
Fibrosis-Related Gene Profiling in Liver Biopsies of PiZZ α1-Antitrypsin Children with Different Clinical Courses

 , , ,
, , ,  , , ,
, , ,  , ,
, , 
Abstract
1. Introduction
2. Results
2.1. Patient Cohorts
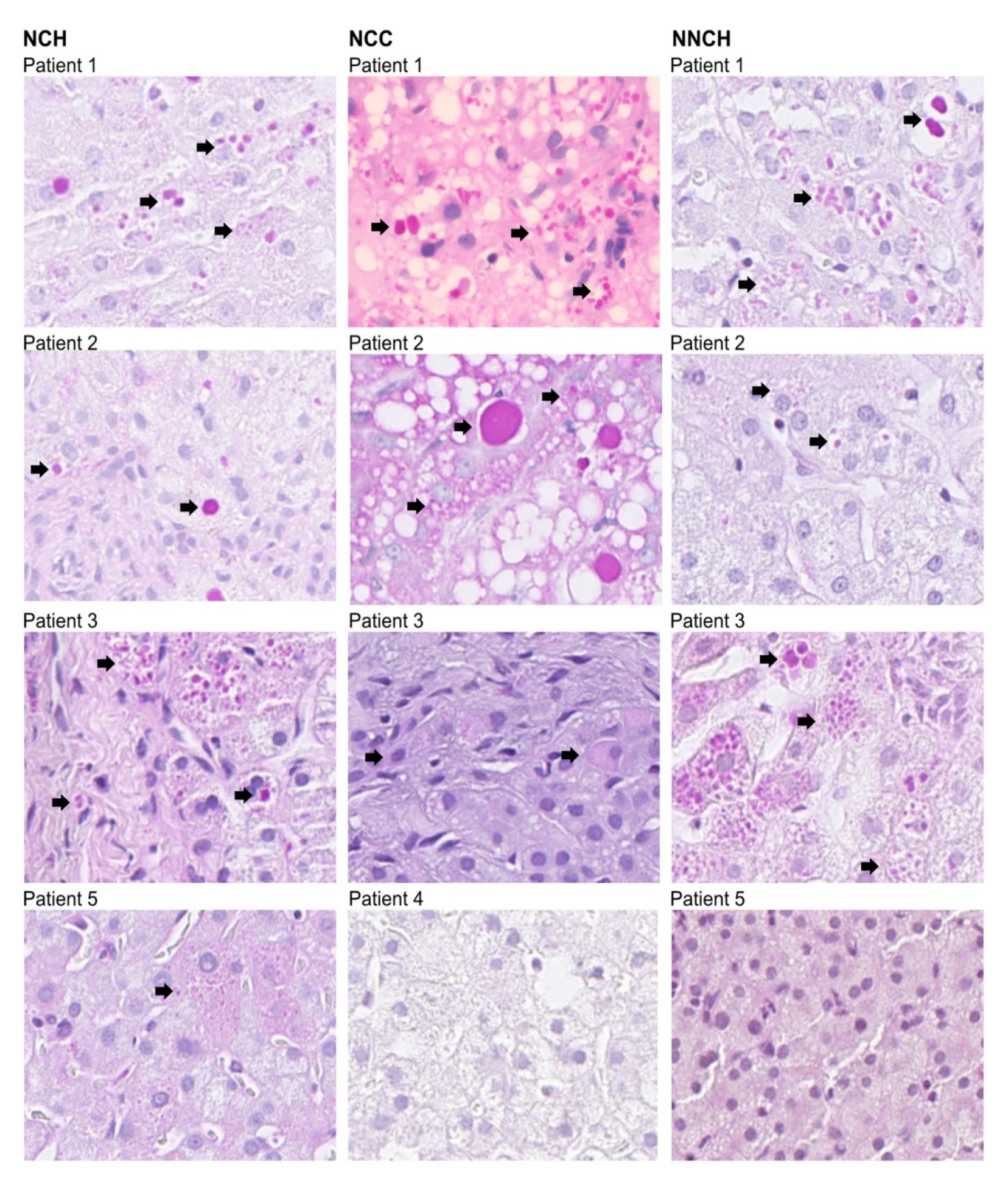
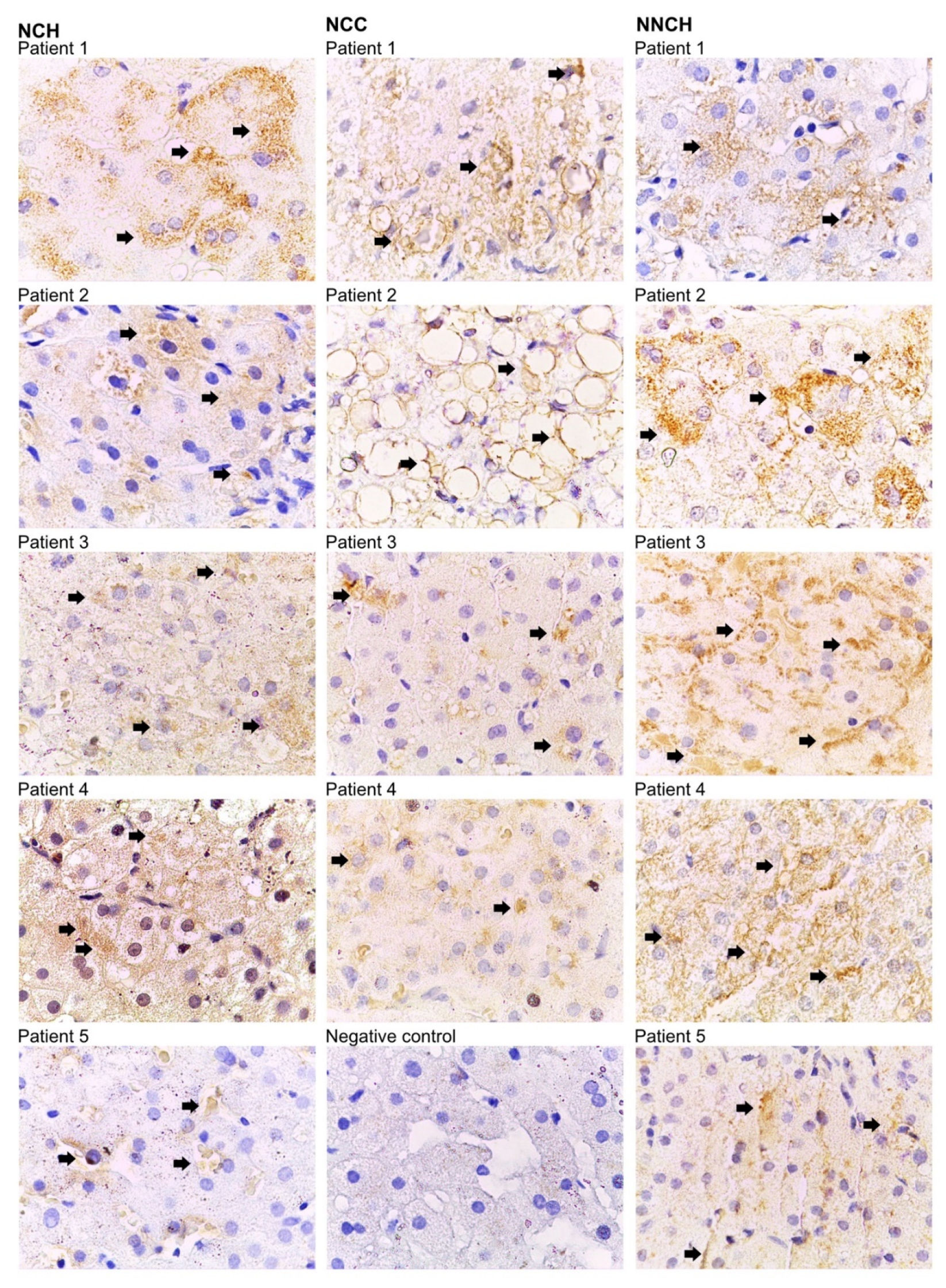
2.2. Immunohistochemistry
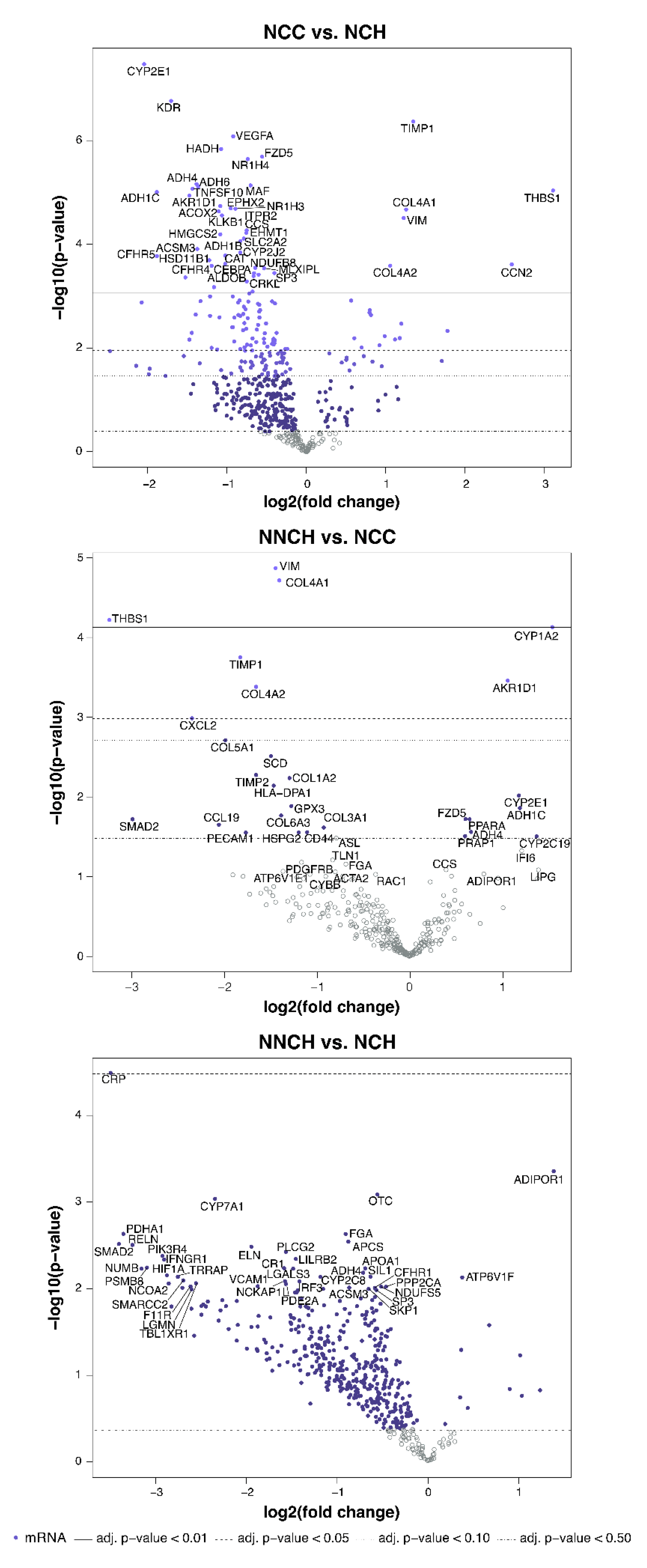
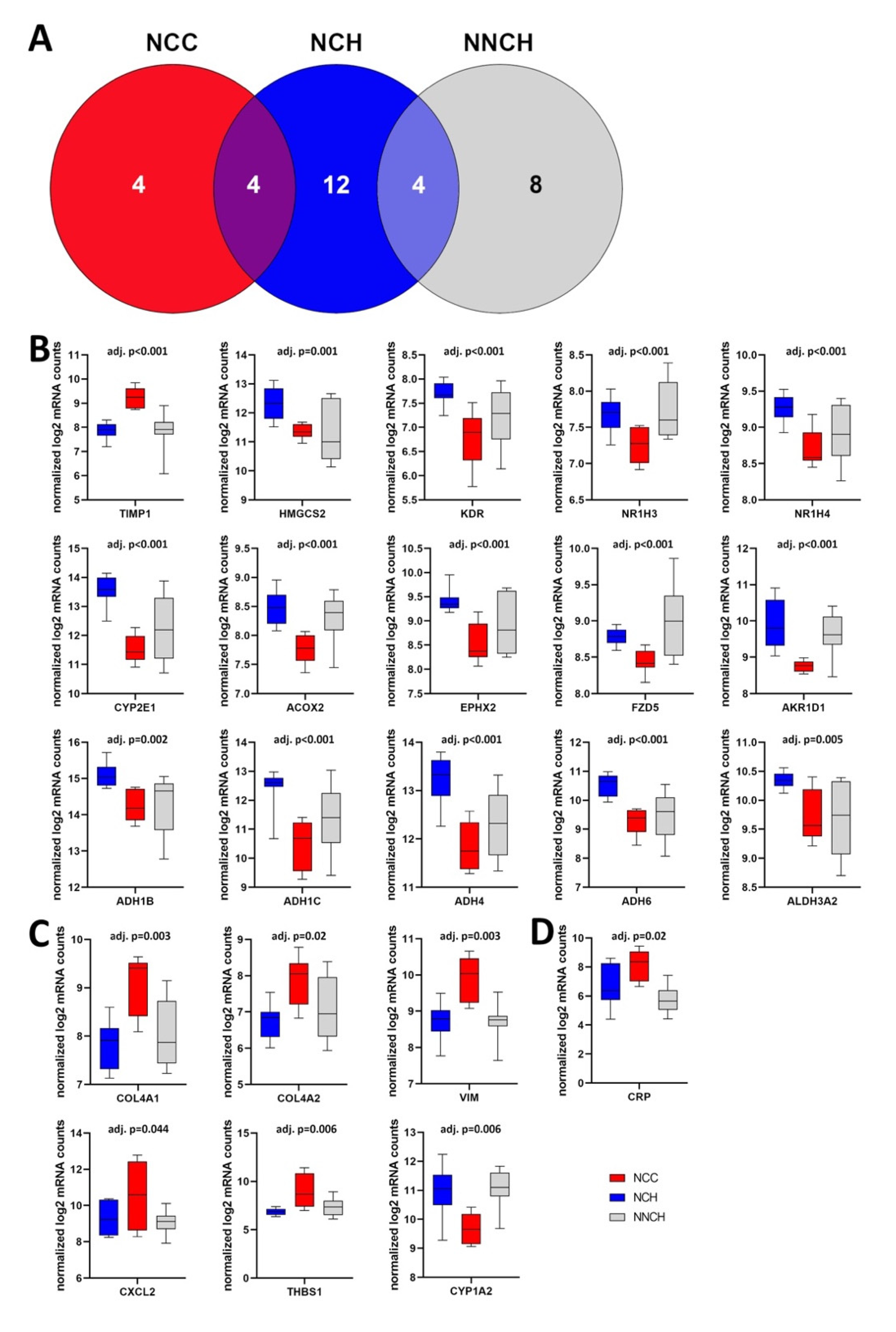
2.3. Targeted Liver Gene Profiling
2.4. Activity of Biological Functions
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Clinical Diagnosis of PiZZ AATD Children
4.3. Liver Biopsies of PiZZ AATD Children
4.4. Immunohistochemistry
4.5. Gene Expression, Biological Pathways, and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Kohnlein, T.; Welte, T. The discovery of alpha1-antitrypsin and its role in health and disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Blanco, I.; de Serres, F.J.; Fernandez-Bustillo, E.; Lara, B.; Miravitlles, M. Estimated numbers and prevalence of PI*S and PI*Z alleles of alpha1-antitrypsin deficiency in European countries. Eur. Respir. J. 2006, 27, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Stoller, J.K.; Smith, P.; Yang, P.; Spray, J. Physical and social impact of alpha 1-antitrypsin deficiency: Results of a survey. Clevel. Clin. J. Med. 1994, 61, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Sveger, T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N. Engl. J. Med. 1976, 294, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Sveger, T. Alpha 1-antitrypsin deficiency in early childhood. Pediatrics 1978, 62, 22–25. [Google Scholar] [CrossRef]
- Sveger, T. The natural history of liver disease in alpha 1-antitrypsin deficient children. Acta Paediatr. Scand. 1988, 77, 847–851. [Google Scholar] [CrossRef]
