
Ca2+-Permeable AMPA Receptors Contribute to Changed Dorsal Horn Neuronal Firing and Inflammatory Pain



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
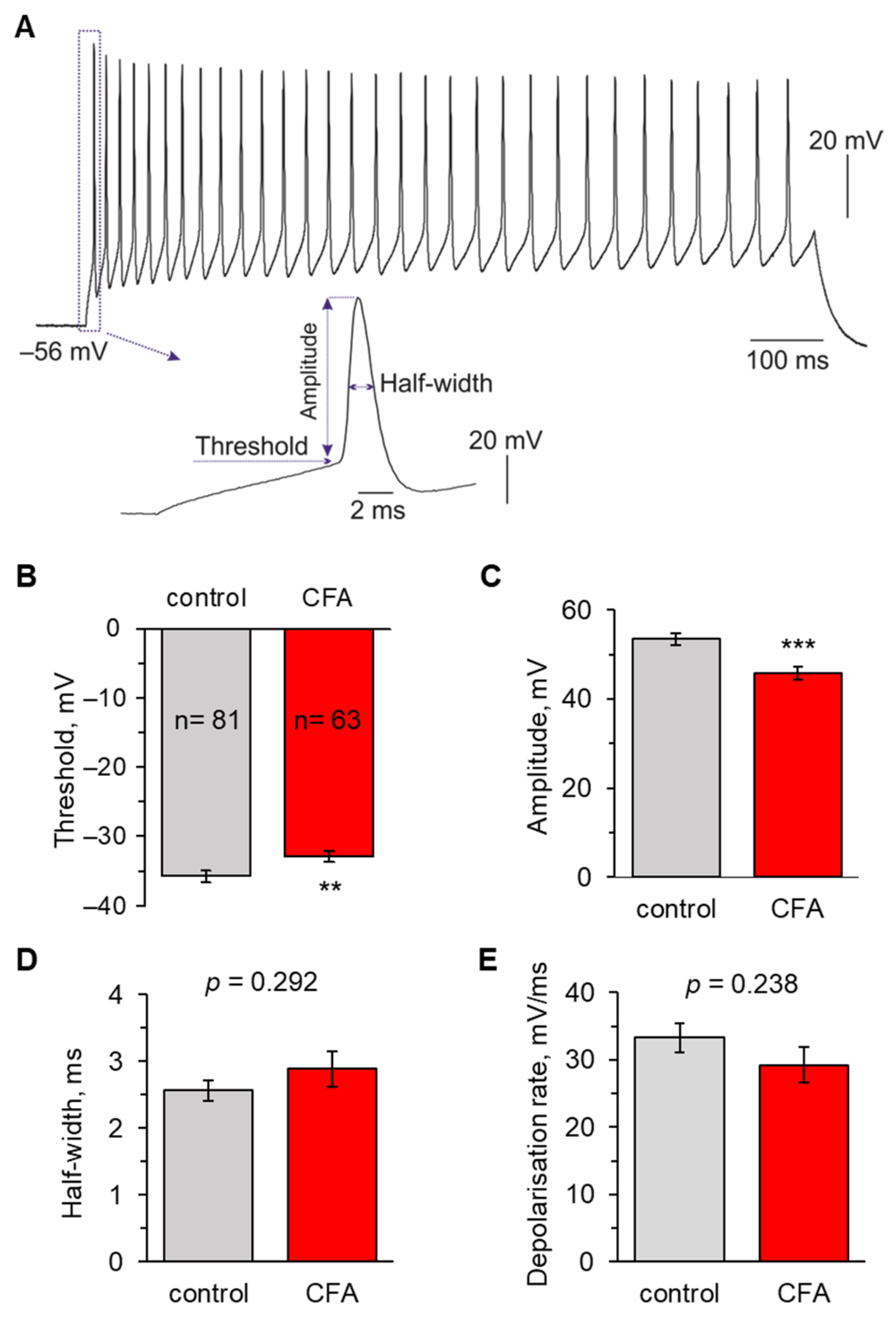
2.1. Peripheral Inflammation Causes Neuronal Hyperexcitability in the Superficial DH
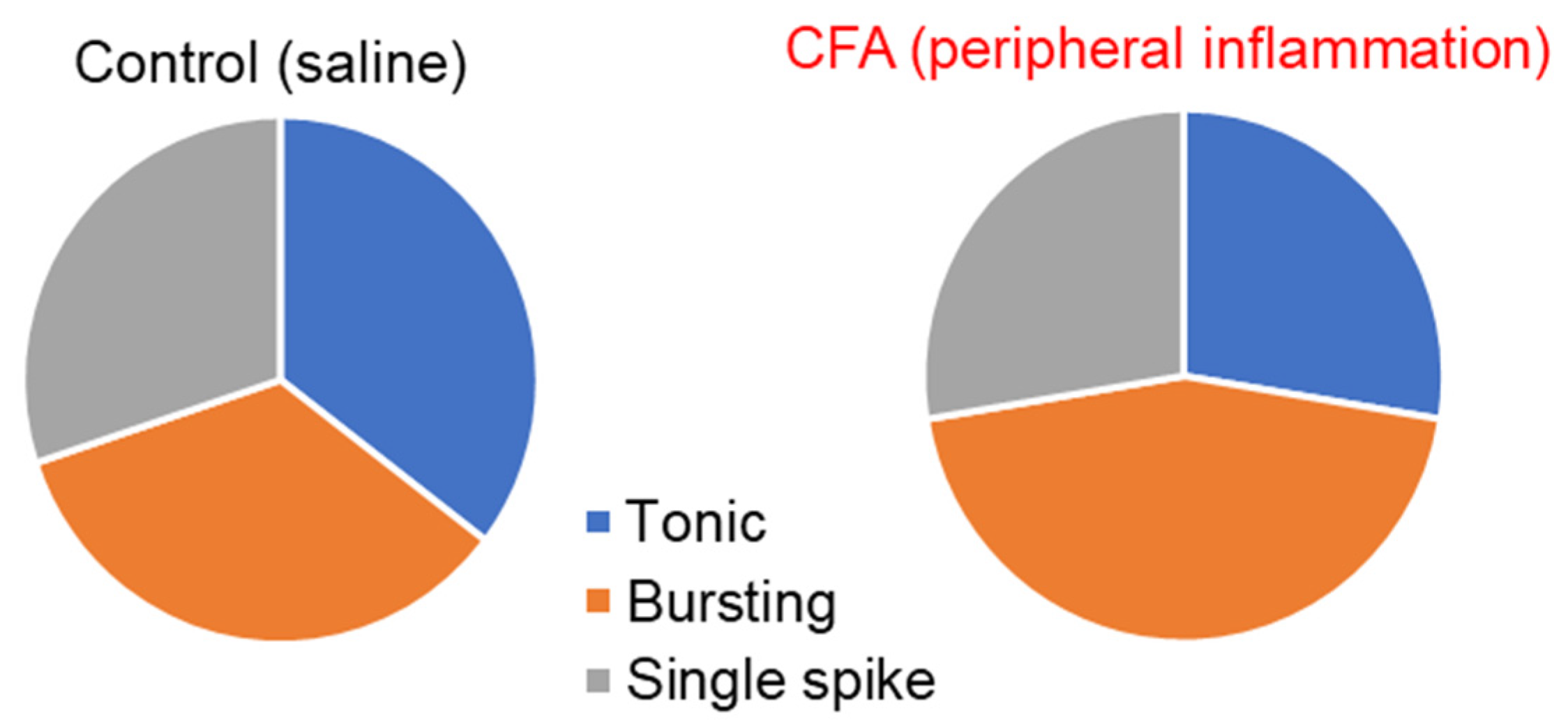
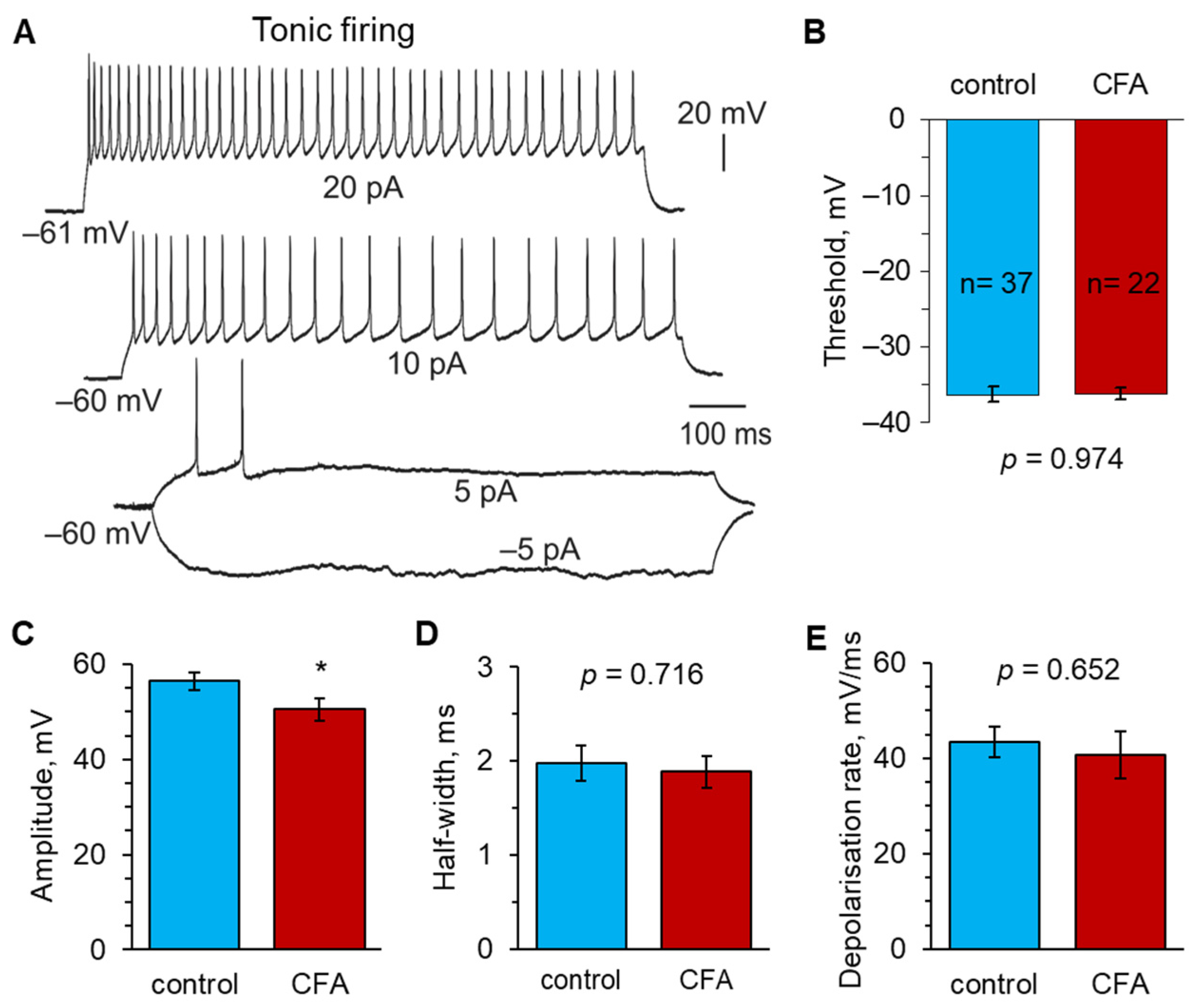
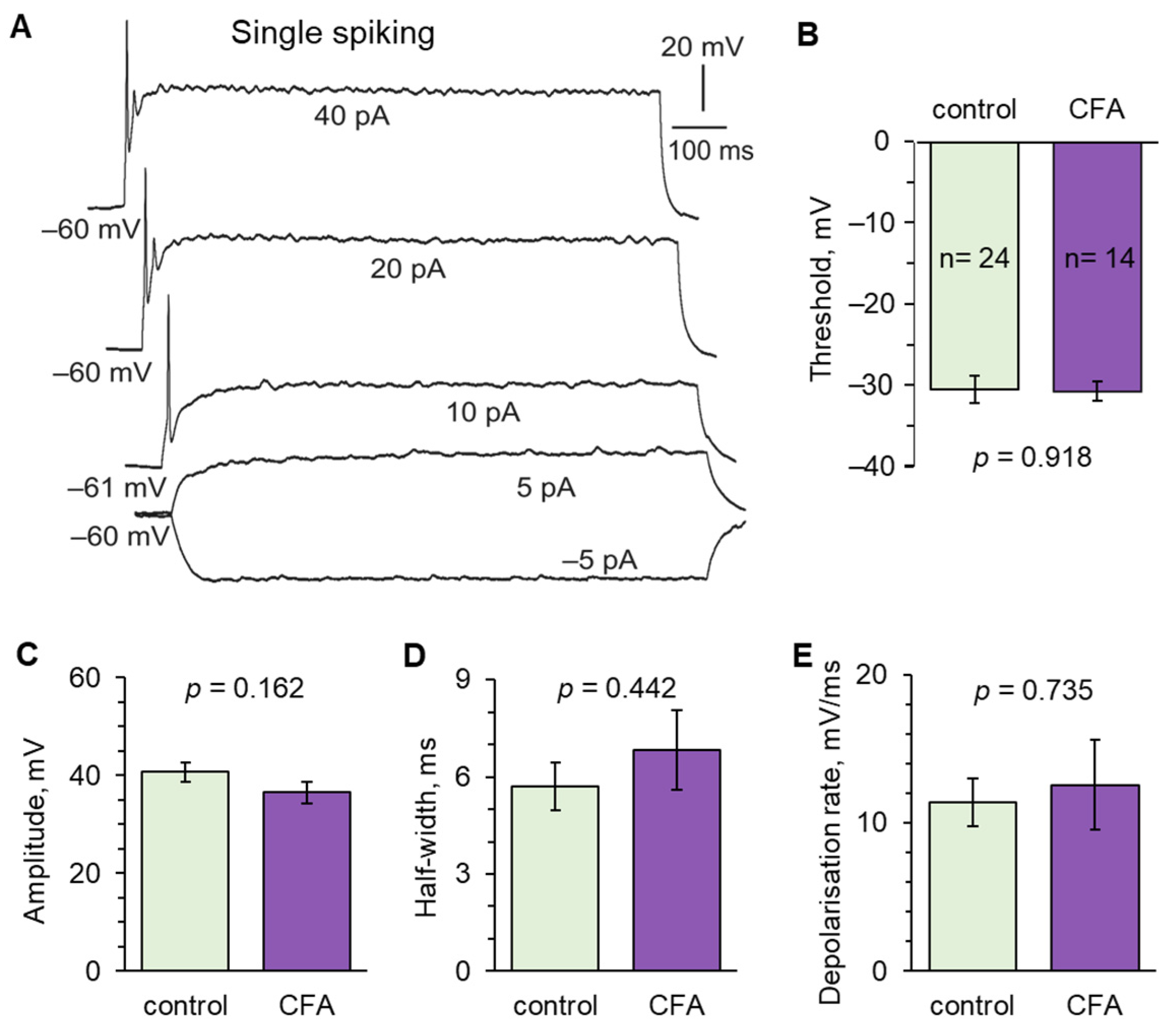
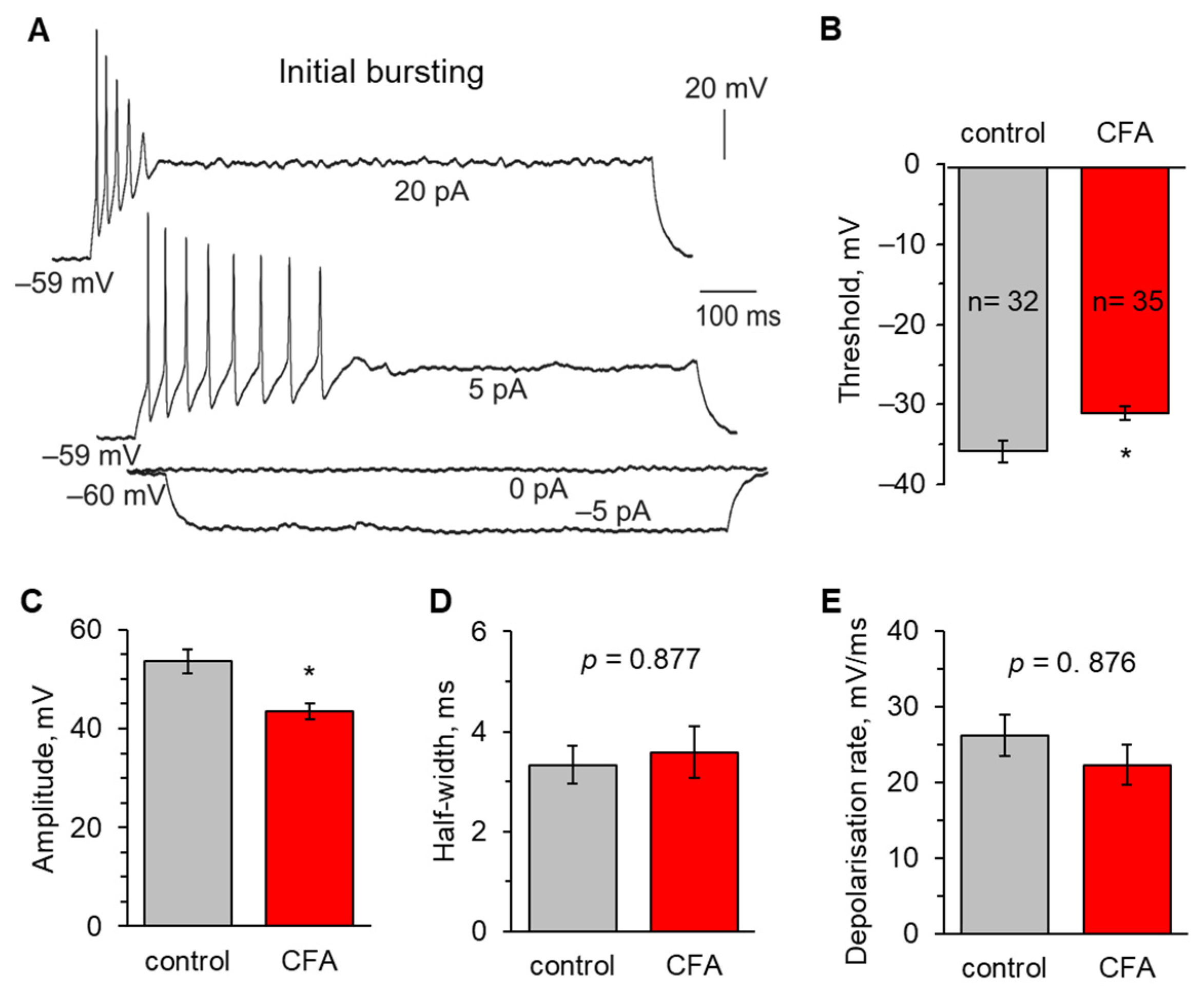
2.2. Peripheral Inflammation Specifically Alters Excitability and AP Parameters in the Lamina I-II DH Neurons
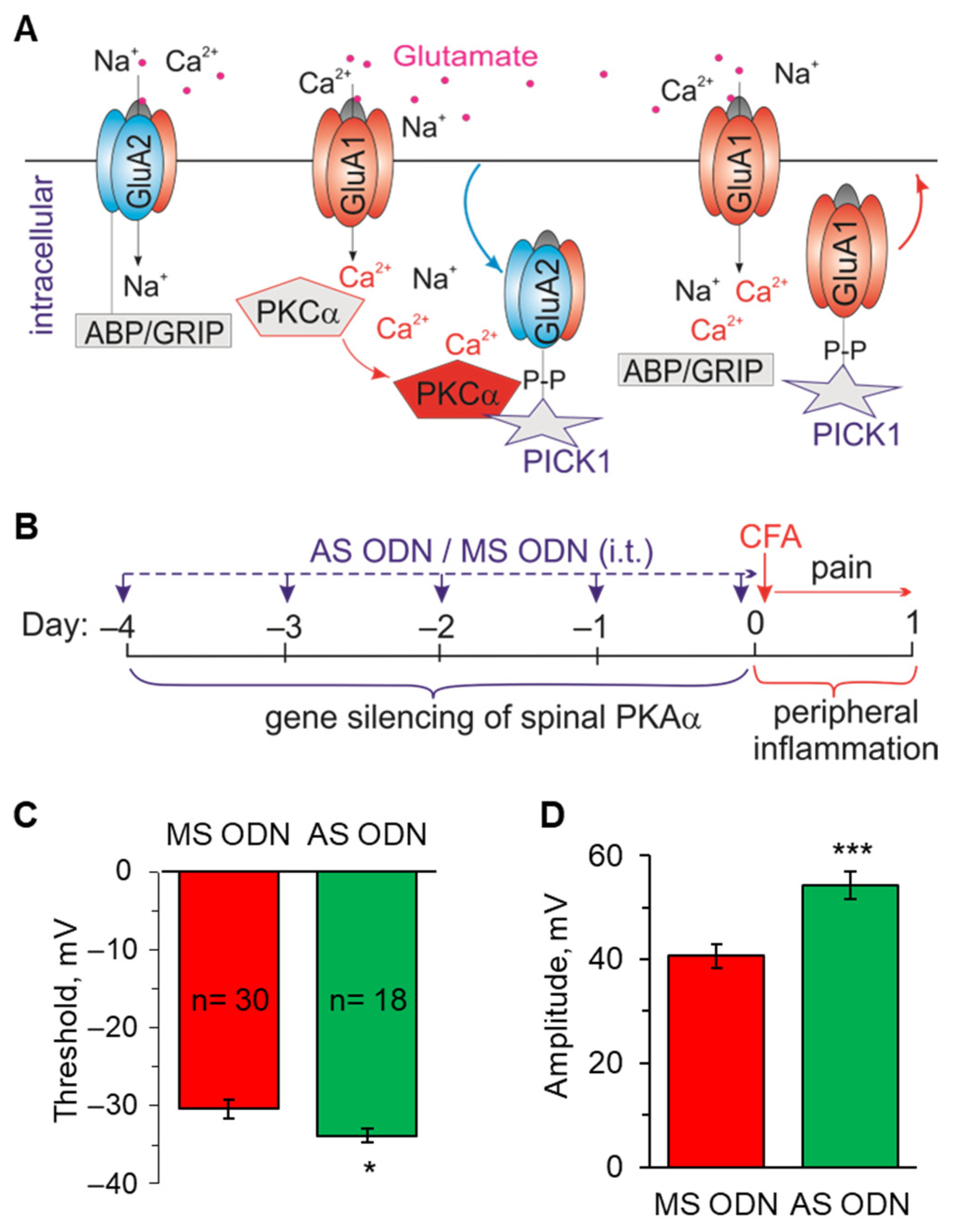
2.3. Knockdown of the Inflammatory-Induced Upregulation of Ca2+-Permeable AMPARs in the Spinal Cord Restores the AP Parameters in Lamina I-II Neurons
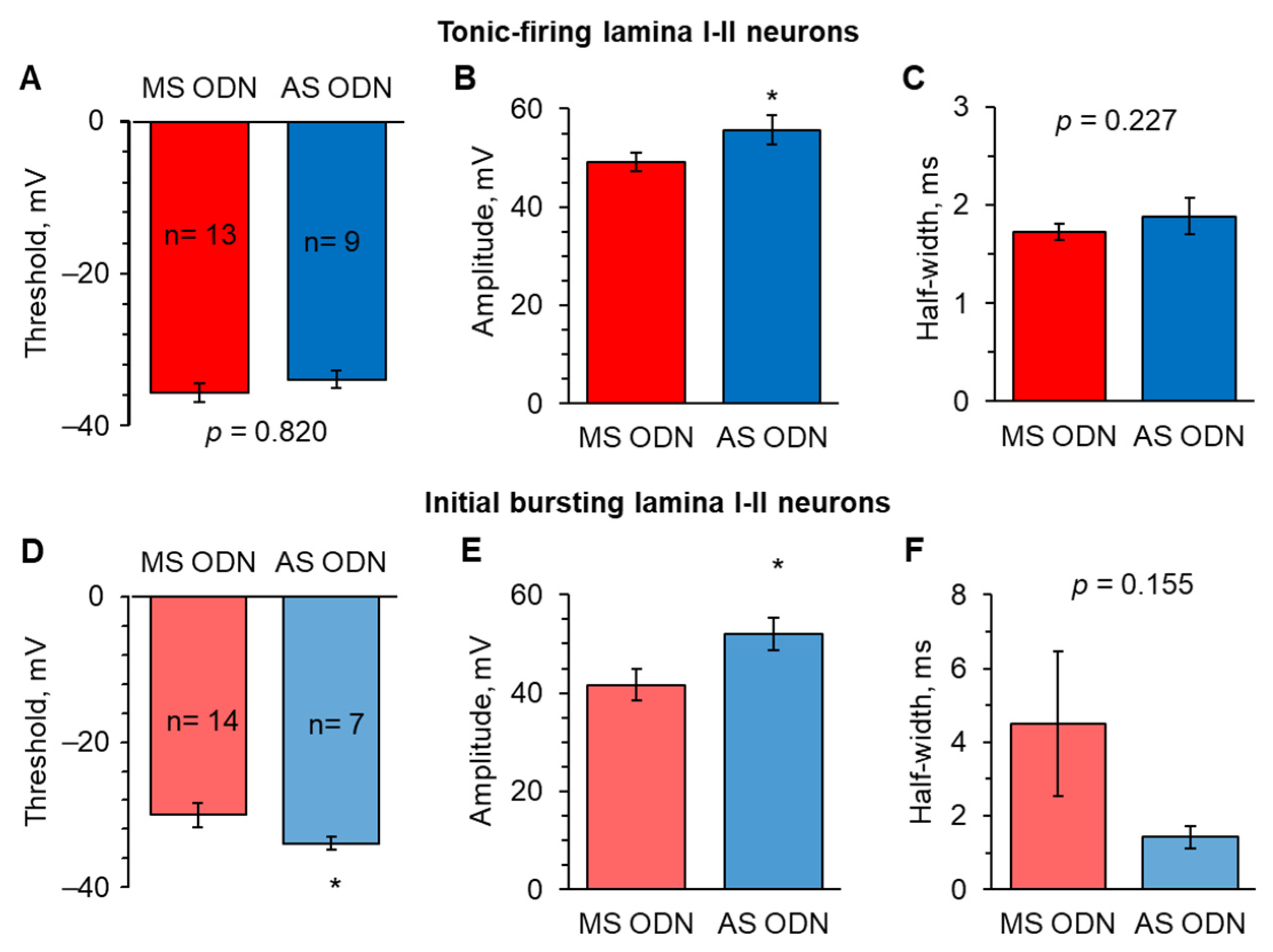
2.4. Knockdown of Upregulated Ca2+-Permeable AMPARs in the Spinal Cord Restores the AP Parameters in Lamina I-II Neuronal Classes in Persistent Peripheral Inflammation
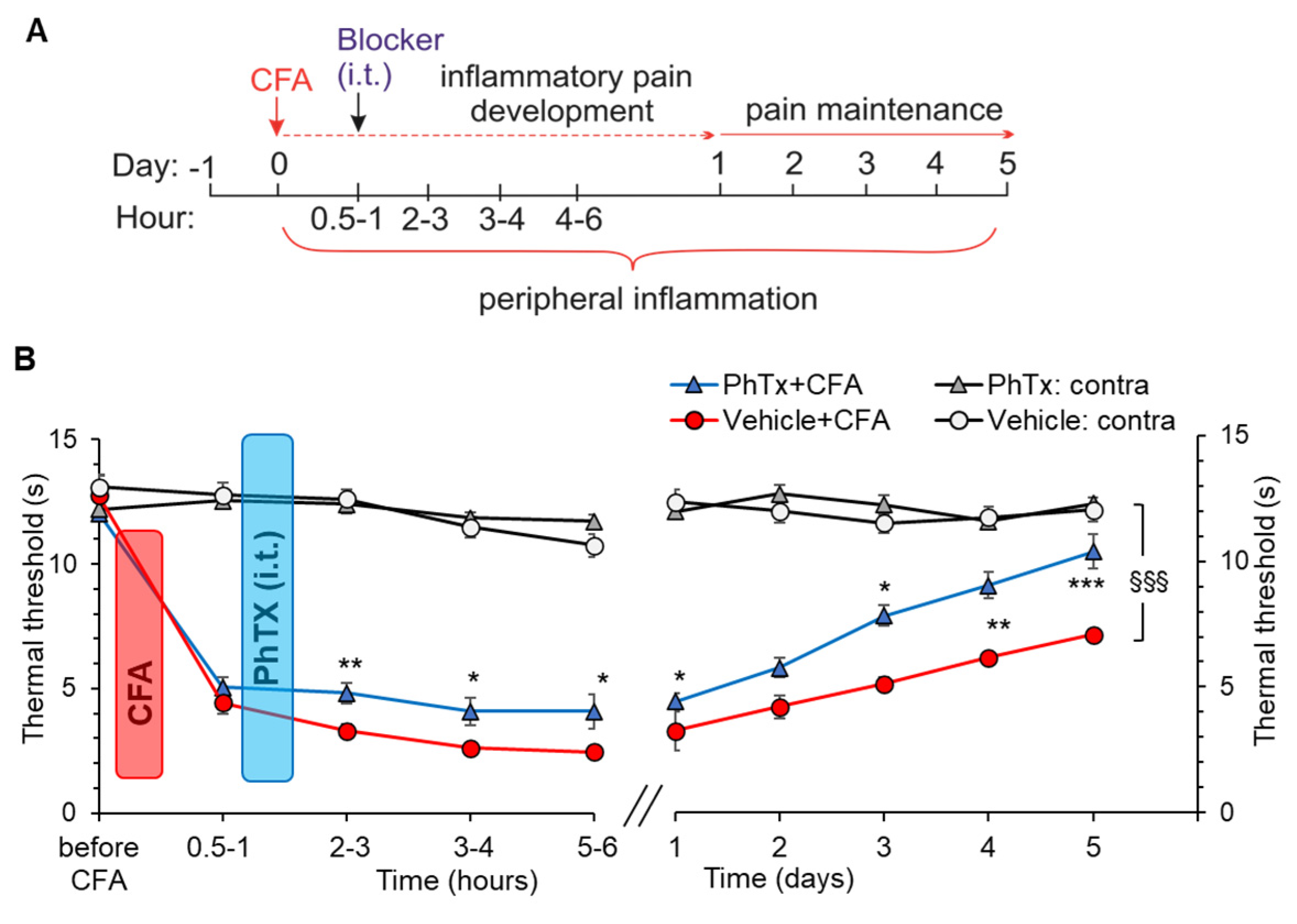
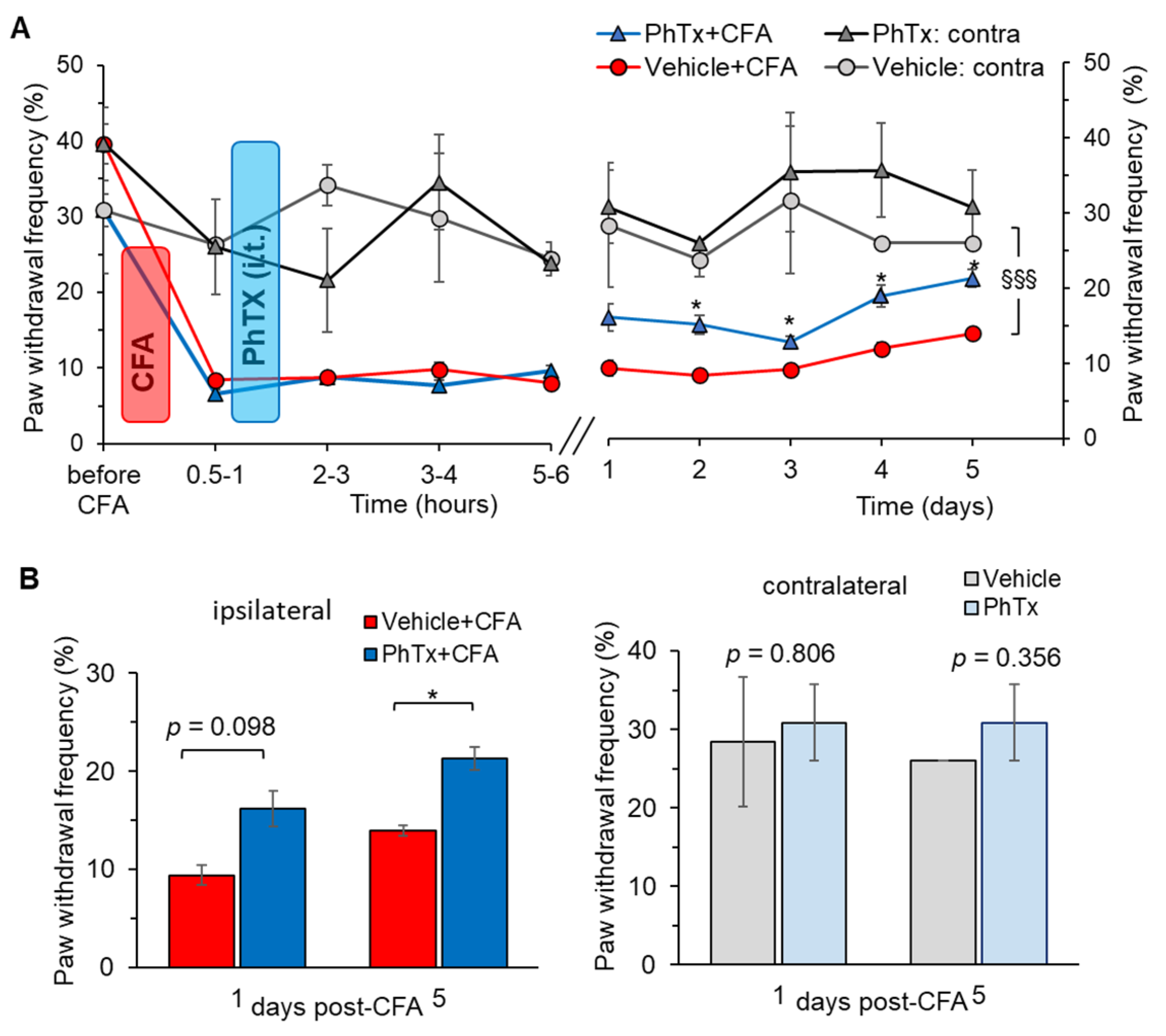
2.5. Selective Inhibition of Spinal Ca2+-Permeable AMPARs Alleviates Inflammatory Pain of Thermal Modality
2.6. Inhibition of Ca2+-Permeable AMPARs in the Spinal Cord Alleviates Inflammatory Pain of Mechanical Modality
3. Discussion
4. Materials and Methods
4.1. Animal Care
4.2. Induction of Peripheral Inflammation
4.3. Surgical Intrathecal Catheter Implantation
4.4. Delivery to the Spinal Cord
4.5. Measuring the Thermal Nociceptive Threshold—The Hargreaves Plantar Test
4.6. Measuring the Thermal Nociceptive Threshold with von Frey Monofilaments
4.7. Knockdown of the PKCα-Dependent Upregulation of Ca2+-Permeable AMPARs in the Dorsal Spinal Cord
4.8. Spinal Cord Slice Preparation
4.9. Electrophysiology
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garry, E.M.; Moss, A.; Rosie, R.; Delaney, A.; Mitchell, R.; Fleetwood-Walker, S.M. Specific involvement in neuropathic pain of AMPA receptors and adapter proteins for the GluR2 subunit. Mol. Cell. Neurosci. 2003, 24, 10–22. [Google Scholar] [CrossRef]
- Gwak, Y.S.; Kang, J.; Leem, J.W.; Hulsebosch, C.E. Spinal AMPA receptor inhibition attenuates mechanical allodynia and neuronal hyperexcitability following spinal cord injury in rats. J. Neurosci. Res. 2007, 85, 2352–2359. [Google Scholar] [CrossRef]
- Hartmann, B.; Ahmadi, S.; Heppenstall, P.A.; Lewin, G.R.; Schott, C.; Borchardt, T.; Seeburg, P.H.; Zeilhofer, H.U.; Sprengel, R.; Kuner, R. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron 2004, 44, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Wang, H.; Amaya, F.; Brenner, G.J.; Cheng, J.K.; Ji, R.R.; Woolf, C.J. Bradykinin enhances AMPA and NMDA receptor activity in spinal cord dorsal horn neurons by activating multiple kinases to produce pain hypersensitivity. J. Neurosci. 2008, 28, 4533–4540. [Google Scholar] [CrossRef]
