] [CrossRef]
- Ji, R.-R.; Chamessian, A.; Zhang, Y.-Q. Pain Regulation by Non-Neuronal Cells and Inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Zelenka, M.; Schäfers, M.; Sommer, C. Intraneural Injection of Interleukin-1β and Tumor Necrosis Factor-Alpha into Rat Sciatic Nerve at Physiological Doses Induces Signs of Neuropathic Pain. Pain 2005, 116, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, K.B.; Agoston, A.T.; Odze, R.D.; Huo, X.; Pham, T.H.; Cipher, D.J.; Castell, D.O.; Genta, R.M.; Souza, R.F.; Spechler, S.J. Association of Acute Gastroesophageal Reflux Disease with Esophageal Histologic Changes. JAMA-J. Am. Med. Assoc. 2016, 315, 2104–2112. [Google Scholar] [CrossRef]
- Souza, R.F.; Huo, X.; Mittal, V.; Schuler, C.M.; Carmack, S.W.; Zhang, H.Y.; Zhang, X.; Yu, C.; Hormi-Carver, K.; Genta, R.M.; et al. Gastroesophageal Reflux Might Cause Esophagitis Through a Cytokine-Mediated Mechanism Rather Than Caustic Acid Injury. Gastroenterology 2009, 137, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Oshima, T.; Fukui, H.; Watari, J.; Miwa, H. Acidic Deoxycholic Acid and Chenodeoxycholic Acid Induce Interleukin-8 Production through P38 Mitogen-Activated Protein Kinase and Protein Kinase A in a Squamous Epithelial Model. J. Gastroenterol. Hepatol. 2013, 28, 823–828. [Google Scholar] [CrossRef]
- Shan, J.; Oshima, T.; Wu, L.; Fukui, H.; Watari, J.; Miwa, H. Interferon γ-Induced Nuclear Interleukin-33 Potentiates the Release of Esophageal Epithelial Derived Cytokines. PLoS ONE 2016, 11, e0151701. [Google Scholar] [CrossRef]
- Huo, X.; Agoston, A.T.; Dunbar, K.B.; Cipher, D.J.; Zhang, X.; Yu, C.; Cheng, E.; Zhang, Q.; Pham, T.H.; Tambar, U.K.; et al. Hypoxia-Inducible Factor-2α Plays a Role in Mediating Oesophagitis in GORD. Gut 2017, 66, 1542–1554. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef]
- Taylor, C.T. Interdependent Roles for Hypoxia Inducible Factor and Nuclear Factor-ΚB in Hypoxic Inflammation. J. Physiol. 2008, 586, 4055–4059. [Google Scholar] [CrossRef]
- Isomoto, H. Elevated Levels of Chemokines in Esophageal Mucosa of Patients with Reflux Esophagitis. Am. J. Gastroenterol. 2003, 98, 551–556. [Google Scholar] [CrossRef]
- Fujino, K.; de la Fuente, S.G.; Takami, Y.; Takahashi, T.; Mantyh, C.R. Attenuation of Acid Induced Oesophagitis in VR-1 Deficient Mice. Gut 2006, 55, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, B.; Medda, B.K.; Schmidt, J.; Zheng, Y.; Zhang, Z.; Shaker, R.; Sengupta, J.N. Altered Expression of P2X3 in Vagal and Spinal Afferents Following Esophagitis in Rats. Histochem. Cell Biol. 2009, 132, 585–597. [Google Scholar] [CrossRef]
- Wardlaw, A.J.; Moqbel, R.; Cromwell, O.; Kay, A.B. Platelet-Activating Factor. A Potent Chemotactic and Chemokinetic Factor for Human Eosinophils. J. Clin. Investig. 1986, 78, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Paterson, W.G.; Kieffer, C.A.; Feldman, M.J.; Miller, D.V.; Morris, G.P. Role of Platelet-Activating Factor in Acid-Induced Esophageal Mucosal Injury. Dig. Dis. Sci. 2007, 52, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Kandulski, A.; Wex, T.; Mönkemüller, K.; Kuester, D.; Fry, L.C.; Roessner, A.; Malfertheiner, P. Proteinase-Activated Receptor-2 in the Pathogenesis of Gastroesophageal Reflux Disease. Am. J. Gastroenterol. 2010, 105, 1934–1943. [Google Scholar] [CrossRef]
