
Primary Liver Cancers: Connecting the Dots of Cellular Studies and Epidemiology with Metabolomics

Abstract
1. Introduction
2. Current Understanding of Liver Cancer Trends, Etiology and Correlates
2.1. Current Epidemiological and Moleular Understanding of Hepatoblastoma (HB)
2.2. Current Epidemiological and Moelcuar Understanding of Cholagniocarinoma (CCA)
2.3. Current Epidemiological and Molecular Understanding of Hepatocellular Carcinoma (HCC)
3. An Integrative Metabolomics Approach to Liver Cancers of Hepatoblastoma (HB), Cholangiocarinoma (CCA) and Hepatocellular Carcinoma (HCC)
3.1. An Integrative Metabolomics Approach to Hepatoblastoma (HB)
3.2. An Integrative Metabolomics Approach to Cholagniocarinoma (CCA)

{kind=link}
{kind=link}
| Liver Cancer | Metabolites |
|---|---|
| Hepatoblastoma (HB) 1 | Lactate, Ammonium, Gammy-glutamyl transferase, Alkaline phosphatase, Methylmalonyl-CoA Mutase MMUT, Methylamlonic acid, Reticulin, Glutamin synthetase, Propionyl CoA, Succinyl CoA, 2-Methylcitrate, Β-catenin expression, Glutamine synthetase expression, Glypican 3 expression, Bile salt export pump expression, Fumarate, Succinate, D-2-Hydroxyglurate (2-HG) |
| Cholangiocarcinoma (CCA) 2 | Bile acids (7β), Phophatidylcholine (PC), 2-Hydroxyglurate 2-HG, CXCl12/SDF-1, HMGB1, IL-6, TGF-β, TNF-α, EGF-like family, FGF-19, HGF, Adrenomedullin, Prostaglandin E2, 17β-Estradiol, miRNA, cfDNA, CYFRA 21-1, MMP, osteopontin, periostin, IL-6 |
| Hepatocellular carcinoma (HCC) 3 | Succinic acid, Fumaric acid, Malic acid, Glucose, Lactic acid, Hypoxanthine, Xanthosine, Adenonsine Monophosphate AMP, Propionylcarnitine, Linoleic acid, Coenzyme Q10, PC (34:2), PC (38:3), PC (36:1), PC (38:2), Glycerol 3-Phosphate, Glycerylphosphorylethanolamine, Glycerophosphocholine, Bile acids, Fatty acids |
3.3. An Integrative Metabolomics Approach to Hepatocellular Carcinoma (HCC)
4. Salient Characteristics of Liver Cancers
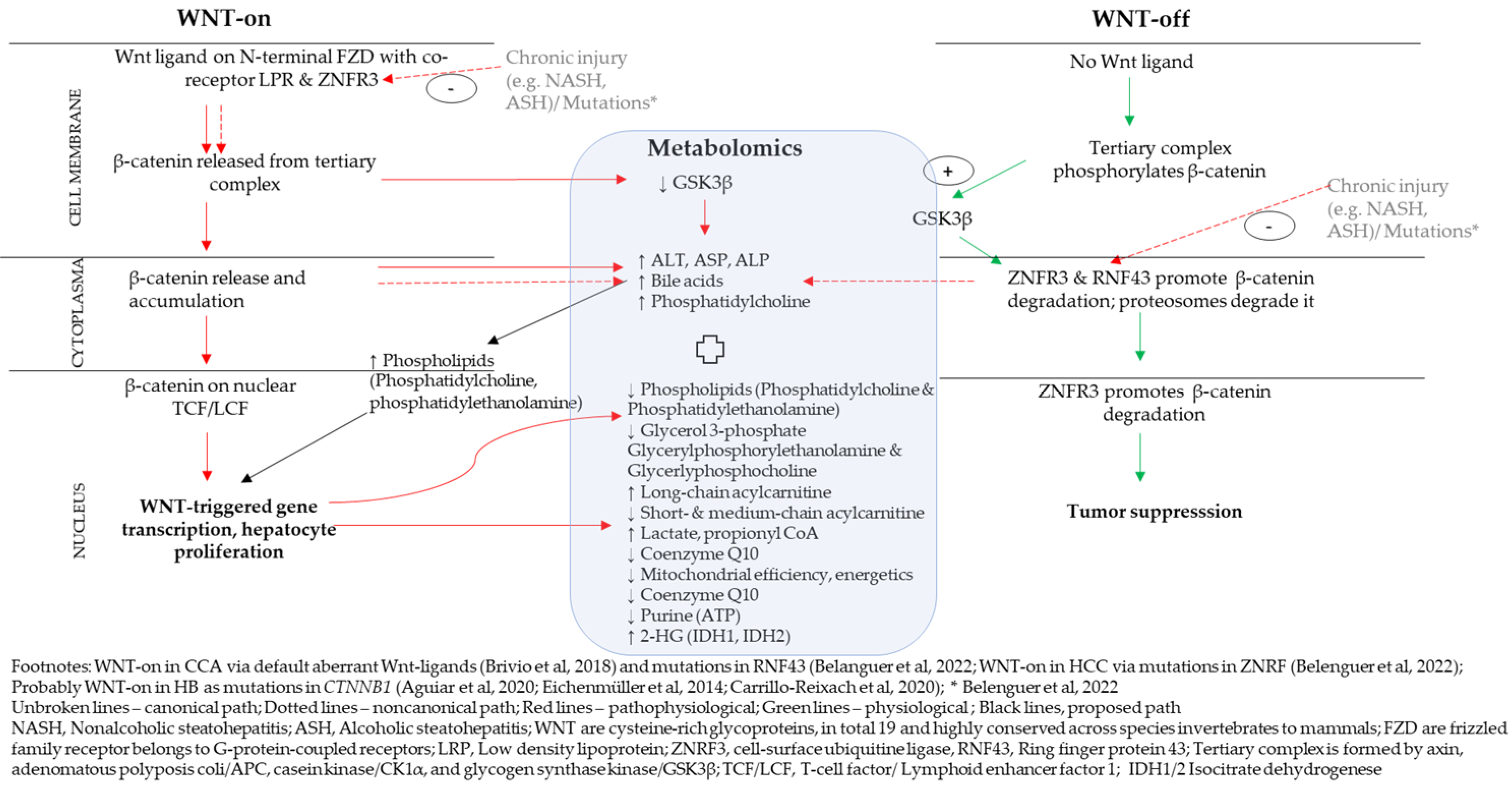
4.1. A Proposed Integrative Link between WNT/β-Catenin Pathway and Metabolomics
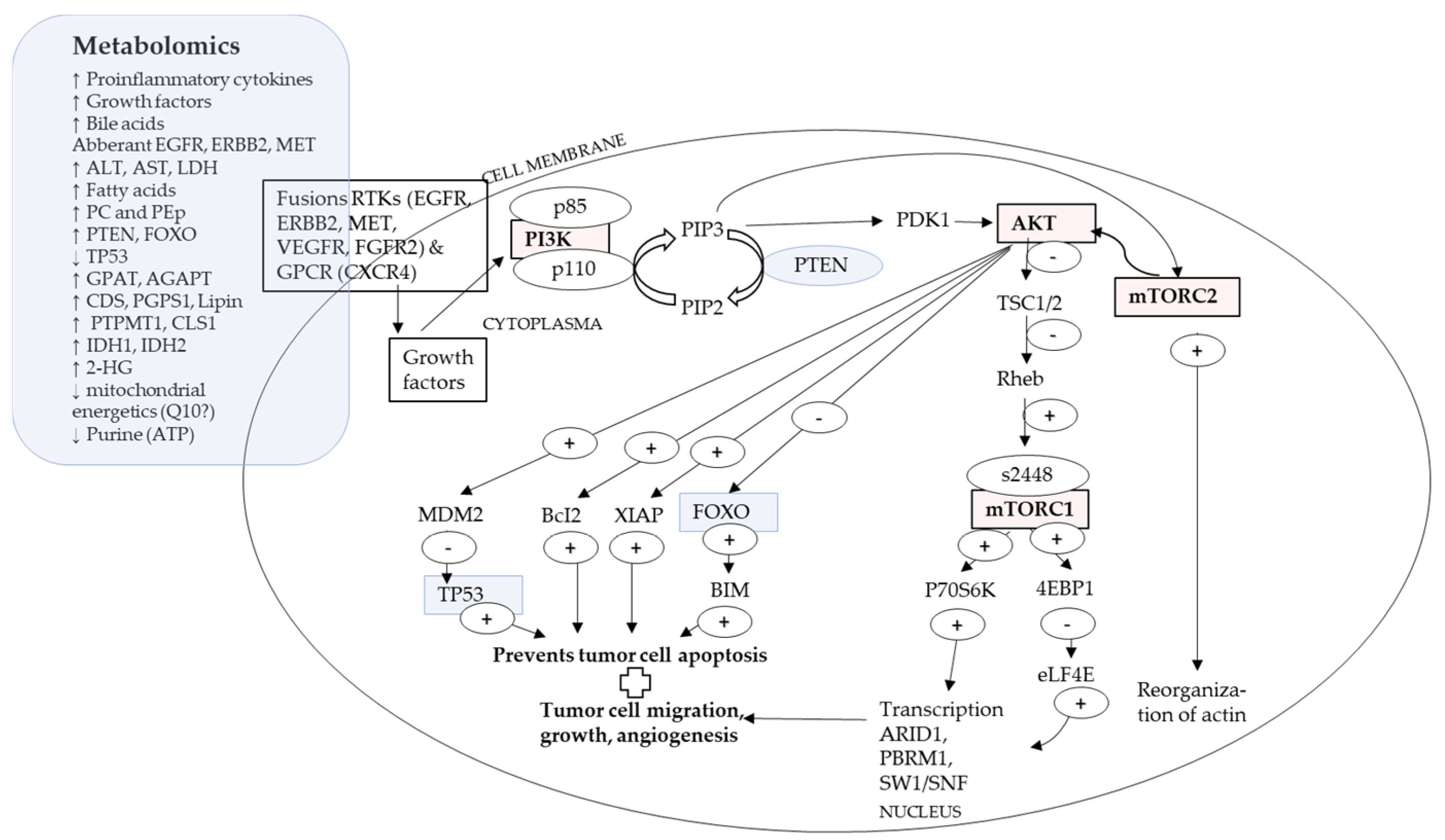
4.2. A Proposed Integrative Link between P13K/AKT/mTOR Pathway and Metabolomics
4.3. What Metabolomics Might Offer for the Investigation of Secondary Liver Cancers
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| 2-HG | D-2-Hydroxyglurate |
| 4EBP1 | Eukaryotic translation initiation factor 4E-binding protein |
| αKG | Alpha Ketoglurate |
| AGPAT | Acylclycerin-3-phosphate-O-acyltransferase |
| AKT | Gene AKT1, AKT2, AKT3 for protein kinase B (PKBα/β/γ) |
| ALT | Alanine transaminase |
| ALP | Alkaline phosphatase |
| AST | Aspartate transaminase |
| AP1 | Activator protein 1, a transcription factor |
| ARID1A | AT-rich interaction domain 1A |
| ATF4 | Activating transcription factor 4 |
| BAP1 | BRCA1 associated protein 1 |
| Bcl2 | B-cell lymphoma 2, a protein, the gene encodes an integral outer Mitochondrial membrane protein that blocks the apoptotic death |
| BRCA1/2 | Breast cancer gene 1 and 2 |
| CDS | Diacylglycerol synthases |
| C/EBPα | CCAAT/Enhancer-binding protein, a Notch signal protein |
| CHK2 | Checkpoint kinase 2, an effector of DNA damage response |
| CL | Cardiolipin |
| CLS1 | Collagenase, Type 1 |
| COX-2 | Cyclooxygenase-2, downstream of TNK-α |
| CSL | Cotranscriptional factors (such as MAML1, RBPJ, p300) |