- Kopach, O.; Krotov, V.; Belan, P.; Voitenko, N. Inflammatory-induced changes in synaptic drive and postsynaptic AMPARs in lamina II dorsal horn neurons are cell-type specific. Pain 2015, 156, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Kohno, T.; Moore, K.A.; Woolf, C.J. Central sensitization and LTP: Do pain and memory share similar mechanisms? Trends Neurosci. 2003, 26, 696–705. [Google Scholar] [CrossRef]
- Woolf, C.J.; Salter, M.W. Neuronal plasticity: Increasing the gain in pain. Science 2000, 288, 1765–1769. [Google Scholar] [CrossRef]
- Kopach, O.; Voitenko, N. Spinal AMPA receptors: Amenable players in central sensitization for chronic pain therapy? Channels 2021, 15, 284–297. [Google Scholar] [CrossRef]
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef]
- Park, J.S.; Yaster, M.; Guan, X.; Xu, J.T.; Shih, M.H.; Guan, Y.; Raja, S.N.; Tao, Y.X. Role of spinal cord alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors in complete Freund’s adjuvant-induced inflammatory pain. Mol. Pain 2008, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Kopach, O.; Kao, S.C.; Petralia, R.S.; Belan, P.; Tao, Y.X.; Voitenko, N. Inflammation alters trafficking of extrasynaptic AMPA receptors in tonically firing lamina II neurons of the rat spinal dorsal horn. Pain 2011, 152, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Voitenko, N.; Petralia, R.S.; Guan, X.; Xu, J.T.; Steinberg, J.P.; Takamiya, K.; Sotnik, A.; Kopach, O.; Huganir, R.L.; et al. Persistent inflammation induces GluR2 internalization via NMDA receptor-triggered PKC activation in dorsal horn neurons. J. Neurosci. 2009, 29, 3206–3219. [Google Scholar] [CrossRef] [PubMed]
- Vikman, K.S.; Rycroft, B.K.; Christie, M.J. Switch to Ca2+-permeable AMPA and reduced NR2B NMDA receptor-mediated neurotransmission at dorsal horn nociceptive synapses during inflammatory pain in the rat. J. Physiol. 2008, 586, 515–527. [Google Scholar] [CrossRef]
- Rycroft, B.K.; Vikman, K.S.; Christie, M.J. Inflammation reduces the contribution of N-type calcium channels to primary afferent synaptic transmission onto NK1 receptor-positive lamina I neurons in the rat dorsal horn. J. Physiol. 2007, 580, 883–894. [Google Scholar] [CrossRef]
- Saegusa, H.; Kurihara, T.; Zong, S.; Kazuno, A.; Matsuda, Y.; Nonaka, T.; Han, W.; Toriyama, H.; Tanabe, T. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. Embo J. 2001, 20, 2349–2356. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, J.; Adams, D.J. Neuronal voltage-gated sodium channel subtypes: Key roles in inflammatory and neuropathic pain. Int. J. Biochem. Cell Biol. 2006, 38, 2005–2010. [Google Scholar] [CrossRef]
- Waxman, S.G.; Zamponi, G.W. Regulating excitability of peripheral afferents: Emerging ion channel targets. Nat. Neurosci. 2014, 17, 153–163. [Google Scholar] [CrossRef]
- Ikeda, H.; Heinke, B.; Ruscheweyh, R.; Sandkühler, J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science 2003, 299, 1237–1240. [Google Scholar] [CrossRef]
- Inquimbert, P.; Bartels, K.; Babaniyi, O.B.; Barrett, L.B.; Tegeder, I.; Scholz, J. Peripheral nerve injury produces a sustained shift in the balance between glutamate release and uptake in the dorsal horn of the spinal cord. Pain 2012, 153, 2422–2431. [Google Scholar] [CrossRef]