- Katzer, D.; Ganschow, R.; Strnad, P.; Hamesch, K. Pi*ZZ-related liver disease in children and adults—Narrative review of the typical presentation and management of alpha-1 antitrypsin deficiency. Dig. Med. Res. 2021, 4, 31. [Google Scholar] [CrossRef]
- Suri, A.; Patel, D.; Teckman, J.H. Alpha-1 Antitrypsin Deficiency Liver Disease. Clin. Liver Dis. 2022, 26, 391–402. [Google Scholar] [CrossRef]
- Wu, Y.; Whitman, I.; Molmenti, E.; Moore, K.; Hippenmeyer, P.; Perlmutter, D.H. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc. Natl. Acad. Sci. USA 1994, 91, 9014–9018. [Google Scholar] [CrossRef]
- Sveger, T. Prospective study of children with alpha 1-antitrypsin deficiency: Eight-year-old follow-up. J. Pediatr. 1984, 104, 91–94. [Google Scholar] [CrossRef]
- Nebbia, G.; Hadchouel, M.; Odievre, M.; Alagille, D. Early assessment of evolution of liver disease associated with alpha 1-antitrypsin deficiency in childhood. J. Pediatr. 1983, 102, 661–665. [Google Scholar] [CrossRef]
- Francavilla, R.; Castellaneta, S.P.; Hadzic, N.; Chambers, S.M.; Portmann, B.; Tung, J.; Cheeseman, P.; Rela, M.; Heaton, N.D.; Mieli-Vergani, G. Prognosis of alpha-1-antitrypsin deficiency-related liver disease in the era of paediatric liver transplantation. J. Hepatol. 2000, 32, 986–992. [Google Scholar] [CrossRef]
- Townsend, S.A.; Edgar, R.G.; Ellis, P.R.; Kantas, D.; Newsome, P.N.; Turner, A.M. Systematic review: The natural history of alpha-1 antitrypsin deficiency, and associated liver disease. Aliment. Pharmacol. Ther. 2018, 47, 877–885. [Google Scholar] [CrossRef]
- Volpert, D.; Molleston, J.P.; Perlmutter, D.H. Alpha1-antitrypsin deficiency-associated liver disease progresses slowly in some children. J. Pediatr. Gastroenterol. Nutr. 2000, 31, 258–263. [Google Scholar] [CrossRef]
- Khodayari, N.; Wang, R.L.; Oshins, R.; Lu, Y.; Millett, M.; Aranyos, A.M.; Mostofizadeh, S.; Scindia, Y.; Flagg, T.O.; Brantly, M. The Mechanism of Mitochondrial Injury in Alpha-1 Antitrypsin Deficiency Mediated Liver Disease. Int. J. Mol. Sci. 2021, 22, 13255. [Google Scholar] [CrossRef]
