Abstract
Obstructive sleep apnea (OSA) is a chronic and highly prevalent condition that is associated with oxidative stress, inflammation, and fibrosis, leading to endothelial dysfunction, arterial stiffness, and vascular insulin resistance, resulting in increased cardiovascular disease and overall mortality rates. To date, OSA remains vastly underdiagnosed and undertreated, with conventional treatments yielding relatively discouraging results for improving cardiovascular outcomes in OSA patients. As such, a better mechanistic understanding of OSA-associated cardiovascular disease (CVD) and the development of novel adjuvant therapeutic targets are critically needed. It is well-established that inappropriate mineralocorticoid receptor (MR) activation in cardiovascular tissues plays a causal role in a multitude of CVD states. Clinical studies and experimental models of OSA lead to increased secretion of the MR ligand aldosterone and excessive MR activation. Furthermore, MR activation has been associated with worsened OSA prognosis. Despite these documented relationships, there have been no studies exploring the causal involvement of MR signaling in OSA-associated CVD. Further, scarce clinical studies have exclusively assessed the beneficial role of MR antagonists for the treatment of systemic hypertension commonly associated with OSA. Here, we provide a comprehensive overview of overlapping mechanistic pathways recruited in the context of MR activation- and OSA-induced CVD and propose MR-targeted therapy as a potential avenue to abrogate the deleterious cardiovascular consequences of OSA.
1. Introduction
Obstructive sleep apnea is a chronic and highly prevalent condition estimated to affect close to 1 billion people worldwide [1]. OSA is characterized by intermittent increases in upper airway resistance in the context of heightened airway collapsibility, ultimately leading to either reductions or altogether cessation of airflow (i.e., hypopneas and apneas, respectively). These events, which usually last 10–30 s, but can last minutes in severe cases [2], lead to important physiological alterations such as intermittent hypoxia (IH), sleep fragmentation (SF), intermittent hypercapnia, and enhanced intrathoracic pressure swings [3,4,5,6,7]. These processes can, in turn, activate and propagate a large array of pathophysiological mechanisms, inducing sustained enhancements of sympathetic activity coupled with parasympathetic withdrawal, disturbances of the hypothalamic–pituitary–adrenal axis, activation of the renin–angiotensin–aldosterone system (RAAS), systemic oxidative stress, both diffuse and localized inflammation and immune dysregulation, and overall hemodynamic stress. As a consequence of these processes, adverse consequences such as endothelial dysfunction, arterial stiffness, vascular insulin resistance, and cardiac remodeling, ultimately causing hypertension and atherosclerosis, manifest over time [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29]. Predictably, OSA has consistently emerged as an independent risk factor for cardiovascular disease (CVD), including coronary artery disease (CAD), stroke, myocardial infarction (MI), congestive heart failure (HF), and several types of arrhythmias, such as atrial fibrillation (AF) [4,6,30,31,32,33,34,35,36]. Unfortunately, the primary line of treatment for OSA, i.e., continuous positive airway pressure (CPAP), has resulted in being disappointingly ineffective in protecting against OSA-induced CVD [37,38,39].
RAAS activation is one of the best-studied hormonal systems involved in the pathophysiology of systemic hypertension and of cardiovascular disease [40,41,42]. Briefly, active renin acts upon angiotensinogen to generate angiotensin I that is cleaved by the angiotensin-converting enzyme to the physiologically active angiotensin II (Ang II). As a main effector of the RAAS system, Ang II exerts many of its effects via type I angiotensin receptors (AT1R) [43]. Another important component of the RAAS, aldosterone, exerts crucial endocrine functions such as regulating fluid volume, sodium, and potassium homeostasis, and primarily acts on mineralocorticoid receptors (MR) in the renal distal tubules [43]. Discovery of an important MR-dependent signaling in the cardiovascular system has resulted in major expansion of the scope of the research on aldosterone-mediated cardiovascular effects [44,45,46]. Indeed, MR activation mediates genomic and non-genomic effects via second messenger systems, and via crosstalk with multiple receptors that underlie downstream vascular biological and (patho)physiological functions, particularly vascular inflammation, oxidative stress, fibrosis, and cardiac remodeling and hypertrophy [47,48]. Clinical and experimental trials have shown that targeted blockade of MR receptors can reduce blood pressure, alleviate oxidative stress and inflammation, improve vascular function and insulin sensitivity, and protect against atherosclerosis, HF, MI, and AF [49,50,51,52,53,54,55,56,57,58]. In other words, MR blockers alleviate a similar spectrum of conditions that are associated with OSA.
Although the relationship between OSA and RAAS activation is not fully understood [59], a bidirectional association exists between the two conditions, whereby OSA can trigger RAAS activation, and the latter can then worsen the prognosis and morbidities associated with OSA [59]. Nevertheless, both conditions share similar mechanisms and processes that underlie cardiovascular disturbances and increase overall mortality, as well as cardiac-specific mortality [5,7,26,44,45]. Despite this rather conspicuous relationship, very few studies have evaluated the effects of MR antagonism in OSA, and of these few studies, most have focused on resistant hypertension in OSA patients [60,61,62]. Of note, these studies have been conducted in light of the disappointing outcomes reported by the few large, randomized control trials (RCTs) that showed minimal to no benefit of CPAP treatment in protecting against OSA-mediated CVD [37,38,39]. Moreover, there are no experimental studies using available OSA models evaluating the role of MR signaling in cardiac and vascular tissues, and the potential benefit of MR antagonism on cardiovascular outcomes. This review focuses on the relationship between the OSA and MR activation, mechanisms shared by both conditions in promoting CVD, and the potential cardiovascular benefits of MR antagonism, thus highlighting MR as a potential therapeutic target for OSA-induced CVD.
2. OSA and CVD
As mentioned above, OSA is a highly prevalent and a serious chronic sleep-related breathing disorder affecting up to one billion people globally [1]. The chronic physiological stresses from recurrent airway obstruction can directly induce cardiovascular morbidity and mortality [5,6,7,26]. Indeed, it is estimated that more than 40% of patients with CVD have OSA [1]. However, OSA remains critically underdiagnosed and undertreated, which raises the risk of developing OSA-related long-term cardiovascular consequences [63]. Several factors hinder adequate OSA screening and diagnosis, including confounding variables and logistical and financial barriers. The gold standard test, polysomnography, can be costly with long waiting times [64,65,66]. Several questionnaires were developed to screen for OSA based on risk factors (male sex, older age, obesity, craniofacial abnormalities, and genetics) [67]. However, these OSA screening questionnaires have limited specificity and poor diagnostic accuracy, especially in patients with underlying CVD [68,69].
OSA has been associated with multiple cardiovascular complications including hypertension, HF, CAD, arrhythmias, and CV mortality [1,5,6,63]. OSA and hypertension often coexist with an estimated 50% of OSA patients having hypertension and 30% of hypertensive patients also having OSA [70,71,72]. In patients with untreated OSA, the risk of developing hypertension is increased 2–3-fold [73], while around 65–80% of patients with resistant hypertension may suffer from OSA [74,75]. The largest impact of OSA on blood pressure is due to dampening of the normal nocturnal fall in blood pressure (dipping) [76]. In HF, The Sleep-Disordered Breathing in Heart Failure (SchlaHF) Registry reported the prevalence of moderate to severe OSA to be around 50% in men and 36% in women with stable, symptomatic heart failure with reduced ejection fraction (HfrEF) [77]. It is also estimated that OSA is present in more than 50% patients with HfpEF [78]. OSA can increase the risk of symptom progression and mortality in HF patients with increased risk of death by 16.1% for each hypoxemic hour (time with oxygen saturation less than 90%) [36,79]. AF has also been associated with OSA with a ranging prevalence between 21% and 74% [35,80]. OSA can independently induce left atrial structural changes and enlargement that predisposes OSA patients to AF [81]. The severity of OSA in directly proportional to increased risk of AF and the presence of OSA has been also shown to inhibit the efficacy of antiarrhythmic treatments in AF patients [82,83]. OSA have been also associated with bradyarrhythmias, supraventricular and ventricular tachyarrhythmias, and sudden cardiac arrest [34,84,85]. It is estimated that 38% to 65% of patients with CAD have OSA and that OSA is present in nearly 50% of patients requiring percutaneous coronary intervention [3,4]. The risk of developing CAD increases substantially in the presence of OSA. In fact, OSA increases the risk for development of MI, coronary revascularization events, or CV death by 2-fold, independent of other risk factors [86]. OSA has been reported to independently enhance the risk of subclinical CAD measured by coronary calcification score and can independently predict the risk of atherosclerotic plaque progression and vulnerability [32,33]. In the SHHS (Sleep Heart Health Study), the all-cause mortality was significantly higher among male patients with severe OSA [87]. Similarly, The MESA (Multiethnic Study of Atherosclerosis) showed that OSA was associated with 2.2 higher incidence of cardiovascular events and a 2.4 increase in mortality over a 7.5-year follow-up period [88].