- Moore, K.A.; Kohno, T.; Karchewski, L.A.; Scholz, J.; Baba, H.; Woolf, C.J. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J. Neurosci. 2002, 22, 6724–6731. [Google Scholar] [CrossRef]
- Ferrini, F.; Perez-Sanchez, J.; Ferland, S.; Lorenzo, L.E.; Godin, A.G.; Plasencia-Fernandez, I.; Cottet, M.; Castonguay, A.; Wang, F.; Salio, C.; et al. Differential chloride homeostasis in the spinal dorsal horn locally shapes synaptic metaplasticity and modality-specific sensitization. Nat. Commun. 2020, 11, 3935. [Google Scholar] [CrossRef]
- Coull, J.A.; Boudreau, D.; Bachand, K.; Prescott, S.A.; Nault, F.; Sík, A.; De Koninck, P.; De Koninck, Y. Trans-Synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 2003, 424, 938–942. [Google Scholar] [CrossRef]
- Cathenaut, L.; Leonardon, B.; Kuster, R.; Inquimbert, P.; Schlichter, R.; Hugel, S. Inhibitory interneurons with differential plasticities at their connections tune excitatory-inhibitory balance in the spinal nociceptive system. Pain 2022, 163, e675–e688. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Taniguchi, W.; Nishida, K.; Nakatsuka, T.; Ito, S. In vivo two-photon imaging of structural dynamics in the spinal dorsal horn in an inflammatory pain model. Eur. J. Neurosci. 2015, 41, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, E.; Yamanaka, H.; Kobayashi, K.; Yagi, H.; Sakagami, M.; Noguchi, K. RhoA/ROCK pathway mediates p38 MAPK activation and morphological changes downstream of P2Y12/13 receptors in spinal microglia in neuropathic pain. Glia 2015, 63, 216–228. [Google Scholar] [CrossRef]
- Peirs, C.; Williams, S.P.; Zhao, X.; Walsh, C.E.; Gedeon, J.Y.; Cagle, N.E.; Goldring, A.C.; Hioki, H.; Liu, Z.; Marell, P.S.; et al. Dorsal Horn Circuits for Persistent Mechanical Pain. Neuron 2015, 87, 797–812. [Google Scholar] [CrossRef] [PubMed]
- Peirs, C.; Williams, S.G.; Zhao, X.; Arokiaraj, C.M.; Ferreira, D.W.; Noh, M.C.; Smith, K.M.; Halder, P.; Corrigan, K.A.; Gedeon, J.Y.; et al. Mechanical Allodynia Circuitry in the Dorsal Horn is Defined by the Nature of the Injury. Neuron 2021, 109, 73–90.e77. [Google Scholar] [CrossRef]
- Duzhyy, D.E.; Voitenko, N.V.; Belan, P.V. Peripheral Inflammation Results in Increased Excitability of Capsaicin-Insensitive Nociceptive DRG Neurons Mediated by Upregulation of ASICs and Voltage-Gated Ion Channels. Front. Cell. Neurosci. 2021, 15, 723295. [Google Scholar] [CrossRef] [PubMed]
- Reichling, D.B.; Levine, J.D. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009, 32, 611–618. [Google Scholar] [CrossRef]
- Malmberg, A.B.; Yaksh, T.L. The effect of morphine on formalin-evoked behaviour and spinal release of excitatory amino acids and prostaglandin E2 using microdialysis in conscious rats. Br. J. Pharmacol. 1995, 114, 1069–1075. [Google Scholar] [CrossRef]
- Matsuda, M.; Huh, Y.; Ji, R.R. Roles of inflammation, neurogenic inflammation, and neuroinflammation in pain. J. Anesth. 2019, 33, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Baccei, M.L. Neonatal tissue injury reduces the intrinsic excitability of adult mouse superficial dorsal horn neurons. Neuroscience 2014, 256, 392–402. [Google Scholar] [CrossRef]