| CTNNB1 | Catenin beta 1 gene |
| CX3CL1/12 | C-X3-C motif chemokine fractalkine ligand 1 gene/ligand 12 gene |
| DLL1 | Delta-like canonical notch ligand 1, a protein coding gene |
| DVL | Disheveled; positive regulator of WNT/β-catenin pathway; DVL is recruited to the plasma membrane and binds to FZD |
| KEAP1 | Kelch-like ECH associated protein 1 |
| ECT2 | Epithelial cell transforming 2 |
| EGFR | Epidermal Growth Factor-Like Receptor (Family), also known as ErbB1 or HER1 |
| eLF4E | Eukaryotic translation initiation factor 4E |
| ERBB2 | V-erb-b2 erythroblastic leukemia viral oncogene homolog 2 |
| ERK | Extracellular-signal-Regulated Kinase, also known as p44/42 |
| EPCA | Early prostate cancer antigen |
| FGFR | Fibroblast growth factor |
| FGFR2 | Fibroblast growth factor rector 2 gene |
| FGF21 | Fibroblast growth factor 21 |
| FOXO4 | Forkhead box protein O4, a protein coded by FOXO4 gene |
| FUT1 | Fucosyltransferase 1 |
| FAK | Focal adhesion kinase, downstream of TNK-α |
| FZD | Frizzled or class F of G protein-coupled receptors |
| GDF15 | Growth differentiation factor 15 of the family TGF-β |
| GPAT | Glycerolphosphate-O-Acyltransferase |
| GPCR | G-protein coupled receptors |
| GSK 3β | Gylcogen synthase kinase 3 beta |
| HER2 | Human epidermal growth factor receptor, scientific name ERBB2, belongs to HMGB1—High-Mobility Group Box 1 or High-Mobility Group Protein 1, an agent to indicate if cell necrosis has set in |
| HGF/c-MET | Hepatocyte growth factor-mesenchymal-epithelial transition factor pathway |
| IDH1/2 | Enzyme isocitrate dehydrogenase and the coding genes (IDH1gene Arg132, IDH2 gene Arg172 point mutation) |
| IL-6 | Interleukin 6 |
| JAK | Janus kinase |
| JAK/STAT3 | Janus kinase (JK)—signal transducer and activator of transcription (STAT) pathway |
| KRT-19 | Creatine 19 protein coding gene |
| LATS1/2 | Large tumor suppressor kinase ½ (Hippo pathway, also indicated in WNT/CTNNB1 pathway) |
| LDH | Lactate dehydrogenase, an enzyme |
| LGR4-5-6 | Leucine-rich repeat-containing G-protein-coupled Receptor (4-5-6) |
| Lipin | A phosphatase, Overproduction of it promotes obesity |
| LRP | Low-density lipoprotein Receptor-related Protein (5/6) |
| LncRNA | Long noncoding RNA |
| Lnc-EGFR | Long noncoding RNA, specifically the long noncoding epidermal growth factor receptor |
| MAML1 | Mastermind-like transcriptional coactivator 1 (of the Notch pathway, with NOTCH1, MAML1, RBPJK (suppressed upon GPR50 knockdown)) |
| MAPK | p38 Mitogen-activated protein kinase |
| MDM2 | Mouse double minute 2 protein coding gene, a proto-oncogene; The gene encodes a nuclear-localized E3 ubiquitin ligase, the encoded protein can promote tumor formation, essential for p53 regulation |
| MET | Protein oncogene, also called c-Met or tyrosine protein kinase Met or hepatocyte growth factor receptor (HGFR) |
| MMP-7/9 | Matrix metalloproteinase with zinc-dependent endopeptidases, and a protein coding gene |
| MOB1 | Monopolar spindle-one-binder protein (1) |
| MST1/2 | Mammmalian STE20-like protein kinase 1/2, regulate lymphocyte development, trafficking, survival and antigen recognition by naïve T cells, MST1/2 and YAP expression increased in platelet-driven growth factor receptor-α 8PDGFRα) |
| mTOR | Mammalian target of rapamycin, immunosuppression with rapamycin |