Repeated nocturnal hypoxic episodes in OSA can initiate a plethora of pathophysiological mechanisms that promote cardiac and vascular disease [3,4,5,6,7,24,26]. OSA-induced IH and hypercapnia elicit chemoreflex-mediated enhancements of sympathetic activity and RAAS activation leading to vasoconstriction and subsequent increases in blood pressure surges and hemodynamic disturbances [19]. IH and sleep fragmentation (SF) are also considered major activators of pro-oxidant and pro-inflammatory pathways leading to systemic inflammation and dysfunction, particularly in the cardiovascular system. All the aforementioned mechanisms contribute to endothelial dysfunction and an enhanced prothrombotic state, initiating the atherosclerotic process and cardiac remodeling [5,7,31]. Additionally, resultant intrathoracic swings can influence cardiac transmural gradients and affect ventricular function, leading to increased wall stress and impaired diastolic function [89]. Evidence from clinical and experimental models of OSA, especially those involving IH exposures, show that lipid, protein, and DNA oxidative stress markers are elevated with activated NADPH oxidases, xanthine oxidase, and mitochondria being major sources [16,17,18,26,90,91,92,93,94,95]. Other studies have also shown enhanced NF-ƙB activation, aggravated proinflammatory cytokine and adhesion molecule production, and profibrotic signaling [13,14,15,16,90,96,97,98,99,100,101,102,103,104,105,106]. Moreover, OSA and experimental IH have shown to disrupt lipid and glucose homeostasis, leading to insulin resistance and metabolic disturbances [11,12,107,108,109,110,111,112]. Collectively, the overwhelming evidence implicates OSA and its preclinical models in the processes favoring the occurrence of atherosclerosis and CVD.
3. OSA Management with CPAP
CPAP is the mainstay treatment for OSA due to its positive impact on symptoms and quality of life, which provides a constant level of positive pressure across inspiration and expiration [113]. Although CPAP treatment has been shown to reduce AHI and mitigate or even abolish the episodic hypoxemia, both of which are predictors of CVD, it failed to reduce the rates of cardiovascular complications in OSA patients [89]. The SAVE (Sleep Apnea Cardiovascular Endpoint) study randomized over 2700 OSA patients with a history of CVD to usual care plus CPAP versus usual care alone and failed to demonstrate conclusive evidence of significant reductions in the primary endpoint (composite CVD) after 3.7 years of follow-up with ongoing CPAP treatment [39]. Similar negative findings were found in The ISAACC (CPAP in Patients With Acute Coronary Syndrome and OSA) and The RICCADSA (Randomized Intervention with CPAP in CAD and OSA) trials, which investigated the effectiveness of CPAP on the putative reduction in CV complications following acute coronary syndrome and PCI, respectively [37,38]. A recent meta-analysis of nine randomized trials showed that CPAP treatment did not prolong survival or reduce cardiovascular events in adults with OSA and CVD [114]. Poor CPAP adherence (minimal adherence being >4 h per night) was identified as the main likely contributor to the negative outcomes of the trials [37,38,114,115]. Moreover, incidents of cardiovascular complications, such as CAD, are substantially increased by OSA, especially in patients without a previous history of CAD [37]. This suggests that advanced atherosclerotic vascular pathological processes are more likely to be irreversible with OSA treatment, a finding that was recapitulated in experimental IH delivered to mice for extended periods of time [116]. Ultimately, the need for additional adjuvant therapies aimed at CVD induced by OSA are needed. In the next sections, we discuss the impact of MR activation in CVD and the potential usefulness of MR antagonists in targeting OSA-induced CVD.
4. MR Structure and Function
MR (NR3C2) is a member of a steroid-activator transcription factor superfamily that preserves structural similarities to the glucocorticoid receptor (NR3C1, GR) and the progesterone receptor [117]. Similar to other nuclear receptors, MR contains DNA- and ligand-binding domains, in addition to an amino terminal domain and hinge region [48]. Under basal conditions, MR is predominantly present in the cytosolic fraction as part of a heterocomplex with multiple chaperones that are important for facilitating ligand binding and cytoplasmic–nuclear translocation [118,119]. To allow DNA binding, MR dissociates from its chaperones and forms homodimers or heterodimers with GR in certain conditions that can then result in different transcriptional responses [120,121]. Once the MR DNA-binding domain binds to hormone response elements, gene transcription ensues. MR can also form complexes with other transcription factors rather than directly bind to DNA itself [122].
Aldosterone is the major mineralocorticoid hormone synthesized in the zona glomerulosa of the adrenal gland under the influence of the RAAS, adrenocorticotropic hormone (ACTH), and extracellular potassium levels [123]. The main function of aldosterone is to regulate the fluid and salt balance via sodium transport machinery in renal epithelial tubular cells [124]. Besides aldosterone, MR has a strong affinity for glucocorticoids that are significantly more abundant in the circulation [125]. However, aldosterone can remain bound to MR for more prolonged periods than cortisol, which stabilizes MR conformation. As a mechanism to confer the specificity of MR to aldosterone, the enzyme 11-beta-hydroxysteroid dehydrogenase type 2 (HSD11B2) is co-expressed with MR and can metabolize cortisol to cortisone, which is unable to bind to or activate MR [126]. Indeed, HSD11B2 deficiency or inhibition can result in hypertension and hypokalemia due to activation of cortisol by renal MR [127]. However, under cellular oxidative stress and inflammation, glucocorticoids can activate MR [128,129]. Thus, distribution of HSD11B2 expression, ligand availability, and cellular redox status can influence the response of the MR to ligand activation.
MR utilizes several mechanisms to promote cellular changes and regulates a multitude of genes, with the majority of such genes being involved in electrolyte homeostasis in renal epithelial tissues [47]. Other MR target genes can be expressed upon stimulation by aldosterone in other tissues, including vascular and cardiac tissues that can affect signal transduction, redox status, cellular structure, adhesion, and migration [130,131]. Due to the slow nature of gene transcription, MR can also trigger faster non-genomic mechanisms, for instance in response to acute changes in fluid homeostasis [47]. MR rapid signaling which occurs through second messenger systems includes: (i) Mitogen-associated protein kinases (MAPK) for cell proliferation or apoptosis and electrolyte handling [132], (ii) Phosphatidylinositide 3-kinases (PI3K) involved in electrolyte handling and vasomotor function [133], (iii) Protein kinase C (PKC) and protein kinase D for renal epithelial cells and cardiomyocyte electrolyte handling [134,135]. Other rapid non-genomic MR-dependent aldosterone effects can be initiated at the plasma membrane where the classical MR is associated with scaffolding proteins to the cytosolic side of the plasma [136]. Striatin and caveolin-1 have been recently identified as candidates for such scaffolding proteins [137,138]. As a result, transactivation of many receptor tyrosine kinases and G-protein-coupled receptors that are located close to the MR scaffolding proteins have been reported [136]. Additionally, MR can initiate crosstalk with other cytosolic signaling pathways affecting genomic signaling, such as nuclear factor of activated T-cells (NFAT) and cAMP-response element-binding protein (CREB) [139,140]. Lastly, genomic MR signaling can be influenced by epigenetic mechanisms and by posttranscriptional regulation by microRNAs [141,142] (Figure 1).
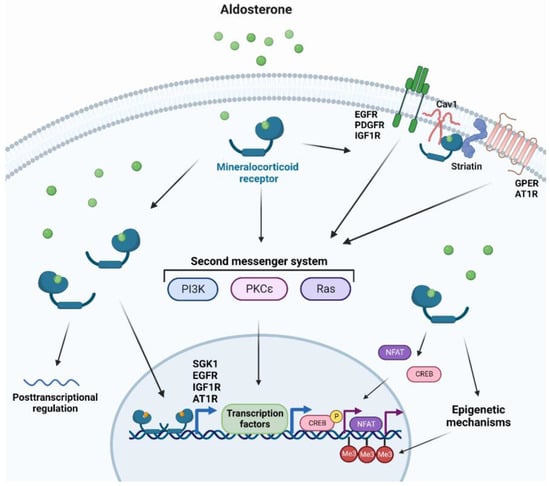
Figure 1.