- Lucas-Romero, J.; Rivera-Arconada, I.; Lopez-Garcia, J.A. Synchronous firing of dorsal horn neurons at the origin of dorsal root reflexes in naïve and paw-inflamed mice. Front. Cell. Neurosci. 2022, 16, 1004956. [Google Scholar] [CrossRef]
- Kim, H.Y.; Jun, J.; Wang, J.; Bittar, A.; Chung, K.; Chung, J.M. Induction of long-term potentiation and long-term depression is cell-type specific in the spinal cord. Pain 2015, 156, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Tadros, M.A.; Zouikr, I.; Hodgson, D.M.; Callister, R.J. Excitability of Rat Superficial Dorsal Horn Neurons Following a Neonatal Immune Challenge. Front. Neurol. 2018, 9, 743. [Google Scholar] [CrossRef]
- Schappacher, K.A.; Xie, W.; Zhang, J.M.; Baccei, M.L. Neonatal vincristine administration modulates intrinsic neuronal excitability in the rat dorsal root ganglion and spinal dorsal horn during adolescence. Pain 2019, 160, 645–657. [Google Scholar] [CrossRef]
- Kopach, O.; Viatchenko-Karpinski, V.; Atianjoh, F.E.; Belan, P.; Tao, Y.X.; Voitenko, N. PKCα is required for inflammation-induced trafficking of extrasynaptic AMPA receptors in tonically firing lamina II dorsal horn neurons during the maintenance of persistent inflammatory pain. J. Pain 2013, 14, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Katano, T.; Furue, H.; Okuda-Ashitaka, E.; Tagaya, M.; Watanabe, M.; Yoshimura, M.; Ito, S. N-ethylmaleimide-sensitive fusion protein (NSF) is involved in central sensitization in the spinal cord through GluR2 subunit composition switch after inflammation. Eur. J. Neurosci. 2008, 27, 3161–3170. [Google Scholar] [CrossRef]
- Kopach, O.; Krotov, V.; Shysh, A.; Sotnic, A.; Viatchenko-Karpinski, V.; Dosenko, V.; Voitenko, N. Spinal PKCα inhibition and gene-silencing for pain relief: AMPAR trafficking at the synapses between primary afferents and sensory interneurons. Sci. Rep. 2018, 8, 10285. [Google Scholar] [CrossRef]
- Choi, J.I.; Svensson, C.I.; Koehrn, F.J.; Bhuskute, A.; Sorkin, L.S. Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain 2010, 149, 243–253. [Google Scholar] [CrossRef]
- Galan, A.; Laird, J.M.; Cervero, F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain 2004, 112, 315–323. [Google Scholar] [CrossRef]
- Sullivan, S.J.; Farrant, M.; Cull-Candy, S.G. TARP γ-2 Is Required for Inflammation-Associated AMPA Receptor Plasticity within Lamina II of the Spinal Cord Dorsal Horn. J. Neurosci. 2017, 37, 6007–6020. [Google Scholar] [CrossRef] [PubMed]
- Kopach, O.; Krotov, V.; Goncharenko, J.; Voitenko, N. Inhibition of Spinal Ca(2+)-Permeable AMPA Receptors with Dicationic Compounds Alleviates Persistent Inflammatory Pain without Adverse Effects. Front. Cell. Neurosci. 2016, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Kopach, O.; Zheng, K.; Dong, L.; Sapelkin, A.; Voitenko, N.; Sukhorukov, G.B.; Rusakov, D.A. Nano-engineered microcapsules boost the treatment of persistent pain. Drug Deliv. 2018, 25, 435–447. [Google Scholar] [CrossRef]