- Bouchecareilh, M. Alpha-1 Antitrypsin Deficiency-Mediated Liver Toxicity: Why Do Some Patients Do Poorly? What Do We Know So Far? Chronic Obstr. Pulm Dis. 2020, 7, 172–181. [Google Scholar] [CrossRef]
- Marek, G.; Collinsworth, A.; Liu, C.; Brantly, M.; Clark, V. Quantitative measurement of the histological features of alpha-1 antitrypsin deficiency-associated liver disease in biopsy specimens. PLoS ONE 2021, 16, e0256117. [Google Scholar] [CrossRef]
- Noordeen, F. Hepatitis B virus infection: An insight into infection outcomes and recent treatment options. Virusdisease 2015, 26, 1–8. [Google Scholar] [CrossRef]
- Tseng, T.C.; Huang, L.R. Immunopathogenesis of Hepatitis B Virus. J. Infect. Dis. 2017, 216, S765–S770. [Google Scholar] [CrossRef]
- Lindblad, D.; Blomenkamp, K.; Teckman, J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology 2007, 46, 1228–1235. [Google Scholar] [CrossRef]
- Schaefer, B.; Haschka, D.; Finkenstedt, A.; Petersen, B.S.; Theurl, I.; Henninger, B.; Janecke, A.R.; Wang, C.Y.; Lin, H.Y.; Veits, L.; et al. Impaired hepcidin expression in alpha-1-antitrypsin deficiency associated with iron overload and progressive liver disease. Hum. Mol. Genet. 2015, 24, 6254–6263. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A.; Strandvik, B. Natural history of children with alpha 1-antitrypsin deficiency and neonatal cholestasis. Acta Paediatr. Scand. 1982, 71, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A.; Strandvik, B. Urinary excretion of tetrahydroxylated bile acids in children with alpha 1-antitrypsin deficiency and neonatal cholestasis. Scand. J. Clin. Lab. Investig. 1984, 44, 387–392. [Google Scholar] [CrossRef]