Cellular responses to MR activation. Aldosterone binding to cytosolic mineralocorticoid receptors forms homodimers and translocates into the nucleus to elicit genomic responses. Cytosolic MR activation can also trigger non-genomic rapid responses through second messenger systems. Additionally, MR activation can transactivate several membrane-bound receptors, crosstalk with other cytosolic pathways, induce or modify epigenetic modifications, and affect posttranslational regulation. AT1R: Type I angiotensin receptor, Cav1: caveolin 1, CREB: cAMP-response element-binding protein, EGFR: epidermal growth factor, GPER: G protein-coupled estrogen receptor 1, IGF1R: insulin-like growth factor receptor 1, Me3: tri-methylation, NFAT: Nuclear factor of activated T-cells, PDGFR: platelet-derived growth factor receptor, PI3K: phosphatidylinositide 3-kinases, PKC: protein kinase C, SGK1: serine/threonine-protein kinase 1.
5. Pathological Mechanisms of MR Activation
Oxidative stress: MR activation can lead to reactive oxygen species (ROS) generation, particularly through the upregulation and activation of NADPH oxidases (NOX) [47]. NOX is a family of membrane-bound enzymes that produces superoxide anions and is abundantly present in endothelial cells, vascular smooth muscle cells (VSMCs), cardiomyocytes, and leukocytes [143,144]. The enhanced ROS production mediated by MR activation can lead to excessive activation of proinflammatory and profibrotic signaling through activator protein 1 (AP-1) and nuclear factor kappa B (NF-ƙB) [145,146]. Aldosterone can rapidly and persistently activate NOX within minutes, suggesting a non-genomic regulation of NOX [147]. Indeed, MR activates NOX through c-Src and downstream Ras-related C3 botulinum toxin substrate (Rac1) activation [148,149]. MR can also increase NOX-dependent ROS production through crosstalk with epidermal growth factor receptor (EGFR) and AT1R [149]. Additionally, MR can upregulate NOX by enhancing synthesis of NOX cytosolic subunits in endothelial cells and cardiomyocytes [150,151]. In animal models of hypertension using aldosterone and salt, the levels of oxidized lipids were elevated in urine, plasma, kidney, and hearts [152,153,154], with levels of 8-isoprostane levels reaching 10-fold higher in hearts of aldosterone-treated animals when compared to controls [153]. Increased superoxide anion production has been also reported in multiple tissues in aldosterone-treated animals [155,156]. Furthermore, the expression of NOX was elevated in hearts and vasculature of aldosterone-treated rats [151,157]. Treatment with the MR antagonist spironolactone lowered free radical production, confirming the involvement of MR in aldosterone-induced oxidative stress [158,159,160,161,162]. In patients with essential hypertension, who often exhibit hyperaldosteronism, the levels of oxidative stress markers were elevated [163,164]. Treatment with spironolactone lowered urinary oxidative stress markers in patients with chronic kidney disease and reduced superoxide anion production in isolated macrophages from patients with congestive HF [165,166].
Inflammation: Persistent MR activation is associated with enhanced inflammatory responses that can eventually lead to tissue remodeling and fibrosis [167,168,169,170]. MR regulates transcription of genes involved in the recruitment of adhesion molecules via platelet-derived growth factor receptor (PDGFR) and c-Src activation [171]. It can also increase production of inflammatory cytokines, including tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6), via regulation of NF-ƙB transcription, which is further enhanced by serine/threonine-protein kinase 1 (SGK1) [172,173]. Constant exposure to aldosterone for 14 days is associated with perivascular and cardiac inflammation, as reflected by leukocyte recruitment, adhesion, and infiltration [174,175]. MR activation induced intracellular adhesion molecule 1 (ICAM1) and P-selectin expression in endothelial cells and in VSMCs and induced vascular endothelial growth factor (VEGF) recruitment of monocytes [176,177,178]. AT1R has been shown to be required for expression of inflammatory genes in the presence of aldosterone in VSMCs since Agtr1a-null mice were protected against aldosterone-induced vascular dysfunction [179,180]. Aldosterone can also induce C-reactive protein (CRP) expression in VSMCs in vitro and in vivo that is prevented by MR antagonism [181]. One of the other mechanisms by which aldosterone induces inflammation is through activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome that can lead to caspase-1-dependent induction of the pro-inflammatory cytokines interleukin 1 beta (IL-1β) and interleukin 18 (IL-18) [182]. Rats infused with aldosterone exhibited increased macrophage infiltration of the kidneys and increased expression of inflammasome markers that were attenuated with immunosuppressant treatment [183]. NLRP3 deletion prevented aldosterone-mediated expression of adhesion molecules and adherence of macrophages to aortic segments in mice [182]. A recent study showed that TNF-α and IL-6 were elevated in patients with primary aldosteronism (PA) when compared to normotensive individuals or to patients with essential hypertension [184]. Treatment of hypertensive patients with eplerenone, an MR antagonist, reduced many proinflammatory mediators, including monocyte chemoattractant protein 1 (MCP-1) and interleukin 8 (IL-8) [185].
Sympathetic activation: MR can be expressed extra-renally in tissues such as the heart, brain, and peripheral sensory neurons [186,187,188]. Despite the well-established role of MR activation in RAAS-induced sympathetic activation, aldosterone has shown to affect the parasympathetic nervous system through inhibition of baroreceptor discharge and bradycardic responses to pressor stimuli [189,190]. It can also activate the sympathetic system and augment its activity by attenuating catecholamine uptake in myocardial tissues [191,192]. In the hypothalamic paraventricular nucleus (PVN) region, MR can contribute to Ang II-induced hypertension [193]. Indeed, central infusion of MR antagonists can prevent neuronal activation in the PVN by Ang II and the associated increases in blood pressure [194]. Moreover, local expression of MR, 11β-HSD2, and aldosterone in the neurons of the cardiac autonomic nervous system has been recently discovered, suggesting a putative modulatory role of MR on cardiac endogenous neuronal activity [195]. Overall, MR may contribute directly to sympathetic activation or parasympathetic inhibition, leading to increased blood pressure and cardiovascular dysfunction.
Endothelial dysfunction: Endothelial dysfunction is the main pathological process preceding atherosclerosis and is manifest as impaired vasodilation and/or enhanced vasoconstriction [196]. In vitro, aldosterone reduces nitric oxide (NO) production in endothelial cells via inhibition of endothelial nitric oxide synthase (eNOS) activity. Aldosterone-induced eNOS inhibition is mediated through increased RhoA kinase activity and consequent protein kinase B (Akt) signaling inhibition, or via increased protein phosphatase 2A (PP2A) activity, leading to eNOS dephosphorylation [157,197]. Vascular endothelial MR activation increases oxidative stress [198]. Aldosterone-induced ROS overproduction can reduce NO bioavailability and lead to the production of the pernicious reactive nitrogen specie peroxynitrite that can oxidize lipids, proteins, and DNA (Figure 2) [199,200]. It can also lead to tetrahydrobiopterin (BH4) depletion, an important cofactor for NO generation by eNOS, resulting in more production of ROS instead of NO [157,201]. Moreover, aortic expression of cyclooxygenase 2 (COX-2) is elevated in aldosterone-treated rats [202,203]. COX-2 can generate vasoactive prostanoids and ROS that not only impair vasodilation but can also enhance vasoconstrictive responses (Figure 2) [203,204]. Aldosterone can also indirectly impair endothelium-dependent vasodilation through glucose-6-phosphate dehydrogenase (G6PD) reduction, exacerbating oxidative stress and impairing NO production [205]. MR activation can also exert NO-independent effects on vasomotor responses. Aldosterone can increase endothelial cell volume and tension though NHE-1 and/or epithelial sodium channel (EnaC) activation, with the latter also affecting eNOS activity and NO production [206,207,208]. However, endothelial response to MR is vastly heterogeneous depending on sex, vascular bed, steroid use, duration of exposure, and environmental context [209,210,211]. For instance, MR deletion in the endothelium had no effect on mesenteric vasomotor responses but significantly mitigated vasoconstrictor responses to endothelin-1 in coronary arteries [210]. Moreover, endothelial MR deletion in females was more protective than in males in the context of diet-induced obesity [212]. The heterogeneity of MR responses extends to signaling through other receptors such as AT1R signaling that is important for aldosterone-mediated endothelial dysfunction. Indeed, inhibiting or knocking out of AT1R in mice blunted endothelial dysfunction after treatment with aldosterone [179]. In humans, there is a strong association between aldosterone levels and impaired flow-mediated dilatation (FMD), a gold-standard indicator of endothelial function [213]. FMD is also inversely correlated with plasma aldosterone and urinary aldosterone secretion in patients with resistant hypertension [214]. In PA patients, FMD is inversely correlated with PA phenotype severity and lower numbers of circulating endothelial progenitor cells that also exhibit reduced migratory potential [215,216,217]. Despite the controversial benefit of MR antagonist on FMD, a recent meta-analysis showed that FMD increased in 11 trials including 570 patients after treatment with MR antagonists (mean difference, 1.18% [95% CI, 0.14 to 2.23], p = 0.03) [218].