- Hughes, D.I.; Sikander, S.; Kinnon, C.M.; Boyle, K.A.; Watanabe, M.; Callister, R.J.; Graham, B.A. Morphological, neurochemical and electrophysiological features of parvalbumin-expressing cells: A likely source of axo-axonic inputs in the mouse spinal dorsal horn. J. Physiol. 2012, 590, 3927–3951. [Google Scholar] [CrossRef]
- Grudt, T.J.; Perl, E.R. Correlations between neuronal morphology and electrophysiological features in the rodent superficial dorsal horn. J. Physiol. 2002, 540, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Kopach, O.; Voitenko, N. Extrasynaptic AMPA receptors in the dorsal horn: Evidence and functional significance. Brain Res. Bull 2013, 93, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Voitenko, N.; Gerber, G.; Youn, D.; Randic, M. Peripheral inflamation-induced increase of AMPA-mediated currents and Ca2+ transients in the presence of cyclothiazide in the rat substantia gelatinosa neurons. Cell Calcium. 2004, 35, 461–469. [Google Scholar] [CrossRef]
- Kopach, O.; Viatchenko-Karpinski, V.; Belan, P.; Voitenko, N. Development of inflammation-induced hyperalgesia and allodynia is associated with the upregulation of extrasynaptic AMPA receptors in tonically firing lamina II dorsal horn neurons. Front. Physiol. 2012, 3, 391. [Google Scholar] [CrossRef]
- Zhuo, M. Ionotropic glutamate receptors contribute to pain transmission and chronic pain. Neuropharmacology 2017, 112, 228–234. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, S.R.; Chen, H.; Zhou, J.J.; Jin, D.; Pan, H.L. Theta-Burst Stimulation of Primary Afferents Drives Long-Term Potentiation in the Spinal Cord and Persistent Pain via α2δ-1-Bound NMDA Receptors. J. Neurosci. 2022, 42, 513–527. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Liu, F.; Lin, J.; Li, Y.L.; Fang, Z.H.; Zhou, C.; Li, C.J.; Shen, J.F. Activation of the N-methyl-D-aspartate receptor contributes to orofacial neuropathic and inflammatory allodynia by facilitating calcium-calmodulin-dependent protein kinase II phosphorylation in mice. Brain Res. Bull 2022, 185, 174–192. [Google Scholar] [CrossRef] [PubMed]
- Tognoli, E.; Proto, P.L.; Motta, G.; Galeone, C.; Mariani, L.; Valenza, F. Methadone for postoperative analgesia: Contribution of N-methyl-D-aspartate receptor antagonism: A randomised controlled trial. Eur. J. Anaesthesiol. 2020, 37, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Pak, D.T. Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu. Rev. Physiol. 2000, 62, 755–778. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Li, T.; Wang, R.; Deng, W.; Pu, H.; Deng, M. Roles of Phosphorylation of N-Methyl-D-Aspartate Receptor in Chronic Pain. Cell. Mol. Neurobiol. 2023, 43, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Huang, L.Y. Alteration in the voltage dependence of NMDA receptor channels in rat dorsal horn neurones following peripheral inflammation. J. Physiol. 2001, 537, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.Y.; Wagner, D.A.; Hsu, M.H.; Leonard, J.P. Evidence for direct protein kinase-C mediated modulation of N-methyl-D-aspartate receptor current. Mol. Pharmacol. 2001, 59, 960–964. [Google Scholar] [CrossRef]