| mTORC1 | mTOR complex 1, made up of mTOR, Raptor, mLST8, PRAS40, extremely sensitive to rapamycin |
| mTORC2 | mTOR complex 2, made up of mTOR, Rictor, Sin1, mLST8, a serine/threonine kinase mTOR less sensitive to rapamycin |
| MUC1/4 | Mucine 1/4 (Protein) |
| NCID | Intracellular domain of Notch |
| NFE2L2 | Nuclear factor, erythroid 2 like bZIP transcription factor 2 |
| NFE2L2/KEAP1 | Nuclear factor, erythroid 2 like bZIP transcription factor 2- Kelch-like ECH associated protein 1 pathway |
| NF-kappaB | Nuclear factor kappa-light-chain-enhancer of activated B-cells protein complex |
| NK | Natural killer cells, of the immune system |
| p300 | Histone acyltransferases, such as, p300 (others: CBP, SAGA, etc.) |
| P72S6K | Ribosomal protein S6 kinase beta-1 |
| PBRM1 | Polybromo 1 gene |
| PC | Phosphatidylcholine (a kind of phospholipid) |
| PCK 1 | Phophoenolpyruvate carboxykinase 1 |
| PDC | Pyruvate dehydrogenase complex |
| PDK1 | Phosphoinositide-dependent kinase-1 |
| PEp | Phosphatidylethanolamine/plasmalogen (a kind of phospholipid) |
| PGPS1 | Phosphatidylglycerophosphate synthase 1 |
| PERK | Protein kinase R (PKR)-like endoplasmic reticulum kinase |
| P13K/AKT/mTOR | Phosphoinsositide-3-kinase (PI3K)/protein kinase B (AKT) (of AGC kinase family)-/mammalian target of rapamycin (mTOR) pathway, PI3K is an heterodimeric enzyme protein involved in lipid synthesis composed of large family of lipids and serine/threonine kinases and has p110 catalytic and p85 regulatory subunits |
| PIK3CA | Phophatidylinsositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (a protein coding gene) |
| PLK1 | Polo like kinase 1 |
| PRKCA/PRKCB | Protein kinase C alpha/beta |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-triphosphate |
| PRPP | Phosphoribosylpyrophosphate |
| PTEN | Phosphatase and tensin homolog, a tumor suppressor protein, which suppresses activation of AKT |
| PTPMT1 | Mitochondrial phosphatase 1 |
| RAF | Serine/Threonine protein kinase, e.g., ARAF, BRAF and CRAF (refer to SMAD) |
| RAS | Renin angiotensin system |
| RAS/ERK/AP1 | Renin angiotensin system-Extracellular-signal-regulated kinase-Activator protein 1 pathway; also RAS/RAF/MEK/MAPK activated RAS-a small G-protein-activated BRAF-activated MEK-activated ERK-MAPK |
| Rheb | Ras homolog enriched in brain, a GTP-binding protein |
| RNF43 | Ring Finger Protein 43 (a protein coding gene) |
| RSPO2 | R-Spondin 2 (a protein coding gene) |
| RTKs | Receptor Tryrosinkinases (also RYKs) |
| SAV1 | Salvador fmailiy WW domain containing protein 1 |
| SDF-1 | Stromal cell-derived factor 1 |
| SMAD | Protein and genes, a tumor suppressor gene SMAD and function as Main signal transducer for receptors of the transforming growth factor, activated by protein serin/threonine kinase (refer to RAF) |
| SP/NK-1R | Substance P and the neurokinin-1 receptor |
| SPTLC1 | Serine Palmityoltransferase, long chain base subunit 1, a protein Encoded by SPLTC1 gene |
| STAT3 | Signal transducer and activator of transcription 3 |
| SW1/SNF | SWitch/Sucrose Non-Fermentable, subfamily of ATP-dependent chromatin remodeling complexes |
| TCF/LEF | T-cell factor-lymphoid enhancer-binding factor transcription Factors |
| TEAD | WWTR1/TAZ-transcriptional enhanced associate domain |
| TERT | Telomerase-reverse-transcriptase |