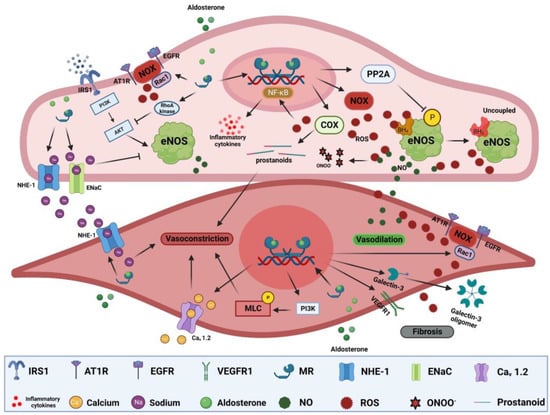
Figure 2.
MR-mediated mechanisms of vascular dysfunction. MR activation in endothelial cells can induce ROS production through activation of NOXs and COXs. ROS react with NO, reducing its bioavailability leading to impaired endothelial-dependent vasodilation, forming a more reactive nitrogen species, ONOO-, oxidizing BH4 to uncouple eNOS that leads to production of ROS instead of NO. Excessive ROS production can also trigger NF-ƙB activation, leading to increased expression of inflammatory cytokines. MR activation can also inhibit eNOS through dephosphorylation by PP2A, impair insulin-dependent signaling, activate NHE-1 and EnaC. In SMCs, MR activation can trigger vasoconstriction through activation of NHE-1 and Cav 1.2 in addition to MLC phosphorylation. Activation of MR can elicit fibrotic signaling through increased ROS production, galectin-3 oligomer formation, and VEGEF signaling. Akt: protein kinase B, AT1R: type I angiotensin receptor, BH4: tetrahydrobiopterin, Cav 1.2: L-type calcium channel alpha 1C, COX2: cyclooxygenase 2, EGFR: epidermal growth factor, EnaC: epithelial sodium channel, eNOS: endothelial nitric oxide synthase, IRS1: insulin receptor substrate 1, MLC: myosin light chain, NHE-1: sodium–hydrogen antiporter 1, NF-ƙB: nuclear factor kappa B, NOX: NADPH oxidases, ONOO-: peroxynitrite, PI3K: phosphatidylinositide 3-kinases, PP2A: protein phosphatase 2A, Rac1: Ras-related C3 botulinum toxin substrate, ROS: reactive oxygen species, VEGFR1: vascular endothelial growth factor receptor 1.
Fibrosis: MR activation can regulate multiple pro-fibrotic and pro-hypertrophic genes via second messenger systems. For instance, MR can induce cardiac fibroblast proliferation and hypertrophy-associated genes as myosin heavy chain beta (MHC-β) through extracellular signal-regulated kinase (ERK) signaling [219]. Additionally, MR activation can promote cardiomyocyte production of connective tissue growth factor (CTGF) via p38 MAPK signaling [220]. AT1R transactivation by MR is also involved in the production of fibrotic and hypertrophic genes including transforming growth factor beta (TGF-β) and alpha-smooth muscle actin (α-SMC) through ERK and c-Jun N-terminal kinase (JNK) signaling [221]. Moreover, cardiac MR transactivation of EGFR can increase sodium–hydrogen antiporter 1 (NHE-1) activity that results in sodium accumulation promoting calcium influx and subsequent facilitation of generation of pro-hypertrophic and pro-fibrotic factors [222]. Genomically, increased SGK1 by MR activation upregulates cardiac CTGF [223]. Hyperaldosteronism in mice exacerbated hypertension-induced fibrosis through activation of inflammation/galectin-3-induced fibrosis and inhibition of antifibrotic factors such as B-type natriuretic peptide (BNP) [224]. Cardiomyocyte-specific MR-null mice after MI showed improved reparative scar formation and reduced infarct size and reactive fibrosis in the viable ventricle wall [225].
Arterial stiffness: Arterial stiffness reflects the attenuation of arterial distensibility due to the combination of smooth muscle cells contraction (active stiffness) and changes of inert components of the vascular wall (passive stiffness) [226]. Arterial stiffness can be assessed by pulse wave velocity (PWV) and by the augmentation index [226]. In animal studies, aldosterone augmented active stiffness by enhancing vascular myogenic tone and vasoconstrictive agent-dependent contraction that was reduced in VSMC knockout mice [227]. Aldosterone can also induce passive stiffness through modulation of the expression of several vascular genes involved in extracellular matrix remodeling including CTGF and metalloproteinases [130]. Furthermore, aldosterone increases collagen deposition in the arterial wall of multiple animal models via several pathways including galectin-3 [228], neutrophil gelatinase-associated lipocalin (NGAL) [229], ENAC [207], and NOX [230] (Figure 2). It has been shown that aldosterone can induce osteogenic phenotype in VSMCs through activation of alkaline phosphatase and NOX, reducing VSMCs autophagy, and induction of inflammatory pathways, thereby increasing calcification of the vascular wall [231,232,233,234]. Spironolactone treatment mitigated the progression of calcification in uremic rats [235]. High aldosterone has been associated with increased PWV in patients newly diagnosed with hypertension, independent of blood pressure [236]. Treatment with spironolactone significantly reduced both PWV and the augmentation index in patients with hypertension [237]. In PA patients, a meta-analysis showed that PWV is higher when compared to matched patients with essential hypertension, but no differences emerged in the augmentation index [238]. Adrenalectomy significantly reduced PWV and augmentation index in patients with PA [239]. A recent meta-analysis showed that MR antagonists reduced PWV in 11 trials including 515 patients when compared to controls (mean difference, −0.75 m/s [95% CI, −1.12 to −0.39], p < 0.00001) independent of blood pressure reduction [218]. MR antagonist treatment also reduced the augmentation index compared with controls in 5 trials including 283 patients (mean difference, −6.74% [95% CI, −10.26 to −3.21], p = 0.0002) [218].
Insulin resistance: Insulin resistance is characterized by the reduced ability of insulin to activate insulin signaling and glucose uptake [240]. Although the main targets of insulin are skeletal muscle, liver, and adipose tissues, insulin receptors are also present in vascular cells, among many other cells [240]. Diminished insulin signaling in vascular tissues leads to reduced glucose uptake and insulin resistance [241]. Animal studies have shown that excessive MR activation impairs insulin signaling and induces vascular stiffness, mainly by aggravating oxidative stress and inflammation [242,243,244,245,246]. In the endothelium, aldosterone can enhance the upregulation and translocation of EnaC to the cell wall, which induces vascular stiffness, impairs insulin-mediated capillary recruitment, and enhances insulin resistance (Figure 2) [245]. MR activation can also activate the mammalian target of the rapamycin/ribosomal protein S6 kinase beta-1 (mTOR/S6K1) signaling pathway, which induces vascular insulin resistance via increased serine phosphorylation insulin receptor substrate 1 (IRS1), leading to reduced translocation of glucose transporter 4 (GLUT4) to the cell membrane and glucose uptake [247]. Thus, reduced insulin signaling can lead to impaired PI3K/Akt signaling and consequent inhibition of eNOS phosphorylation and NO production, and the hyperinsulinemia associated with such events can increase the release of vasoconstrictive substances, ultimately leading to impaired vasodilation and enhanced vasoconstriction (Figure 2) [246,247]. MR blockade with spironolactone or with mice harboring endothelial-specific MR knockout abolished diet-induced aortic and mesenteric arterial stiffness and microvascular dysfunction, improved insulin-mediated eNOS activity, and suppressed endothelial EnaC activation [210,242,245]. Furthermore, spironolactone decreased the expression of multiple inflammatory cytokines and decreased oxidative stress markers with accompanying increases in vascular insulin sensitivity in female mice fed a Western diet [242]. Despite the promising results from preclinical data on spironolactone and eplerenone on glucose metabolism insulin sensitivity, clinical studies have shown no benefit from MR antagonism on glucose metabolism in relevant patient cohorts. Data from the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) clinical study showed that MR antagonists may be involved in increased risk for the development of diabetes [248]. Additionally, a recent meta-analysis of 18 trials assessing spironolactone effects on fasting glucose and insulin, hemoglobin A1c, and homeostatic model assessment (HOMA)-insulin resistance (IR) showed that spironolactone increased HbA1c but had no clear effect on fasting glucose, HOMA-IR, and insulin levels [249]. However, newer non-steroidal and more selective MR antagonists may prove to have a favorable impact on glucose and insulin [250].