- Dent, J. Microscopic Esophageal Mucosal Injury in Nonerosive Reflux Disease. Clin. Gastroenterol. Hepatol. 2007, 5, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Tobey, N.A.; Carson, J.L.; Alkiek, R.A.; Orlando, R.C. Dilated Intercellular Spaces: A Morphological Feature of Acid Reflux- Damaged Human Esophageal Epithelium. Gastroenterology 1996, 111, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Triantos, C.; Koukias, N.; Karamanolis, G.; Thomopoulos, K. Changes in the Esophageal Mucosa of Patients with Non Erosive Reflux Disease: How Far Have We Gone? World J. Gastroenterol. 2015, 21, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Coull, J.A.M.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; de Koninck, Y. BDNF from Microglia Causes the Shift in Neuronal Anion Gradient Underlying Neuropathic Pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef]
- Ansel, J.C.; Brown, J.R.; Payan, D.G.; Brown, M.A. Substance P Selectively Activates TNF-Alpha Gene Expression in Murine Mast Cells. J. Immunol. 1993, 150, 4478–4485. [Google Scholar] [CrossRef]
- Hosoi, J.; Murphy, G.F.; Egan, C.L.; Lerner, E.A.; Grabbe, S.; Asahina, A.; Granstein, R.D. Regulation of Langerhans Cell Function by Nerves Containing Calcitonin Gene-Related Peptide. Nature 1993, 363, 159–163. [Google Scholar] [CrossRef] [PubMed]
- de Bortoli, N.; Tolone, S.; Frazzoni, M.; Martinucci, I.; Sgherri, G.; Albano, E.; Ceccarelli, L.; Stasi, C.; Bellini, M.; Savarino, V.; et al. Gastroesophageal Reflux Disease, Functional Dyspepsia and Irritable Bowel Syndrome: Common Overlapping Gastrointestinal Disorders. Ann. Gastroenterol. 2018, 31, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Diolaiti, D.; Bernardoni, R.; Trazzi, S.; Papa, A.; Porro, A.; Bono, F.; Herbert, J.M.; Perini, G.; della Valle, G. Functional Cooperation between TrkA and P75NTR Accelerates Neuronal Differentiation by Increased Transcription of GAP-43 and P21(CIP/WAF) Genes via ERK1/2 and AP-1 Activities. Exp. Cell Res. 2007, 313, 2980–2992. [Google Scholar] [CrossRef] [PubMed]
- Barbara, G.; Stanghellini, V.; de Giorgio, R.; Cremon, C.; Cottrell, G.S.; Santini, D.; Pasquinelli, G.; Morselli-Labate, A.M.; Grady, E.F.; Bunnett, N.W.; et al. Activated Mast Cells in Proximity to Colonic Nerves Correlate with Abdominal Pain in Irritable Bowel Syndrome. Gastroenterology 2004, 126, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Peiris, M.; Bulmer, D.C.; Baker, M.D.; Boundouki, G.; Sinha, S.; Hobson, A.; Lee, K.; Aziz, Q.; Knowles, C.H. Human Visceral Afferent Recordings: Preliminary Report. Gut 2011, 60, 204–208. [Google Scholar] [CrossRef]
- Hockley, J.R.F.; Barker, K.H.; Taylor, T.S.; Callejo, G.; Husson, Z.M.; Bulmer, D.C.; Smith, E.S.J. Acid and Inflammatory Sensitisation of Naked Mole-Rat Colonic Afferent Nerves. Mol. Pain 2020, 16, 1744806920903150. [Google Scholar] [CrossRef]
- Mamet, J.; Baron, A.; Lazdunski, M.; Voilley, N. Proinflammatory Mediators, Stimulators of Sensory Neuron Excitability via the Expression of Acid-Sensing Ion Channels. J. Neurosci. 2002, 22, 10662–10670. [Google Scholar] [CrossRef]
- Silva, R.O.; Bingana, R.D.; Sales, T.M.A.L.; Moreira, R.L.R.; Costa, D.V.S.; Sales, K.M.O.; Brito, G.A.C.; Santos, A.A.; Souza, M.Â.N.; Soares, P.M.G.; et al. Role of TRPV1 Receptor in Inflammation and Impairment of Esophageal Mucosal Integrity in a Murine Model of Nonerosive Reflux Disease. Neurogastroenterol. Motil. 2018, 30, e13340. [Google Scholar] [CrossRef]