| TGF-β | Transforming growth factor-β, a pleiotropic cytokine three forms TGF-β1, TGF-β2, TGF-β3) (see GDF15) |
| TNF-α | Tumor necrosis factor-α, a pleiotropic cytokine belonging to the TNF superfamily, predominantly expressed by macrophages, T and B lymphocytes, and natural killer (NK) cells |
| TSC1/2 | Tuberous sclerosis complex 1 and 2 |
| TP53 | Protein 53 |
| VEGF | Vascular endothelial growth factor |
| VEGFR | VEGF receptor |
| WNT | Wingless-related integration site (identified first in Drosophila) |
| WNT/β-catenin | WNT gene- β-catenin pathway, the canonical WNT pathway Involves nuclear translocation of β-catenin and activation of target genes via T-cell factor/lymphoid enhancer-binding factor (TCF/LEF) transcription factors, controlling cell proliferation The noncanonical WNT pathway is independent of β-catenin and TCF/LEF (e.g., WNT/Ca2+ pathway) and regulates cell polarity and migration. Both WNT pathways are mutually regulated. |
| XIAP | X-linked inhibitor of apoptosis protein |
| YAP/TAZ | Transcriptional regulators YAP (Yes-associated protein 1) and TAZ |
| ZNRF3 | Cell-surface transmembrane E3 ubiquitin ligase zinc and ring finger (a protein coding gene, which with RNF43 and RSPO2 are considered as a master switch that determines specification by similarity) |
Appendix A
| Steatosis 1 | Cancers 2 |
| AFP | |
| ALDH3A2 | |
| ALDH7A1 | |
| ALDOC | |
| ANGPT4 | |
| ARID1A | |
| ATF4 | |
| ATM | |
| AURKB | |
| AXIN1 | |
| AXIN2 | |
| BAP1 | |
| BEX1 | |
| BICCI1 | |
| BIRC5 | |
| BRAF | BRAF |
| BRCA1 | |
| BRCA2 | |
| BUB1 | |
| CCND1 | |
| CCND2 | |
| CDC2 | |
| CREB3L4 | |
| CRTC2 | |
| CTNNB1 | |
| CYP (CYP1A1, FYP2E1) | |
| DLG7 | |
| DLK1 | |
| ECT2 | |
| EGFR | |
| EFNA1 | |
| ERBB1-3 | |
| FGF12 | |
| FGFR4 | FGFR2, FGFR1-3 |
| GLUL | |
| GPI | |
| GNA13 | |
| HDAC2 | |
| IGF2 | |
| IKBKG | |
| INSR | |
| ITGAV | |
| KDM | |
| KMT2C | |
| KRAS | |
| MAP2K2 | |
| MAP3K7 | |
| METLL1 | |
| MEG3 | |
| MGEA5 | |
| MML | |
| MYC | |
| NDN | |
| NFKB1 | |
| NPM1 | |
| OGT | |
| PBRM1 | |
| PDGFB | |
| PEG3 | |
| PEG10 | |
| PFKL | |
| PHKA1 | |
| PHKB | |
| PIK3CA | |
| PPHLN1 | |
| PLK1 | |
| PPP1CA | |
| PRKACA(CAMP activated) | |
| PRKCA-PRKCB (activated Ca & Diacylglycerol) | |
| PTEN | |
| RHBG | |
| RNF43 | |
| RRAS2 | |
| RPS6KA2 | |
| SMAD4 | |
| SMARCA | |
| STK11 | |
| TACC3 | |
| TACSTD1 | |
| TAPBP | |
| TP53 | |
| VEGFB | |
| ZNRF3 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.; Gogineni, V.; Saeian, K. Epidemiology of Primary and Secondary Liver Cancers. Semin. Interv. Radiol. 2006, 23, 047–063. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, T.F.M.; Rivas, M.P.; Costa, S.; Maschietto, M.; Rodrigues, T.; de Barros, J.S.; Barbosa, A.C.; Valieris, R.; Fernandes, G.R.; Bertola, D.R.; et al. Insights Into the Somatic Mutation Burden of Hepatoblastomas From Brazilian Patients. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Eichenmüller, M.; Trippel, F.; Kreuder, M.; Beck, A.; Schwarzmayr, T.; Häberle, B.; Cairo, S.; Leuschner, I.; von Schweinitz, D.; Strom, T.M.; et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J. Hepatol. 2014, 61, 1312–1320. [Google Scholar] [CrossRef]
- Amathieu, R.; Triba, M.N.; Goossens, C.; Bouchemal, N.; Nahon, P.; Savarin, P.; Le Moyec, L. Nuclear magnetic resonance based metabolomics and liver diseases: Recent advances and future clinical applications. World J. Gastroenterol. 2016, 22, 417–426. [Google Scholar] [CrossRef]