- Scott, D.B.; Blanpied, T.A.; Ehlers, M.D. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 2003, 45, 755–767. [Google Scholar] [CrossRef]
- Koga, K.; Descalzi, G.; Chen, T.; Ko, H.G.; Lu, J.; Li, S.; Son, J.; Kim, T.; Kwak, C.; Huganir, R.L.; et al. Coexistence of two forms of LTP in ACC provides a synaptic mechanism for the interactions between anxiety and chronic pain. Neuron 2015, 85, 377–389. [Google Scholar] [CrossRef]
- Bhangoo, S.K.; Swanson, G.T. Kainate receptor signaling in pain pathways. Mol. Pharmacol. 2013, 83, 307–315. [Google Scholar] [CrossRef]
- Qiu, C.S.; Wyhe, L.L.; Sasaki, M.; Sakai, R.; Swanson, G.T.; Gereau, R.W.t. Antinociceptive effects of MSVIII-19, a functional antagonist of the GluK1 kainate receptor. Pain 2011, 152, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, D.J.; Belle, M.D.; Cheunsuang, O.; Stewart, A.; Morris, R. Morphology of inhibitory and excitatory interneurons in superficial laminae of the rat dorsal horn. J. Physiol. 2007, 584, 521–533. [Google Scholar] [CrossRef]
- Polgár, E.; Durrieux, C.; Hughes, D.I.; Todd, A.J. A quantitative study of inhibitory interneurons in laminae I-III of the mouse spinal dorsal horn. PLoS ONE 2013, 8, e78309. [Google Scholar] [CrossRef]
- Bice, T.N.; Beal, J.A. Quantitative and neurogenic analysis of neurons with supraspinal projections in the superficial dorsal horn of the rat lumbar spinal cord. J. Comp. Neurol. 1997, 388, 565–574. [Google Scholar] [CrossRef]
- Bice, T.N.; Beal, J.A. Quantitative and neurogenic analysis of the total population and subpopulations of neurons defined by axon projection in the superficial dorsal horn of the rat lumbar spinal cord. J. Comp. Neurol. 1997, 388, 550–564. [Google Scholar] [CrossRef]
- Cervero, F.; Laird, J.M.; García-Nicas, E. Secondary hyperalgesia and presynaptic inhibition: An update. Eur. J. Pain 2003, 7, 345–351. [Google Scholar] [CrossRef]
- Yasaka, T.; Tiong, S.Y.; Hughes, D.I.; Riddell, J.S.; Todd, A.J. Populations of inhibitory and excitatory interneurons in lamina II of the adult rat spinal dorsal horn revealed by a combined electrophysiological and anatomical approach. Pain 2010, 151, 475–488. [Google Scholar] [CrossRef]
- Punnakkal, P.; von Schoultz, C.; Haenraets, K.; Wildner, H.; Zeilhofer, H.U. Morphological, biophysical and synaptic properties of glutamatergic neurons of the mouse spinal dorsal horn. J. Physiol. 2014, 592, 759–776. [Google Scholar] [CrossRef]
- Santos, S.F.; Rebelo, S.; Derkach, V.A.; Safronov, B.V. Excitatory interneurons dominate sensory processing in the spinal substantia gelatinosa of rat. J. Physiol. 2007, 581, 241–254. [Google Scholar] [CrossRef]
- Albuquerque, C.; Lee, C.J.; Jackson, A.C.; MacDermott, A.B. Subpopulations of GABAergic and non-GABAergic rat dorsal horn neurons express Ca2+-permeable AMPA receptors. Eur. J. Neurosci. 1999, 11, 2758–2766. [Google Scholar] [CrossRef]