- Woodland, P.; Aktar, R.; Mthunzi, E.; Lee, C.; Peiris, M.; Preston, S.L.; Blackshaw, L.A.; Sifrim, D. Distinct Afferent Innervation Patterns within the Human Proximal and Distal Esophageal Mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G525–G531. [Google Scholar] [CrossRef]
- Woodland, P.; Shen Ooi, J.L.; Grassi, F.; Nikaki, K.; Lee, C.; Evans, J.A.; Koukias, N.; Triantos, C.; McDonald, S.A.; Peiris, M.; et al. Superficial Esophageal Mucosal Afferent Nerves May Contribute to Reflux Hypersensitivity in Nonerosive Reflux Disease. Gastroenterology 2017, 153, 1230–1239. [Google Scholar] [CrossRef]
- Krarup, A.L.; Ny, L.; Åstrand, M.; Bajor, A.; Hvid-Jensen, F.; Hansen, M.B.; Simrén, M.; Funch-Jensen, P.; Drewes, A.M. Randomised Clinical Trial: The Efficacy of a Transient Receptor Potential Vanilloid 1 Antagonist AZD1386 in Human Oesophageal Pain. Aliment. Pharmacol. Ther. 2011, 33, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Tobey, N.A.; Argote, C.M.; Awayda, M.S.; Vanegas, X.C.; Orlando, R.C. Effect of Luminal Acidity on the Apical Cation Channel in Rabbit Esophageal Epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G796–G805. [Google Scholar] [CrossRef]
- Tobey, N.A.; Hosseini, S.S.; Argote, C.M.; Dobrucali, A.M.; Awayda, M.S.; Orlando, R.C. Dilated Intercellular Spaces and Shunt Permeability in Nonerosive Acid-Damaged Esophageal Epithelium. Am. J. Gastroenterol. 2004, 99, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Barlow, W.J.; Orlando, R.C. The Pathogenesis of Heartburn in Nonerosive Reflux Disease: A Unifying Hypothesis. Gastroenterology 2005, 128, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Farré, R.; Fornari, F.; Blondeau, K.; Vieth, M.; de Vos, R.; Bisschops, R.; Mertens, V.; Pauwels, A.; Tack, J.; Sifrim, D. Acid and Weakly Acidic Solutions Impair Mucosal Integrity of Distal Exposed and Proximal Non-Exposed Human Oesophagus. Gut 2010, 59, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Bortolotti, M.; Fabbri, A.; Areni, A.; Cenacchi, G.; Scialpi, C.; Miglioli, M.; di Febo, G. Reversibility of GERD Ultrastructural Alterations and Relief of Symptoms after Omeprazole Treatment. Am. J. Gastroenterol. 2005, 100, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Tobey, N.A.; Gambling, T.M.; Vanegas, X.C.; Carson, J.L.; Orlando, R.C. Physicochemical Basis for Dilated Intercellular Spaces in Non-Erosive Acid-Damaged Rabbit Esophageal Epithelium. Dis. Esophagus 2008, 21, 757–764. [Google Scholar] [CrossRef]
- Woodland, P.; Lee, C.; Duraysami, Y.; Farré, R.; Dettmar, P.; Sifrim, D. Assessment and Protection of Esophageal Mucosal Integrity in Patients with Heartburn without Esophagitis. Am. J. Gastroenterol. 2013, 108, 535–543. [Google Scholar] [CrossRef]
- Kessing, B.F.; Bredenoord, A.J.; Weijenborg, P.W.; Hemmink, G.J.M.; Loots, C.M.; Smout, A.J.P.M. Esophageal Acid Exposure Decreases Intraluminal Baseline Impedance Levels. Am. J. Gastroenterol. 2011, 106, 2093–2097. [Google Scholar] [CrossRef]
- Woodland, P.; Al-Zinaty, M.; Yazaki, E.; Sifrim, D. In Vivo Evaluation of Acid-Induced Changes in Oesophageal Mucosa Integrity and Sensitivity in Non-Erosive Reflux Disease. Gut 2013, 62, 1256–1261. [Google Scholar] [CrossRef]