- Turcotte, L.M.; Georgieff, M.K.; Ross, J.A.; Feusner, J.H.; Tomlinson, G.E.; Malogolowkin, M.H.; Krailo, M.D.; Miller, N.; Fonstad, R.; Spector, L.G. Neonatal medical exposures and characteristics of low birth weight hepatoblastoma cases: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 2018–2023. [Google Scholar] [CrossRef]
- Zhang, Y.; Solinas, A.; Cairo, S.; Evert, M.; Chen, X.; Calvisi, D.F. Molecular Mechanisms of Hepatoblastoma. Semin. Liver Dis. 2021, 41, 028–041. [Google Scholar] [CrossRef]
- Reynolds, P.; Urayama, K.Y.; Von Behren, J.; Feusner, J. Birth characteristics and hepatoblastoma risk in young children. Cancer 2004, 100, 1070–1076. [Google Scholar] [CrossRef]
- Cairo, S.; Armengol, C.; De Reyniès, A.; Wei, Y.; Thomas, E.; Renard, C.-A.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar] [CrossRef]
- Carrillo-Reixach, J.; Torrens, L.; Simon-Coma, M.; Royo, L.; Domingo-Sàbat, M.; Abril-Fornaguera, J.; Akers, N.; Sala, M.; Ragull, S.; Arnal, M.; et al. Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J. Hepatol. 2020, 73, 328–341. [Google Scholar] [CrossRef]
- Albrecht, J.H. An Epigenetic Switch Between Differentiation and Proliferation in Hepatoblastoma. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1875–1876. [Google Scholar] [CrossRef]
- Forny, P.; Hochuli, M.; Rahman, Y.; Deheragoda, M.; Weber, A.; Baruteau, J.; Grunewald, S. Liver neoplasms in methylmalonic aciduria: An emerging complication. J. Inherit. Metab. Dis. 2019, 42, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Stahle, J.A.; Vunta, H.; Reddy, C.C.; Prabhu, K.S. Regulation of expression of apolipoprotein A-I by selenium status in human liver hepatoblastoma cells. Eur. J. Nutr. 2009, 48, 283. [Google Scholar] [CrossRef]
- Vandraas, K.F.; Vikanes, A.V.; Stoer, N.C.; Troisi, R.; Stephansson, O.; Sorensen, H.T.; Vangen, D.; Magnus, P.; Grjibovski, A.M.; Grotmol, T. Hyperemesis gravidarum and risk of cancer in offspring, a Scandinaivan registry-based nested case-control study. BMC Cancer 2015, 15, 398. [Google Scholar] [CrossRef]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Consensus Statement. Chlolangiocarcinoma 2020: The next horizon in mechanisms and management. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar]
- Parkin, D.M.; Ohshima, H.; Srivatanakul, P.; Vatanasapt, V. Cholangiocarcinoma: Epidemiology, mechanisms of carcinogenesis and prevention. Cancer Epidemiol. Biomark. Prev 1993, 2, 537–544. [Google Scholar]
- de Martel, C.; Plummer, M.; Franceschi, S. Cholangiocarcinoma: Descriptive epidemiology and risk factors. Gastroenterol. Clin. Biol. 2010, 34, 173–180. [Google Scholar] [CrossRef]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and Genetic Characterization of Cholangiocarcinoma Identifies Therapeutic Targets for Tyrosine Kinase Inhibitors. Gastroenterology 2012, 142, 1021–1031.e15. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.-F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.