- Hantman, A.W.; van den Pol, A.N.; Perl, E.R. Morphological and physiological features of a set of spinal substantia gelatinosa neurons defined by green fluorescent protein expression. J. Neurosci. 2004, 24, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Albisetti, G.W.; Ganley, R.P.; Pietrafesa, F.; Werynska, K.; Magalhaes de Sousa, M.; Sipione, R.; Scheurer, L.; Bösl, M.R.; Pelczar, P.; Wildner, H.; et al. Inhibitory Kcnip2 neurons of the spinal dorsal horn control behavioral sensitivity to environmental cold. Neuron 2023, 111, 92–105.e5. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.O.; Hegedüs, K.; Wildner, H.; Zeilhofer, H.U.; Antal, M. Morphological and neurochemical characterization of glycinergic neurons in laminae I-IV of the mouse spinal dorsal horn. J. Comp. Neurol. 2022, 530, 607–626. [Google Scholar] [CrossRef]
- Gradwell, M.A.; Boyle, K.A.; Browne, T.J.; Bell, A.M.; Leonardo, J.; Peralta Reyes, F.S.; Dickie, A.C.; Smith, K.M.; Callister, R.J.; Dayas, C.V.; et al. Diversity of inhibitory and excitatory parvalbumin interneuron circuits in the dorsal horn. Pain 2022, 163, e432–e452. [Google Scholar] [CrossRef] [PubMed]
- Zeilhofer, H.U.; Wildner, H.; Yévenes, G.E. Fast synaptic inhibition in spinal sensory processing and pain control. Physiol. Rev. 2012, 92, 193–235. [Google Scholar] [CrossRef]
- Petitjean, H.; Pawlowski, S.A.; Fraine, S.L.; Sharif, B.; Hamad, D.; Fatima, T.; Berg, J.; Brown, C.M.; Jan, L.Y.; Ribeiro-da-Silva, A.; et al. Dorsal Horn Parvalbumin Neurons Are Gate-Keepers of Touch-Evoked Pain after Nerve Injury. Cell Rep. 2015, 13, 1246–1257. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Cheng, L.; Bourane, S.; Britz, O.; Padilla, C.; Garcia-Campmany, L.; Krashes, M.; Knowlton, W.; Velasquez, T.; Ren, X.; et al. Identification of spinal circuits transmitting and gating mechanical pain. Cell 2014, 159, 1417–1432. [Google Scholar] [CrossRef]
- Cheng, L.; Duan, B.; Huang, T.; Zhang, Y.; Chen, Y.; Britz, O.; Garcia-Campmany, L.; Ren, X.; Vong, L.; Lowell, B.B.; et al. Identification of spinal circuits involved in touch-evoked dynamic mechanical pain. Nat. Neurosci. 2017, 20, 804–814. [Google Scholar] [CrossRef]
- Chen, Y.; Balasubramanyan, S.; Lai, A.Y.; Todd, K.G.; Smith, P.A. Effects of sciatic nerve axotomy on excitatory synaptic transmission in rat substantia gelatinosa. J. Neurophysiol. 2009, 102, 3203–3215. [Google Scholar] [CrossRef]
- Lu, Y.; Perl, E.R. Modular organization of excitatory circuits between neurons of the spinal superficial dorsal horn (laminae I and II). J. Neurosci. 2005, 25, 3900–3907. [Google Scholar] [CrossRef]
- Alba-Delgado, C.; El Khoueiry, C.; Peirs, C.; Dallel, R.; Artola, A.; Antri, M. Subpopulations of PKCγ interneurons within the medullary dorsal horn revealed by electrophysiologic and morphologic approach. Pain 2015, 156, 1714–1728. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Miao, X.; Liang, L.; Abdus-Saboor, I.; Olson, W.; Fleming, M.S.; Ma, M.; Tao, Y.X.; Luo, W. Identification of Early RET+ Deep Dorsal Spinal Cord Interneurons in Gating Pain. Neuron 2016, 91, 1137–1153. [Google Scholar] [CrossRef]