- Beuers, U.; Hohenester, S.; de Buy Wenniger, L.J.; Kremer, A.E.; Jansen, P.L.; Elferink, R.P. The biliary HCO(3)(-) umbrella: A unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010, 52, 1489–1496. [Google Scholar] [CrossRef]
- Lefeuvre, C.; Roux, M.; Blanchard, S.; Le Guillou-Guillemette, H.; Boursier, J.; Lunel-Fabiani, F.; Jeannin, P.; Pivert, A.; Ducancelle, A. Analysis of hepatic fibrosis markers in the serum of chronic hepatitis B patients according to basal core promoter/precore mutants. Sci. Rep. 2022, 12, 10261. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.; Chen, Y.; Sohel, H.; Ke, X.; Chen, J.; Li, Y.X. The correlation and role analysis of COL4A1 and COL4A2 in hepatocarcinogenesis. Aging 2020, 12, 204–223. [Google Scholar] [CrossRef]
- Shin, H.J.; Gil, M.; Lee, I.S. Association of Elevated Expression Levels of COL4A1 in Stromal Cells with an Immunosuppressive Tumor Microenvironment in Low-Grade Glioma, Pancreatic Adenocarcinoma, Skin Cutaneous Melanoma, and Stomach Adenocarcinoma. J. Pers. Med. 2022, 12, 534. [Google Scholar] [CrossRef]
- Paulin, D.; Lilienbaum, A.; Kardjian, S.; Agbulut, O.; Li, Z. Vimentin: Regulation and pathogenesis. Biochimie 2022, 197, 96–112. [Google Scholar] [CrossRef]
- Wang, W.M.; Zhang, W.S.; Yang, Z.G. Vimentin (VIM) predicts advanced liver fibrosis in chronic hepatitis B patients: A random forest-derived analysis. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 5164–5177. [Google Scholar] [CrossRef]
- Min-DeBartolo, J.; Schlerman, F.; Akare, S.; Wang, J.; McMahon, J.; Zhan, Y.; Syed, J.; He, W.; Zhang, B.; Martinez, R.V. Thrombospondin-I is a critical modulator in non-alcoholic steatohepatitis (NASH). PLoS ONE 2019, 14, e0226854. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Turpin, C.P.; Wang, S. Role of thrombospondin 1 in liver diseases. Hepatol. Res. 2017, 47, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Tissue inhibitors of metalloproteinases in liver fibrosis. Int. J. Biochem. Cell Biol. 1997, 29, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S.; Subramaniyam, D.; Piitulainen, E.; Kohnlein, T.; Sveger, T. Plasma levels of TIMP-1 are higher in 34-year-old individuals with severe alpha1-antitrypsin deficiency. Thorax 2010, 65, 937. [Google Scholar] [CrossRef]
- Kaserman, J.E.; Werder, R.B.; Wang, F.; Matte, T.; Higgins, M.I.; Dodge, M.; Lindstrom-Vautrin, J.; Hinds, A.; Bullitt, E.; Caballero, I.S.; et al. Modeling of Alpha-1 Antitrypsin Deficiency with Syngeneic Human iPSC-Hepatocytes Reveals Metabolic Dysregulation and Cellular Heterogeneity in PiMZ and PiZZ Hepatocytes. bioRxiv 2022, arXiv:2022.2002.2001.478663. [Google Scholar] [CrossRef]
- Piccolo, P.; Annunziata, P.; Soria, L.R.; Attanasio, S.; Barbato, A.; Castello, R.; Carissimo, A.; Quagliata, L.; Terracciano, L.M.; Brunetti-Pierri, N. Down-regulation of hepatocyte nuclear factor-4alpha and defective zonation in livers expressing mutant Z alpha1-antitrypsin. Hepatology 2017, 66, 124–135. [Google Scholar] [CrossRef]
- Goodman, R.P.; Markhard, A.L.; Shah, H.; Sharma, R.; Skinner, O.S.; Clish, C.B.; Deik, A.; Patgiri, A.; Hsu, Y.H.; Masia, R.; et al. Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature 2020, 583, 122–126. [Google Scholar] [CrossRef]
- Nikolaou, N.; Gathercole, L.L.; Marchand, L.; Althari, S.; Dempster, N.J.; Green, C.J.; van de Bunt, M.; McNeil, C.; Arvaniti, A.; Hughes, B.A.; et al. AKR1D1 is a novel regulator of metabolic phenotype in human hepatocytes and is dysregulated in non-alcoholic fatty liver disease. Metabolism 2019, 99, 67–80. [Google Scholar] [CrossRef]
- Nikolaou, N.; Gathercole, L.L.; Kirkwood, L.; Dunford, J.E.; Hughes, B.A.; Gilligan, L.C.; Oppermann, U.; Penning, T.M.; Arlt, W.; Hodson, L.; et al. AKR1D1 regulates glucocorticoid availability and glucocorticoid receptor activation in human hepatoma cells. J. Steroid Biochem. Mol. Biol. 2019, 189, 218–227. [Google Scholar] [CrossRef]