-X.; et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Sihrota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Brivio, S.; Cadmuro, M.; Fabris, L.; Strazzabosco, M. Molecular mechansims driving cholangiocarcinoma invasiveness: An overview. Gene Expr. 2018, 18, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Villaneuva, A.; Friedman, S.L.; Llovet, J.M. Liver cancer cell of origin, molecular class and effects on patient prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Stavraka, C.; Rush, H.; Ross, P. Combined hepatocellular cholangiocarcinoma (cHCC-CC): An update of genetics, molecular biology, and therapeutic interventions. J Hepatocell. Carcinoma 2019, 6, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70.e3. [Google Scholar] [CrossRef]
- Pinero, F.; Dirchwolf, M.; Pessoa, M.G. Biomarkers in hepatocellular carcinoma; diagnosis, prognosis and treatment response assessment. Cells 2020, 9, 1370. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Nga, H.; Tian, J.; Yi, H.-S. Mitochondrial Metabolic Signatures in Hepatocellular Carcinoma. Cells 2021, 10, 1901. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Zhang, C.; Guo, W.; Xu, Y.; Sharma, R.; Chen, Z.-S.; Zheng, Y.-C.; Wang, N.; et al. Epigenetic regulation of ferroptosis via ETS1/miR-23a-3p/ACSL4 axis mediates sorafenib resistance in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 3. [Google Scholar] [CrossRef]
- Loong, J.H.; Wong, T.-L.; Tong, M.; Sharma, R.; Zhou, L.; Ng, K.-Y.; Yu, H.-J.; Li, C.-H.; Man, K.; Lo, C.-M.; et al. Glucose deprivation–induced aberrant FUT1-mediated fucosylation drives cancer stemness in hepatocellular carcinoma. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Belenguer, G.; Mastrogiovanni, G.; Pacini, C.; Hall, Z.; Dowbaj, A.M.; Arnes-Benito, R.; Sljukic, A.; Prior, N.; Kakava, S.; Bradshaw, C.R.; et al. RNF43/ZNRF3 loss predisposes to hepatocellu-larcarcinoma by impairing liver regeneration and altering the liver lipid metabolic ground-state. Nat. Commun. 2022, 13, 334–353. [Google Scholar] [CrossRef]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondinsensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef]
- de Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef]
- Annunziato, S.; Sun, T.; Tchorz, J.S. The RSPO-LGR4/5-ZNRF3/RNF43 module in liver homeostasis, regeneration, and disease. Hepatology. 2022, 76, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Forgues, M.; Ye, Q.-H.; Jia, H.-L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.-X.; Tang, Z.-Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef]
- Gregori, S.; Mangia, P.; Bacchetta, R.; Tresoldi, E.; Kolbinger, F.; Traversari, C.; Carballido, J.M.; de Vries, J.E.; Korthäuer, U.; Roncarolo, M.-G. An anti-CD45RO/RB monoclonal antibody modulates T cell responses via induction of apoptosis and gen-eration of regulatory T cells. J. Exp. Med. 2005, 201, 1293–1305. [Google Scholar] [CrossRef]
- Kong, K.F.; Fu, G.; Zhang, Y.; Yokosuka, T.; Casas, J.; Canonigo-Balancio, A.J.; Becart, S.; Kim, G.; Yates, J.R.; Kronenberg, M.; et al. Protein kinase C-eta controls CTLA-4-mediated regulatory T cell function. Nat. Immunol. 2014, 15, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Tang, J.; Chen, Y.; Deng, L.; Ji, J.; Xie, Y.; Wang, K.; Jia, W.; Chu, W.-M.; Sun, B. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat. Commun. 2017, 8, 15129–15144. [Google Scholar] [CrossRef]