- Sorkin, L.S.; Yaksh, T.L.; Doom, C.M. Mechanical allodynia in rats is blocked by a Ca2+ permeable AMPA receptor antagonist. Neuroreport 1999, 10, 3523–3526. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, L.S.; Yaksh, T.L.; Doom, C.M. Pain models display differential sensitivity to Ca2+-permeable non-NMDA glutamate receptor antagonists. Anesthesiology 2001, 95, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Pogatzki, E.M.; Niemeier, J.S.; Sorkin, L.S.; Brennan, T.J. Spinal glutamate receptor antagonists differentiate primary and secondary mechanical hyperalgesia caused by incision. Pain 2003, 105, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Pogatzki, E.M.; Zahn, P.K.; Brennan, T.J. Effect of pretreatment with intrathecal excitatory amino acid receptor antagonists on the development of pain behavior caused by plantar incision. Anesthesiology 2000, 93, 489–496. [Google Scholar] [CrossRef]
- Nozaki-Taguchi, N.; Yaksh, T.L. Pharmacology of spinal glutamatergic receptors in post-thermal injury-evoked tactile allodynia and thermal hyperalgesia. Anesthesiology 2002, 96, 617–626. [Google Scholar] [CrossRef]
- Taylor, B.K.; Sinha, G.P.; Donahue, R.R.; Grachen, C.M.; Morón, J.A.; Doolen, S. Opioid receptors inhibit the spinal AMPA receptor Ca(2+) permeability that mediates latent pain sensitization. Exp. Neurol. 2019, 314, 58–66. [Google Scholar] [CrossRef]
- Gangadharan, V.; Wang, R.; Ulzhöfer, B.; Luo, C.; Bardoni, R.; Bali, K.K.; Agarwal, N.; Tegeder, I.; Hildebrandt, U.; Nagy, G.G.; et al. Peripheral calcium-permeable AMPA receptors regulate chronic inflammatory pain in mice. J. Clin. Investig. 2011, 121, 1608–1623. [Google Scholar] [CrossRef]
- Kopach, O.; Krotov, V.; Voitenko, N. Atlanto-occipital catheterization of young rats for long-term drug delivery into the lumbar subarachnoid space combined with in vivo testing and electrophysiology in situ. J. Neurosci. Methods 2017, 290, 125–132. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopach, O.; Dobropolska, Y.; Belan, P.; Voitenko, N. Ca2+-Permeable AMPA Receptors Contribute to Changed Dorsal Horn Neuronal Firing and Inflammatory Pain. Int. J. Mol. Sci. 2023, 24, 2341. https://doi.org/10.3390/ijms24032341
Kopach O, Dobropolska Y, Belan P, Voitenko N. Ca2+-Permeable AMPA Receptors Contribute to Changed Dorsal Horn Neuronal Firing and Inflammatory Pain. International Journal of Molecular Sciences. 2023; 24(3):2341. https://doi.org/10.3390/ijms24032341
Chicago/Turabian StyleKopach, Olga, Yulia Dobropolska, Pavel Belan, and Nana Voitenko. 2023. "Ca2+-Permeable AMPA Receptors Contribute to Changed Dorsal Horn Neuronal Firing and Inflammatory Pain" International Journal of Molecular Sciences 24, no. 3: 2341. https://doi.org/10.3390/ijms24032341
APA StyleKopach, O., Dobropolska, Y., Belan, P., & Voitenko, N. (2023). Ca2+-Permeable AMPA Receptors Contribute to Changed Dorsal Horn Neuronal Firing and Inflammatory Pain. International Journal of Molecular Sciences, 24(3), 2341. https://doi.org/10.3390/ijms24032341