- Farré, R.; Blondeau, K.; Clement, D.; Vicario, M.; Cardozo, L.; Vieth, M.; Mertens, V.; Pauwels, A.; Silny, J.; Jimenez, M.; et al. Evaluation of Oesophageal Mucosa Integrity by the Intraluminal Impedance Technique. Gut 2011, 60, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Farré, R.; de Vos, R.; Geboes, K.; Verbecke, K.; Berghe, P.V.; Depoortere, I.; Blondeau, K.; Tack, J.; Sifrim, D. Critical Role of Stress in Increased Oesophageal Mucosa Permeability and Dilated Intercellular Spaces. Gut 2007, 56, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Demaude, J.; Salvador-Cartier, C.; Fioramonti, J.; Ferrier, L.; Bueno, L. Phenotypic Changes in Colonocytes Following Acute Stress or Activation of Mast Cells in Mice: Implications for Delayed Epithelial Barrier Dysfunction. Gut 2006, 55, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Brown, B.E.; Crumrine, D.; Chang, S.; Man, M.Q.; Elias, P.M.; Feingold, K.R. Mechanisms by Which Psychologic Stress Alters Cutaneous Permeability Barrier Homeostasis and Stratum Corneum Integrity. J. Investig. Dermatol. 2005, 124, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.V.; Yuan, P.Q.; Wang, L.; Peng, Y.L.; Chen, C.Y.; Taché, Y. Identification and Characterization of Multiple Corticotropin-Releasing Factor Type 2 Receptor Isoforms in the Rat Esophagus. Endocrinology 2007, 148, 1675–1687. [Google Scholar] [CrossRef]
- Aziz, Q.; Fass, R.; Gyawali, C.P.; Miwa, H.; Pandolfino, J.E.; Zerbib, F. Esophageal Disorders. Gastroenterology 2016, 150, 1368–1379. [Google Scholar] [CrossRef]
- Naliboff, B.D.; Mayer, M.; Fass, R.; Fitzgerald, L.Z.; Chang, L.; Bolus, R.; Mayer, E.A. The Effect of Life Stress on Symptoms of Heartburn. Psychosom. Med. 2004, 66, 426–434. [Google Scholar] [CrossRef]
- Sarkar, S.; Aziz, Q.; Woolf, C.J.; Hobson, A.R.; Thompson, D.G. Contribution of Central Sensitisation to the Development of Non-Cardiac Chest Pain. Lancet 2000, 356, 1154–1159. [Google Scholar] [CrossRef]
- Hobson, A.R.; Aziz, Q. Brain Processing of Esophageal Sensation in Health and Disease. Gastroenterol. Clin. 2004, 33, 69–91. [Google Scholar] [CrossRef]
- Moiseff, R.; Olson, N.; Suriawinata, A.A.; Rothstein, R.I.; Lisovsky, M. CD8 T-Cell–Predominant Lymphocytic Esophagitis Is One of the Major Patterns of Lymphocytic Inflammation in Gastroesophageal Reflux Disease. Arch. Pathol. Lab. Med. 2021, 145, 1138–1143. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ustaoglu, A.; Woodland, P. Sensory Phenotype of the Oesophageal Mucosa in Gastro-Oesophageal Reflux Disease. Int. J. Mol. Sci. 2023, 24, 2502. https://doi.org/10.3390/ijms24032502
Ustaoglu A, Woodland P. Sensory Phenotype of the Oesophageal Mucosa in Gastro-Oesophageal Reflux Disease. International Journal of Molecular Sciences. 2023; 24(3):2502. https://doi.org/10.3390/ijms24032502
Chicago/Turabian StyleUstaoglu, Ahsen, and Philip Woodland. 2023. "Sensory Phenotype of the Oesophageal Mucosa in Gastro-Oesophageal Reflux Disease" International Journal of Molecular Sciences 24, no. 3: 2502. https://doi.org/10.3390/ijms24032502
APA StyleUstaoglu, A., & Woodland, P. (2023). Sensory Phenotype of the Oesophageal Mucosa in Gastro-Oesophageal Reflux Disease. International Journal of Molecular Sciences, 24(3), 2502. https://doi.org/10.3390/ijms24032502