- Silk, J.D.; Abbott, R.J.M.; Adams, K.J.; Bennett, A.D.; Brett, S.; Cornforth, T.V.; Crossland, K.L.; Figueroa, D.J.; Jing, J.; O’Connor, C.; et al. Engineering cancer antigen-specific T cells to overcome the immunosuppressive effects of TGF-β. J. Immunol. 2022, 208, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.S.; Akter, S.; Ramproshad, S.; Mondal, B.; Alam Riaz, T.; Islam, M.T.; Khan, I.N.; Docea, A.O.; Calina, D.; Sharifi-Rad, J.; et al. Targeting Ras-ERK cascade by bioactive natural products for potential treatment of cancer: An updated overview. Cancer Cell Int. 2022, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Fernández-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Gregersen, N. The specific inhibition of the pyruvate dehydrogenase complex from pig kidney by propionyl-CoA and iso-valeryl-Co-A. Biochem. Med. 1981, 26, 20–27. [Google Scholar] [CrossRef]
- Cheema-Dhadli, S.; Leznoff, C.C.; Halperin, M.L. Effect of 2-methylcitrate on citrate metabolism: Implications for the man-agement of patients with propionic academia and methylmalonic aciduria. Pediatr. Res. 1975, 12, 905–908. [Google Scholar] [CrossRef]
- Jungermann, K.; Kietzmann, T. Oxygen: Modulator of metabolic zonation and disease of the liver. Hepatology 2000, 31, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer pro-gression and treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Murali, M.G.; Shailender, G.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Morland, C.; Gonzalez, S.V.; Rise, F.; Storm-Mathisen, J.; Gundesen, V.; Hassel, B. Propionate increases neu-ronal histone acetylation, but is metabolized oxidatively by glia. Relevance for propionic academia. J. Neurochem. 2007, 101, 806–814. [Google Scholar] [CrossRef]
- Sharif, A.W.; Williams, H.R.; Lampejo, T.; Khan, S.A.; Bansi, D.S.; Westaby, D.; Thillainayagam, A.V.; Thomas, H.C.; Cox, I.J.; Taylor-Robinson, S.D. Metabolic profiling of bile in cholangiocarcinoma using in vitro magnetic resonance spectroscopy. Hpb 2010, 12, 396–402. [Google Scholar] [CrossRef]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Lee, M.; Yu, G.; Yoo, H.; Kim, J.; Yoon, B.; Choi, Y.; Kim, D. ANXA8 Down-regulation by EGF-FOXO4 Signaling Is Involved in Cell Scattering and Tumor Metastasis of Cholangiocarcinoma. Gastroenterology 2009, 137, 1138–1150.e9. [Google Scholar] [CrossRef]
- Ferrarini, A.; Di Poto, C.; He, S.; Tu, C.; Vargehse, R.S.; Balla, A.K.; Jayatilake, M.; Li, Z.; Ghaffari, K.; Fan, Z.; et al. Metabolomic analysis of liver tissues for characterization of hepato-cellular carcinoma. J. Proteome. Res. 2019, 18, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, N.; Gao, L.; Xu, Y.J.; Huang, C.; Yu, K.; Ling, Q.; Cheng, Q.; Chen, S.; Zhu, M.; et al. Ac-etylcarnitine Is a Candidate Diagnostic and Prognostic Biomarker of Hepatocellular Carcinoma. Cancer Res. 2016, 76, 2912–2920. [Google Scholar] [CrossRef] [PubMed]
- Lulli, M.; Del Coco, L.; Mello, T.; Sukowati, C.; Madiai, S.; Gragnani, L.; Forte, P.; Fanizzi, F.P.; Mazzocca, A.; Rombouts, K.; et al. DNA Damage Response Protein CHK2 Regulates Metabolism in Liver Cancer. Cancer Res. 2021, 81, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Huang, C.; Li, N.; Zou, L.; Chia, S.E.; Chen, S.; Yu, K.; Ling, Q.; Cheng, Q.; et al. Comparison of hepatic and serum lipid signatures in hepatocellular carcinoma patients leads to the discovery of diagnostic and prognostic biomarkers. Oncotarget 2017, 9, 5032–5043. [Google Scholar] [CrossRef]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep Learning–Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12. [Google Scholar] [CrossRef]