- Abshagen, K.; Konig, M.; Hoppe, A.; Muller, I.; Ebert, M.; Weng, H.; Holzhutter, H.G.; Zanger, U.M.; Bode, J.; Vollmar, B.; et al. Pathobiochemical signatures of cholestatic liver disease in bile duct ligated mice. BMC Syst. Biol. 2015, 9, 83. [Google Scholar] [CrossRef]
- Fukushima, S.; Okuno, H.; Shibatani, N.; Nakahashi, Y.; Seki, T.; Okazaki, K. Effect of biliary obstruction and internal biliary drainage on hepatic cytochrome P450 isozymes in rats. World J. Gastroenterol. 2008, 14, 2556–2560. [Google Scholar] [CrossRef]
- Villeneuve, J.P.; Pichette, V. Cytochrome P450 and liver diseases. Curr. Drug Metab. 2004, 5, 273–282. [Google Scholar] [CrossRef]
- Keitel, V.; Droge, C.; Haussinger, D. Targeting FXR in Cholestasis. Handb Exp. Pharmacol. 2019, 256, 299–324. [Google Scholar] [CrossRef]
- Ramos Pittol, J.M.; Milona, A.; Morris, I.; Willemsen, E.C.L.; van der Veen, S.W.; Kalkhoven, E.; van Mil, S.W.C. FXR Isoforms Control Different Metabolic Functions in Liver Cells via Binding to Specific DNA Motifs. Gastroenterology 2020, 159, 1853–1865. [Google Scholar] [CrossRef]
- Czubkowski, P.; Thompson, R.J.; Jankowska, I.; Knisely, A.S.; Finegold, M.; Parsons, P.; Cielecka-Kuszyk, J.; Strautnieks, S.; Pawlowska, J.; Bull, L.N. Progressive familial intrahepatic cholestasis—Farnesoid X receptor deficiency due to NR1H4 mutation: A case report. World J. Clin. Cases 2021, 9, 3631–3636. [Google Scholar] [CrossRef]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Beaven, S.W.; Wroblewski, K.; Wang, J.; Hong, C.; Bensinger, S.; Tsukamoto, H.; Tontonoz, P. Liver X receptor signaling is a determinant of stellate cell activation and susceptibility to fibrotic liver disease. Gastroenterology 2011, 140, 1052–1062. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Koganti, L.; Muchenditsi, A.; Pendyala, V.S.; Huso, D.; Hankin, J.; Murphy, R.C.; Huster, D.; Merle, U.; Mangels, C.; et al. Activation of liver X receptor/retinoid X receptor pathway ameliorates liver disease in Atp7B(-/-) (Wilson disease) mice. Hepatology 2016, 63, 1828–1841. [Google Scholar] [CrossRef]