Cardiac electrical remodeling: MR activity can influence cardiomyocyte electrolyte handling, the action potential, and cardiac contractility [47]. Low-voltage-activated T-type channels that regulate rapid activation and slow deactivation, and L-type dihydropyridine channels that activate slower but more rapidly deactivate than T-type channels, are both crucial for pacemaker activity and action potential propagation [251]. MR activation increases calcium current through both of these types of calcium channels [251,252]. Indeed, cardiomyocyte calcium status is linked to transmembrane sodium concentrations [253]. Aldosterone can raise sodium influx rapidly via multiple channels via transactivation of many second messenger systems, such as increased EGFR-dependent NHE-1 activation [222,254]. In addition to increasing sodium influx, NHE-1 can regulate intracellular pH and cell volume, leading to cardiomyocyte alkalinization increasing myofilaments response to calcium [255]. At the structural level, enhanced calcium load can induce profibrotic pathways in atrial tissues, leading to atrial remodeling and dilatation and consequent atrial fibrillation [256]. Cardiac overexpression of MR in mice increases action potential duration and ventricular arrhythmia due to aberrant release of calcium from the sarcoplasmic reticulum [257]. MR knockdown with interfering RNA blocked slow force response accompanied by reduced NHE-1 activity [258].
6. Cardiovascular Consequences of MR Activation
Coronary artery disease: CAD is the consequence of the atherosclerotic processes that are driven by activated endothelium and macrophage recruitment followed by progressive engulfment of macrophages with oxidized LDL, forming foam cells. Thus, vascular oxidative stress and inflammation are very crucial pathological stimuli for plaque formation, the major cause for CAD (Figure 3) [259]. Aldosterone can activate endothelial cells and stimulate VSMCs, increasing leukocyte and monocyte adhesion and infiltration [176,178,260]. Once in the intima media layer, monocytes differentiate into M1 macrophages characterized by a proinflammatory action that is facilitated by aldosterone [175,182]. In Apolipoprotein E (ApoE) knockout mice, aldosterone infusion increased the infiltration of macrophages in the atherosclerotic plaque, increased plaque size and lipid accumulation in the plaque [176,261,262]. MR antagonism reduced plaque size, lipid peroxides, and oxidation of LDL within the plaque [263,264]. Moreover, specific MR deletion in VSMC improved left ventricular function after MI via preservation of coronary flow reserve [265]. In humans, studies have shown a clear association between aldosterone levels and atherosclerosis in the coronary arteries [266]. Elevated aldosterone levels are independently associated with an increased risk of acute cardiac ischemic events, higher rates of cardiovascular events, cardiovascular mortality, and overall mortality in patients with acute MI [267,268]. Patients with PA exhibit more pronounced vascular inflammatory phenotype when compared to patients with essential hypertension [269,270]. Results from a meta-analysis showed that patients with PA are at a 1.77-fold higher risk of developing coronary artery disease compared to patients with essential hypertension [271]. Adrenalectomy and treatment with MR antagonists reduced the risk of cardiovascular disease in PA patients [272]. In large, randomized control trials, the REMINDER trial (Impact of Eplerenone on Cardiovascular Outcomes in Patients Post MI) showed that eplerenone reduced the primary composite outcome (cardiovascular events and mortality, reduced left ventricular function, and prolonged hospitalization) [273]. In the ALBATROSS trial, (Aldosterone Lethal Effects Blocked in Acute MI Treated With or Without Reperfusion to Improve Outcome and Survival at Six Months Follow-Up), intravenous canrenone administration followed by oral spironolactone for 6 months did not improve the composite primary outcome, but exerted a benefit in mortality among patients with MI with ST-segment elevation [274]. In the DIDELIO-DKD trial (Finerenone in Reducing Kidney Failure and Disease Progression in Diabetic Kidney Disease), finerenone reduced the risk of the composite cardiovascular outcome compared with placebo in patients with or without cardiovascular disease [275].
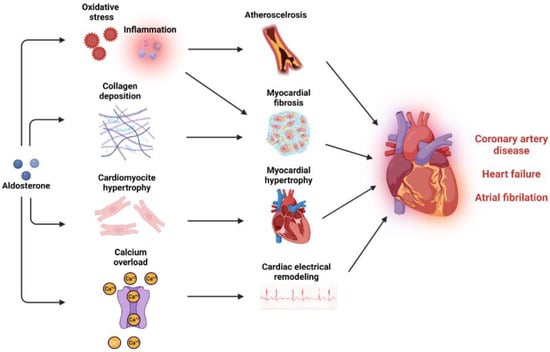
Figure 3.
Mineralocorticoid activation and cardiovascular disease. MR activation can induce multiple pathological mechanisms that promote atherosclerosis and myocardial dysfunction and can lead to coronary artery disease, heart failure, and atrial fibrillation.
Heart Failure: Hemodynamic overload and neurohumoral mechanisms can lead to cardiac hypertrophy and fibrosis. In the presence of MI, the loss of cardiomyocytes exceeding the cardiac regenerative capacity can lead to fibrotic tissue formation and consequent ventricular remodeling [276]. Excessive MR stimulation plays an integral role in both situations, leading to left ventricular systolic and diastolic dysfunction, heart failure, and increased overall cardiovascular mortality (Figure 3) [277]. In murine models, aldosterone infusion increases perivascular and interstitial fibrosis through oxidative stress and inflammatory pathways and alteration of extracellular matrix deposition through metalloproteinases inhibition and increased collagen deposition [278,279,280,281,282,283]. Direct effects of aldosterone on cardiac hypertrophy are mediated via increased blood pressure and hemodynamic overload [284]. Aldosterone infusion increases myocardial hypertrophy through cardiotropin-1, plasminogen activator inhibitor 1 (PAI-1), and circadian clock proteins [285,286,287]. Most of these mechanisms can lead to cardiac fibrosis and contribute further to ventricular hypertrophy and heart failure [285,287,288]. Patients with PA exhibit enhanced signs of cardiac fibrosis and increased rates of left ventricular mass and hypertrophy [271,289,290]. There is a correlation between the severity of left ventricular hypertrophy and the level of autonomous aldosterone secretion [291]. Systolic and diastolic function are also compromised in PA patients who display increased risk of heart failure than patients with essential hypertension [271,292,293]. MR antagonist treatment and adrenalectomy partially reverse left ventricular hypertrophy and restore diastolic function [272]. MR antagonists have been evaluated in patients with heart failure with preserved ejection fraction (HfpEF) and HfrEF [294]. In patients with HfrEF, the EPHESUS trial (Eplerenone Post-Acute MI Heart Failure Efficacy and Survival Study) and the RALES trial (Randomized Aldactone Evaluation Study) have demonstrated that the addition of up to 50 mg of eplerenone and 25 mg of spironolactone to the standard therapy significantly reduces cardiac and overall mortality [295,296]. The ARTS-HF phase 2b trial (Mineralocorticoid Receptor antagonist Tolerability Study-HF) compared the efficacy of finerenone vs. eplerenone in patients with HfrEF and diabetes or chronic kidney disease, showing similar reduction effects on natriuretic peptide with a greater safety profile [297]. Spironolactone improved diastolic function in patients with HfpEF and diastolic dysfunction [298,299]. In the TOPCAT trial (Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist), spironolactone did not improve the primary composite outcome of cardiovascular mortality [300]. However, a reduced primary outcome composite after a post hoc analysis was present only in patients with resistant hypertension [300]. A recent individual-patient-data meta-analysis of three large RCTs showed that spironolactone improved cardiac structure and function of patients with HfpEF [301].