- Ericksen, R.E.; Lim, S.L.; McDonnell, E.; Shuen, W.H.; Vadiveloo, M.; White, P.J.; Ding, Z.; Kwok, R.; Lee, P.; Radda, G.K.; et al. Loss of BCAA Catabolism during Carcinogenesis Enhances mTORC1 Activity and Promotes Tumor Development and Progression. Cell Metab. 2019, 29, 1151–1165.e6. [Google Scholar] [CrossRef]
- Horn, S.R.; Stoltzfus, K.C.; Lehrer, E.J.; Dawson, L.A.; Tchelebi, L.; Gusani, N.J.; Sharma, N.K.; Chen, H.; Trifiletti, D.M.; Zaorsky, N.G. Epidemiology of liver metastases. Cancer Epidemiol. 2020, 67, 101760. [Google Scholar] [CrossRef]
- Ganesan, R.; Yoon, S.J.; Suk, K.T. Microbiome and Metabolomics in Liver Cancer: Scientific Technology. Int. J. Mol. Sci. 2022, 24, 537. [Google Scholar] [CrossRef]
- Brodt, P. Role of the Microenvironment in Liver Metastasis: From Pre- to Prometastatic Niches. Clin. Cancer Res. 2016, 22, 5971–5982. [Google Scholar] [CrossRef] [PubMed]
- Berkemeyer, S. The straight line hypothesis elaborated: Case reference obesity, an argument for acidosis, oxidative stress, and disease conglomeration? Med. Hypotheses 2010, 75, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Berkemeyer, S. Acid-base balance and weight gain: Are there crucial links via protein and organic acids in understanding obesity? Med. Hypotheses 2009, 73, 347–356. [Google Scholar] [CrossRef] [PubMed]


| Genes (Pathways) | Intermediaries/Metabolites | Cell Signal Metabolites |
|---|---|---|
| ARID12, PBRM1, SW1/SNF | STAT3, JAK (JAK/STAT signaling) | IL-6 |
| ARID12, PBRM1, SW1/SNF | RAS-MAPK Pathway, KRAS2, BRAF, MEK1/MEK2, ERK1/ERK2 | EGFR (EGF), ERBB2, MET (HGF), VEGFR (VEGF), FGFR2 fusions, CXCR4 (SDF1) |
| ARID12, PBRM1, SW1/SNF, (BAP1) | P13K-AKT, PI3K, AKT, mTOR | EGFR (EGF), ERBB2, MET (HGF), VEGFR (VEGF), FGFR2 fusions, CXCR4 (SDF1) |
| p300, MAML1, CSL, NCID (Notch pathway) | NCID | NOTCH1-3, JAG1-2, DLL1 |
| Β-Catenin, TCF/LEF (WNT/β-catenin pathway) | Β-Catenin, GSK 3β, AXIN, APC, CK1α, DVL, (Ubiquitone) | WNT (LRP5/LRP6) |
| YAP/TAZ, TEAD (Hippo pathway) | YAP/TAZ, LATS1/2, MOB1, SAV1, MST1/2, Cytoplasm: Citrate, Isocitrate, α-KG, 2-HC via IDH12 (Hippo pathway) | RTKs, GPCRs, (Extracellular matrix stiffness) |
| YAP/TAZ, TEAD (Hippo pathway) | Mitochondria: Citrate, isocitrate, α-KG, 2-HC via IDH2 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berkemeyer, S. Primary Liver Cancers: Connecting the Dots of Cellular Studies and Epidemiology with Metabolomics. Int. J. Mol. Sci. 2023, 24, 2409. https://doi.org/10.3390/ijms24032409
Berkemeyer S. Primary Liver Cancers: Connecting the Dots of Cellular Studies and Epidemiology with Metabolomics. International Journal of Molecular Sciences. 2023; 24(3):2409. https://doi.org/10.3390/ijms24032409
Chicago/Turabian StyleBerkemeyer, Shoma. 2023. "Primary Liver Cancers: Connecting the Dots of Cellular Studies and Epidemiology with Metabolomics" International Journal of Molecular Sciences 24, no. 3: 2409. https://doi.org/10.3390/ijms24032409
APA StyleBerkemeyer, S. (2023). Primary Liver Cancers: Connecting the Dots of Cellular Studies and Epidemiology with Metabolomics. International Journal of Molecular Sciences, 24(3), 2409. https://doi.org/10.3390/ijms24032409