- Asif, S.; Kim, R.Y.; Fatica, T.; Sim, J.; Zhao, X.; Oh, Y.; Denoncourt, A.; Cheung, A.C.; Downey, M.; Mulvihill, E.E.; et al. Hmgcs2-mediated ketogenesis modulates high-fat diet-induced hepatosteatosis. Mol. Metab. 2022, 61, 101494. [Google Scholar] [CrossRef]
- Alonso-Pena, M.; Espinosa-Escudero, R.; Herraez, E.; Briz, O.; Cagigal, M.L.; Gonzalez-Santiago, J.M.; Ortega-Alonso, A.; Fernandez-Rodriguez, C.; Bujanda, L.; Calvo Sanchez, M.; et al. Beneficial effect of ursodeoxycholic acid in patients with acyl-CoA oxidase 2 (ACOX2) deficiency-associated hypertransaminasemia. Hepatology 2022, 76, 1259–1274. [Google Scholar] [CrossRef]
- Batts, K.P.; Ludwig, J. Chronic hepatitis. An update on terminology and reporting. Am. J. Surg. Pathol. 1995, 19, 1409–1417. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Group | Gender | Age at Biopsy (Years) | Clinical Status (Last Contact) | Liver Function (Last Contact) | Other Organs | Hepatocytes | Fibrosis | Portal Tract | Inflammation | Fibrosis (Total) | F/4 (After Tx) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fibrosis | Bile Duct Proliferation | |||||||||||
| NCH | F | 10 | 0 | 0 | 0 | PAS-D+ | ++ | ++ | + | |||
| F | 1 | 0 | 0 | 0 | PAS-D++ | ++ | ++ | (+) | ||||
| M | 4 | 0 | 0 | 0 | PAS-D+++ | ++ | ++ | ++ | ||||
| F | 5 | 0 | 0 | 0 | PAS-D+ | +++ | +++ | + | ++ | |||
| M | 8 | 0 | 0 | 0 | PAS-D+++ | + | + | + | ||||
| NCC | M | 3 | MOF | failure | MOF | PAS-D+ steatosis | + | ++++ | + | ++++ | Tx | |
| F | 10 | MOF | failure | MOF | PAS-D + | +++ | ++++ | ++ | ++++ | Tx | ||
| F | 5 | MOF | failure | MOF | PAS-D + | ++++ | +++ | + | ++++ | Tx | ||