Atrial fibrillation: Like all tachyarrhythmias, the onset and maintenance of AF is dependent on three key mechanisms: automaticity, triggered activity, and re-entry. Automaticity refers to when an excitable tissue spontaneously depolarizes, triggered activity refers to when additional impulses triggered by afterdepolarizations relate to calcium overload, and re-entry refers to when waves of depolarization circle around an obstacle reinitiating continuously [256]. Experimental studies show that aldosterone has the ability to alter the electrophysiological properties of cardiomyocytes, enhancing cytosolic calcium load and promoting cardiac arrythmias (Figure 3) [302,303]. Chronic aldosterone infusion in rats enhances P-wave duration, activation time of the right atrium, and atrial anisotropy of conduction, facilitating re-entry mechanisms and AF stabilization [304]. Furthermore, remodeling of atrial tissue through increasing fibroblast proliferation and collagen deposition in the atrium contributed further to AF maintenance, increasing the time of spontaneous conversion [304,305]. MR antagonism in a tachy-paced sheep model partially reduced atrial fibrosis and dilatation with subsequent reduction in AF progression [306]. In patients with PA, the risk of AF is 3.52-fold higher than patients with essential hypertension [271]. It is estimated that 42% of AF patients have PA which warrants the screening of these patients for PA [307,308]. Although adrenalectomy reduces AF incident risks in PA patients, the use of MR antagonists failed to reduce the risk of AF [309]. The EMPHASIS-HF trial has shown that eplerenone significantly reduced the new-onset AF in patients with HfrEF [310]. The RACE-3 trial (Routine Versus Aggressive Upstream Rhythm Control for Prevention of Early Atrial Fibrillation in Heart Failure) showed that in the intervention group (MR antagonists, statins, angiotensin converting enzyme inhibitors and/or receptor blockers, and cardiac rehabilitation) significantly improved sinus rhythm maintenance in HF patients with AF when compared to conventional therapy. Interestingly, 85% of patients undergoing interventional therapy used MR antagonists versus only 4% in the conventional therapy group [311]. As a result, the European Society of Cardiology Guidelines of 2020 have introduced MR antagonists as a potential non-antiarrhythmic medication for AF therapy [312].
7. Is MR a Mediator of OSA-Induced CVD?
As mentioned earlier, MR activation-induced CVD can be induced through multiple mechanisms similar to those influenced by OSA, while OSA can directly activate RAAS and elevate renin, Ang II, and aldosterone levels [5,7,26,47,313]. Thus, it is plausible that OSA can mediate not only resistant hypertension and PA, but also atherosclerosis and CVD through MR activation, at least partially (Figure 4). The Endocrine Society has noted OSA in the presence of hypertension as one of the groups with high prevalence of PA, and now recommends screening of PA among this group of patients [314]. Experimental IH and SF have been shown to increase plasma levels of aldosterone and renin and stimulated release of ACTH from the pituitary and enhance cortisol secretion [315,316,317]. Both ACTH and RAAS act synergistically to regulate aldosterone pulse wave [318,319]. While elevated cortisol concentrations control the pulse amplitude during the daytime, RAAS plays a substantial role at night when plasma cortisol concentration is low [318]. Additionally, it is well established that obesity is a major risk factor for OSA and that 70% of OSA patients are obese [320]. The dysfunctional adipose tissue in obesity is considered an important source of RAAS hormone secretion that is independent from the classical RAAS activation [62,321]. It has been demonstrated that adipocytes can release adipokines responsible for stimulation for aldosterone secretion from adrenocortical cells [316,322]. Overall, evidence points to OSA-mediated RAAS activation. However, it is also suggested that excess aldosterone can worsen the clinical course of OSA potentially through increased salt and water retention, leading to rostral fluid shifts and para-pharyngeal edema and exacerbated upper airway obstruction and collapsibility, further worsening the severity of OSA [323,324,325]. Aldosterone can also act centrally to increase brain RAAS activity and oxidative stress, which may cause abnormal regulation of central breathing mechanisms, worsening OSA prognosis [326]. Thus, early OSA diagnosis and management and MR inhibition are crucial for preventing or ameliorating OSA-induced CVD.
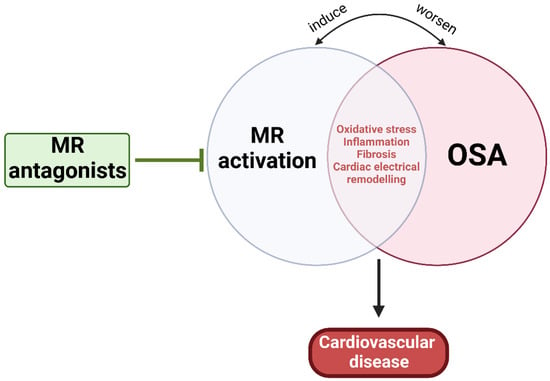
Figure 4.
Pathophysiological relationship between OSA and MR activation. MR activation and OSA share multiple mechanisms that can lead to CVD, suggesting the potential benefit of MR antagonism.
Several studies have evaluated the use of MR receptor antagonists on resistant hypertension in OSA. Two RCTs reported positive modulation in OSA severity and lowering blood pressure using spironolactone, but due to the small effect size and risk of bias, no definitive conclusions can be drawn from the available data [60,61]. Other studies used eplerenone in OSA and found that the treatment reduced blood pressure, arterial stiffness, and left ventricular hypertrophy in OSA patients [327,328]. These promising results showcase the significance of MR antagonists, not only as an antihypertensive medication, but also as a treatment for OSA-induced CVD. Conversely, several studies with small sample size and short treatment duration with CPAP did not reveal the anticipated reduction in the RAAS components [319,329,330,331,332,333], further justifying the need to include MR antagonists as an add-on adjuvant therapy in OSA patients. However, there is a lack of experimental data and of large RCTs that have assessed the benefits and safety of MR antagonists as part of long-term treatment of OSA-induced CVD. As described earlier, MR activation shares multiple pathophysiological mechanisms with OSA, all of which can be targeted by MR antagonism [7,313]. Extensive experimental IH and SF studies are needed, evaluating multiple in vivo and in vitro cardiovascular parameters while using different MR antagonists as well as specific transgenic animals with cell-specific targeted disruption of MR, to better understand the mutually dependent effects and interactions between RAAS and OSA and further explore the viability of MR-targeted treatment on OSA cardiovascular outcomes.
8. Conclusions
OSA is a chronic and highly prevalent condition that is associated with vascular dysfunction, cardiac remodeling, and overall cardiovascular risk and mortality. MR is essential for fluid homeostasis and balance. However, excessive MR activation in cardiovascular tissues may promote atherosclerosis and cardiac dysfunction through similar mechanisms induced by OSA. OSA can trigger RAAS activation and subsequent excessive MR activation. Thus, enhanced MR activation in OSA patients could contribute to OSA-induced CVD. However, MR influences on CVD in OSA has been insufficiently explored. To this effect, experimental studies assessing the impact of IH and SF in MR transgenic animals with vascular-specific deletion of MR are essential to elucidate the role of MR in OSA-induced CVD. Furthermore, the use of MR antagonists in OSA in the context of clinical trials in focused experimental settings may also provide insights into the effectiveness of MR antagonism in mitigating OSA-mediated CVD. Therefore, MR could prove to be a valuable putative target for improving cardiovascular outcomes in OSA patients.
Author Contributions
M.B., S.B.B. and D.G. wrote and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
M.B. is the recipient of the American Thoracic Society Unrestricted Research Grant. SBB is supported by NIH grant HL136386. D.G. is supported by NIH grant AG061824, and by Tier 2 and TRIUMPH grants from the University of Missouri.