| F | 15 | MOF | failure | MOF | PAS-D - | ++ | ++++ | +++ | ++++ | Tx | ||
| NNCH | M | 2 | 0 | 0 | 0 | PAS-D++ | ++ | + | ++ | |||
| F | 4 | 0 | 0 | 0 | PAS-D++ | + | + | + | ||||
| F | 12 | 0 | 0 | 0 | PAS-D- | + | ||||||
| 19 | PAS-D+ | + | ||||||||||
| M | 6 | 0 | 0 | 0 | Vacuoli | + | ||||||
| M | 4 | 0 | 0 | 0 | PAS-D- | + | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamp, J.C.; Kappe, N.N.; Moro, C.F.; Fuge, J.; Kuehnel, M.P.; Wrenger, S.; Welte, T.; Hoek, B.v.; Jonigk, D.D.; Khedoe, P.P.S.J.; et al. Fibrosis-Related Gene Profiling in Liver Biopsies of PiZZ α1-Antitrypsin Children with Different Clinical Courses. Int. J. Mol. Sci. 2023, 24, 2485. https://doi.org/10.3390/ijms24032485
Kamp JC, Kappe NN, Moro CF, Fuge J, Kuehnel MP, Wrenger S, Welte T, Hoek Bv, Jonigk DD, Khedoe PPSJ, et al. Fibrosis-Related Gene Profiling in Liver Biopsies of PiZZ α1-Antitrypsin Children with Different Clinical Courses. International Journal of Molecular Sciences. 2023; 24(3):2485. https://doi.org/10.3390/ijms24032485
Chicago/Turabian StyleKamp, Jan C., Naomi N. Kappe, Carlos Fernández Moro, Jan Fuge, Mark P. Kuehnel, Sabine Wrenger, Tobias Welte, Bart van Hoek, Danny D. Jonigk, Padmini P. S. J. Khedoe, and et al. 2023. "Fibrosis-Related Gene Profiling in Liver Biopsies of PiZZ α1-Antitrypsin Children with Different Clinical Courses" International Journal of Molecular Sciences 24, no. 3: 2485. https://doi.org/10.3390/ijms24032485
APA StyleKamp, J. C., Kappe, N. N., Moro, C. F., Fuge, J., Kuehnel, M. P., Wrenger, S., Welte, T., Hoek, B. v., Jonigk, D. D., Khedoe, P. P. S. J., Strnad, P., Björnstedt, M., Stolk, J., Janciauskiene, S., & Nemeth, A. (2023). Fibrosis-Related Gene Profiling in Liver Biopsies of PiZZ α1-Antitrypsin Children with Different Clinical Courses. International Journal of Molecular Sciences, 24(3), 2485. https://doi.org/10.3390/ijms24032485