Acknowledgments
Figures were created with BioRender.com (accessed on 8 December 2022). This work was supported by the use of resources and facilities at the Harry S. Truman Memorial Veterans Hospital in Columbia, MO.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| α-SMC | Alpha-smooth muscle actin |
| AF | Atrial fibrillation |
| AHI | Apnea/hypopnea index |
| Akt | Protein kinase B |
| AP-1 | Activator protein 1 |
| Ang II | Angiotensin II |
| AT1R | Type I angiotensin receptors |
| BH4 | Tetrahydrobiopterin |
| BNP | B-type natriuretic peptide |
| CAD | Coronary artery disease |
| Cav1 | Caveolin 1 |
| Cav 1.2 | L-type calcium channel alpha 1C |
| COX-2 | Cyclooxygenase 2 |
| CPAP | Continuous positive airway pressure |
| CREB | cAMP-response element-binding protein |
| CRP | C-reactive protein |
| CTGF | Connective tissue growth factor |
| EGFR | Epidermal growth factor receptor |
| ENaC | Epithelial sodium channel |
| eNOS | Endothelial nitric oxide synthase |
| ERK | Extracellular signal-regulated kinase |
| FMD | Flow-mediated dilatation |
| GLUT4 | Glucose transporter 4 |
| GPER | G protein-coupled estrogen receptor 1 |
| GR | Glucocorticoid receptor |
| HF | Heart failure |
| HfpEF | Heart failure with preserved ejection fraction |
| HfrEF | Heart failure with reduced ejection fraction |
| HOMA | Homeostatic model assessment |
| HSD11B2 | 11-beta-hydroxysteroid dehydrogenase type 2 |
| ICAM1 | Intracellular adhesion molecule 1 |
| IGF1R | Insulin-like growth factor receptor 1 |
| IH | Intermittent hypoxia |
| IL1β | Interleukin 1 beta |
| IL-6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| IL-18 | Interleukin 18 |
| IR | Insulin resistance |
| IRS1 | Insulin receptor substrate 1 |
| JNK | c-Jun N-terminal kinase |
| MAPK | Mitogen-associated protein kinases |
| MCP-1 | Monocyte chemoattractant protein 1 |
| MHC-β | Myosin heavy chain beta |
| MI | Myocardial infarction |
| MLC | Myosin light chain |
| mTOR | Mammalian target of rapamycin |
| NGAL | Neutrophil gelatinase-associated lipocalin |
| NHE-1 | Sodium–hydrogen antiporter 1 |
| NFAT | Nuclear factor of activated T-cells |
| NF-ƙB | Nuclear factor kappa B |
| NLRP3 | NLR family pyrin domain containing 3 |
| NO | Nitric oxide |
| NOX | NADPH oxidases |
| OSA | Obstructive sleep apnea |
| PA | Primary aldosteronism |
| PAI-1 | Plasminogen activator inhibitor 1 |
| PDGFR | Platelet-derived growth factor receptor |
| PI3K | Phosphatidylinositide 3-kinases |
| PKC | Protein kinase C |
| PP2A | Protein phosphatase 2A |
| PWV | Pulse wave velocity |
| PVN | Paraventricular nucleus |
| RAAS | Renin–angiotensin–aldosterone system |
| Rac1 | Ras-related C3 botulinum toxin substrate |
| RCT | Randomized control trial |
| ROS | Reactive oxygen species |
| S6K1 | Ribosomal protein S6 kinase beta 1 |
| SF | Sleep fragmentation |
| SGK1 | Serine/threonine-protein kinase 1 |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| VEGF | Vascular endothelial growth factor |
| VEGFR1 | Vascular endothelial growth factor receptor 1 |
| VSMCs | Vascular smooth muscle cells |
References
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip, M.S.M.; Morrell, M.J.; Nunez, C.M.; Patel, S.R.; Penzel, T.; Pepin, J.L.; et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: A literature-based analysis. Lancet Respir. Med. 2019, 7, 687–698. [Google Scholar] [CrossRef]
- Kapur, V.K.; Auckley, D.H.; Chowdhuri, S.; Kuhlmann, D.C.; Mehra, R.; Ramar, K.; Harrod, C.G. Clinical Practice Guideline for Diagnostic Testing for Adult Obstructive Sleep Apnea: An American Academy of Sleep Medicine Clinical Practice Guideline. J. Clin. Sleep Med. 2017, 13, 479–504. [Google Scholar] [CrossRef]
- Javaheri, S.; Barbe, F.; Campos-Rodriguez, F.; Dempsey, J.A.; Khayat, R.; Javaheri, S.; Malhotra, A.; Martinez-Garcia, M.A.; Mehra, R.; Pack, A.I.; et al. Sleep Apnea: Types, Mechanisms, and Clinical Cardiovascular Consequences. J. Am. Coll Cardiol. 2017, 69, 841–858. [Google Scholar] [CrossRef]
- Bradley, T.D.; Floras, J.S. Obstructive sleep apnoea and its cardiovascular consequences. Lancet 2009, 373, 82–93. [Google Scholar] [CrossRef]
- Golbidi, S.; Badran, M.; Ayas, N.; Laher, I. Cardiovascular consequences of sleep apnea. Lung 2012, 190, 113–132. [Google Scholar] [CrossRef]
- Badran, M.; Yassin, B.A.; Fox, N.; Laher, I.; Ayas, N. Epidemiology of Sleep Disturbances and Cardiovascular Consequences. Can. J. Cardiol. 2015, 31, 873–879. [Google Scholar] [CrossRef]
- Badran, M.; Ayas, N.; Laher, I. Insights into obstructive sleep apnea research. Sleep Med. 2014, 15, 485–495. [Google Scholar] [CrossRef]
- Carreras, A.; Zhang, S.X.; Peris, E.; Qiao, Z.; Gileles-Hillel, A.; Li, R.C.; Wang, Y.; Gozal, D. Chronic sleep fragmentation induces endothelial dysfunction and structural vascular changes in mice. Sleep 2014, 37, 1817–1824. [Google Scholar] [CrossRef]
- Farre, R.; Almendros, I.; Martinez-Garcia, M.A.; Gozal, D. Experimental Models to Study End-Organ Morbidity in Sleep Apnea: Lessons Learned and Future Directions. Int. J. Mol. Sci. 2022, 23, 14430. [Google Scholar] [CrossRef]
- Khalyfa, A.; Wang, Y.; Zhang, S.X.; Qiao, Z.; Abdelkarim, A.; Gozal, D. Sleep fragmentation in mice induces nicotinamide adenine dinucleotide phosphate oxidase 2-dependent mobilization, proliferation, and differentiation of adipocyte progenitors in visceral white adipose tissue. Sleep 2014, 37, 999–1009. [Google Scholar] [CrossRef]
- Li, J.; Thorne, L.N.; Punjabi, N.M.; Sun, C.K.; Schwartz, A.R.; Smith, P.L.; Marino, R.L.; Rodriguez, A.; Hubbard, W.C.; O’Donnell, C.P.; et al. Intermittent hypoxia induces hyperlipidemia in lean mice. Circ. Res. 2005, 97, 698–706. [Google Scholar] [CrossRef]
- Ip, M.S.; Lam, B.; Ng, M.M.; Lam, W.K.; Tsang, K.W.; Lam, K.S. Obstructive sleep apnea is independently associated with insulin resistance. Am. J. Respir. Crit. Care Med. 2002, 165, 670–676. [Google Scholar] [CrossRef]
- Kheirandish-Gozal, L.; Gozal, D. Obstructive Sleep Apnea and Inflammation: Proof of Concept Based on Two Illustrative Cytokines. Int. J. Mol. Sci. 2019, 20, 459. [Google Scholar] [CrossRef]
- Gozal, D.; Kheirandish-Gozal, L. Cardiovascular morbidity in obstructive sleep apnea: Oxidative stress, inflammation, and much more. Am. J. Respir Crit. Care Med. 2008, 177, 369–375. [Google Scholar] [CrossRef]
- Gileles-Hillel, A.; Alonso-Alvarez, M.L.; Kheirandish-Gozal, L.; Peris, E.; Cordero-Guevara, J.A.; Teran-Santos, J.; Martinez, M.G.; Jurado-Luque, M.J.; Corral-Penafiel, J.; Duran-Cantolla, J.; et al. Inflammatory markers and obstructive sleep apnea in obese children: The NANOS study. Mediators Inflamm. 2014, 2014, 605280. [Google Scholar] [CrossRef]
- Dyugovskaya, L.; Lavie, P.; Lavie, L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am. J. Respir Crit. Care Med. 2002, 165, 934–939. [Google Scholar] [CrossRef]
- Maniaci, A.; Iannella, G.; Cocuzza, S.; Vicini, C.; Magliulo, G.; Ferlito, S.; Cammaroto, G.; Meccariello, G.; De Vito, A.; Nicolai, A.; et al. Oxidative Stress and Inflammation Biomarker Expression in Obstructive Sleep Apnea Patients. J. Clin. Med. 2021, 10, 277. [Google Scholar] [CrossRef]
- Jun, J.; Reinke, C.; Bedja, D.; Berkowitz, D.; Bevans-Fonti, S.; Li, J.; Barouch, L.A.; Gabrielson, K.; Polotsky, V.Y. Effect of intermittent hypoxia on atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 2010, 209, 381–386. [Google Scholar] [CrossRef]
- Hakim, F.; Gozal, D.; Kheirandish-Gozal, L. Sympathetic and catecholaminergic alterations in sleep apnea with particular emphasis on children. Front. Neurol. 2012, 3, 7. [Google Scholar] [CrossRef]
- Badran, M.; Yassin, B.A.; Lin, D.T.S.; Kobor, M.S.; Ayas, N.; Laher, I. Gestational intermittent hypoxia induces endothelial dysfunction, reduces perivascular adiponectin and causes epigenetic changes in adult male offspring. J. Physiol. 2019, 597, 5349–5364. [Google Scholar] [CrossRef]
- Badran, M.; Khalyfa, A.; Ericsson, A.; Puech, C.; McAdams, Z.; Bender, S.B.; Gozal, D. Gut microbiota mediate vascular dysfunction in a murine model of sleep apnea: Effect of probiotics. Eur. Respir. J. 2022, 61, 2200002. [Google Scholar] [CrossRef]
- Badran, M.; Gozal, D. PAI-1: A Major Player in the Vascular Dysfunction in Obstructive Sleep Apnea? Int. J. Mol. Sci. 2022, 23, 5516. [Google Scholar] [CrossRef]
- Badran, M.; Golbidi, S.; Devlin, A.; Ayas, N.; Laher, I. Chronic intermittent hypoxia causes endothelial dysfunction in a mouse model of diet-induced obesity. Sleep Med. 2014, 15, 596–602. [Google Scholar] [CrossRef]
- Badran, M.; Golbidi, S.; Ayas, N.; Laher, I. Nitric Oxide Bioavailability in Obstructive Sleep Apnea: Interplay of Asymmetric Dimethylarginine and Free Radicals. Sleep Disord. 2015, 2015, 387801. [Google Scholar] [CrossRef]
- Badran, M.; Bender, S.B.; Khalyfa, A.; Padilla, J.; Martinez-Lemus, L.A.; Gozal, D. Temporal changes in coronary artery function and flow velocity reserve in mice exposed to chronic intermittent hypoxia. Sleep 2022, 45, zsac131. [Google Scholar] [CrossRef]
- Badran, M.; Ayas, N.; Laher, I. Cardiovascular complications of sleep apnea: Role of oxidative stress. Oxid. Med. Cell Longev. 2014, 2014, 985258. [Google Scholar] [CrossRef]
- Badran, M.; Abuyassin, B.; Golbidi, S.; Ayas, N.; Laher, I. Alpha Lipoic Acid Improves Endothelial Function and Oxidative Stress in Mice Exposed to Chronic Intermittent Hypoxia. Oxid. Med. Cell Longev. 2019, 2019, 4093018. [Google Scholar] [CrossRef]
- Badran, M.; Abuyassin, B.; Golbidi, S.; Ayas, N.; Laher, I. Uncoupling of Vascular Nitric Oxide Synthase Caused by Intermittent Hypoxia. Oxid. Med. Cell Longev. 2016, 2016, 2354870. [Google Scholar] [CrossRef]
- Badran, M.; Abuyassin, B.; Ayas, N.; Laher, I. Intermittent hypoxia impairs uterine artery function in pregnant mice. J. Physiol. 2019, 597, 2639–2650. [Google Scholar] [CrossRef]
- Porto, F.; Sakamoto, Y.S.; Salles, C. Association between Obstructive Sleep Apnea and Myocardial Infarction: A Systematic Review. Arq. Bras. Cardiol. 2017, 108, 361–369. [Google Scholar] [CrossRef]
- Saeed, S.; Romarheim, A.; Solheim, E.; Bjorvatn, B.; Lehmann, S. Cardiovascular remodeling in obstructive sleep apnea: Focus on arterial stiffness, left ventricular geometry and atrial fibrillation. Expert Rev. Cardiovasc. Ther. 2022, 20, 455–464. [Google Scholar] [CrossRef]
- Nakashima, H.; Kurobe, M.; Minami, K.; Furudono, S.; Uchida, Y.; Amenomori, K.; Nunohiro, T.; Takeshita, S.; Maemura, K. Effects of moderate-to-severe obstructive sleep apnea on the clinical manifestations of plaque vulnerability and the progression of coronary atherosclerosis in patients with acute coronary syndrome. Eur. Heart J. Acute Cardiovasc. Care 2015, 4, 75–84. [Google Scholar] [CrossRef]
- Hao, W.; Wang, X.; Fan, J.; Zeng, Y.; Ai, H.; Nie, S.; Wei, Y. Association between apnea-hypopnea index and coronary artery calcification: A systematic review and meta-analysis. Ann. Med. 2021, 53, 302–317. [Google Scholar] [CrossRef]
- Hoffstein, V.; Mateika, S. Cardiac arrhythmias, snoring, and sleep apnea. Chest 1994, 106, 466–471. [Google Scholar] [CrossRef]
- Gami, A.S.; Pressman, G.; Caples, S.M.; Kanagala, R.; Gard, J.J.; Davison, D.E.; Malouf, J.F.; Ammash, N.M.; Friedman, P.A.; Somers, V.K. Association of atrial fibrillation and obstructive sleep apnea. Circulation 2004, 110, 364–367. [Google Scholar] [CrossRef]
- Oldenburg, O.; Lamp, B.; Faber, L.; Teschler, H.; Horstkotte, D.; Topfer, V. Sleep-disordered breathing in patients with symptomatic heart failure: A contemporary study of prevalence in and characteristics of 700 patients. Eur. J. Heart Fail 2007, 9, 251–257. [Google Scholar] [CrossRef]
- Sanchez-de-la-Torre, M.; Sanchez-de-la-Torre, A.; Bertran, S.; Abad, J.; Duran-Cantolla, J.; Cabriada, V.; Mediano, O.; Masdeu, M.J.; Alonso, M.L.; Masa, J.F.; et al. Effect of obstructive sleep apnoea and its treatment with continuous positive airway pressure on the prevalence of cardiovascular events in patients with acute coronary syndrome (ISAACC study): A randomised controlled trial. Lancet Respir. Med. 2020, 8, 359–367. [Google Scholar] [CrossRef]
- Peker, Y.; Glantz, H.; Eulenburg, C.; Wegscheider, K.; Herlitz, J.; Thunstrom, E. Effect of Positive Airway Pressure on Cardiovascular Outcomes in Coronary Artery Disease Patients with Nonsleepy Obstructive Sleep Apnea. The RICCADSA Randomized Controlled Trial. Am. J. Respir. Crit. Care Med. 2016, 194, 613–620. [Google Scholar] [CrossRef]
- McEvoy, R.D.; Antic, N.A.; Heeley, E.; Luo, Y.; Ou, Q.; Zhang, X.; Mediano, O.; Chen, R.; Drager, L.F.; Liu, Z.; et al. CPAP for Prevention of Cardiovascular Events in Obstructive Sleep Apnea. N. Engl. J. Med. 2016, 375, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Nehme, A.; Zibara, K. Efficiency and specificity of RAAS inhibitors in cardiovascular diseases: How to achieve better end-organ protection? Hypertens. Res. 2017, 40, 903–909. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Bharadwaj, D.; Prasad, G.; Grechko, A.V.; Sazonova, M.A.; Orekhov, A.N. Renin-Angiotensin System in Pathogenesis of Atherosclerosis and Treatment of CVD. Int. J. Mol. Sci. 2021, 22, 6702. [Google Scholar] [CrossRef] [PubMed]
- Pacurari, M.; Kafoury, R.; Tchounwou, P.B.; Ndebele, K. The Renin-Angiotensin-aldosterone system in vascular inflammation and remodeling. Int. J. Inflam. 2014, 2014, 689360. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Strawn, W.B. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am. J. Cardiol. 2006, 98, 121–128. [Google Scholar] [CrossRef] [PubMed]
- DuPont, J.J.; Jaffe, I.Z. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: The role of the mineralocorticoid receptor in the vasculature. J. Endocrinol. 2017, 234, T67–T82. [Google Scholar] [CrossRef] [PubMed]
- McCurley, A.; Jaffe, I.Z. Mineralocorticoid receptors in vascular function and disease. Mol. Cell Endocrinol. 2012, 350, 256–265. [Google Scholar] [CrossRef]
- Gorini, S.; Kim, S.K.; Infante, M.; Mammi, C.; La Vignera, S.; Fabbri, A.; Jaffe, I.Z.; Caprio, M. Role of Aldosterone and Mineralocorticoid Receptor in Cardiovascular Aging. Front. Endocrinol. 2019, 10, 584. [Google Scholar] [CrossRef]
- Ong, G.S.; Young, M.J. Mineralocorticoid regulation of cell function: The role of rapid signalling and gene transcription pathways. J. Mol. Endocrinol. 2017, 58, R33–R57. [Google Scholar] [CrossRef]
- Pippal, J.B.; Fuller, P.J. Structure-function relationships in the mineralocorticoid receptor. J. Mol. Endocrinol. 2008, 41, 405–413. [Google Scholar] [CrossRef]
- Alexandre, J.; Dolladille, C.; Douesnel, L.; Font, J.; Dabrowski, R.; Shavit, L.; Legallois, D.; Funck-Brentano, C.; Champ-Rigot, L.; Ollitrault, P.; et al. Effects of Mineralocorticoid Receptor Antagonists on Atrial Fibrillation Occurrence: A Systematic Review, Meta-Analysis, and Meta-Regression to Identify Modifying Factors. J. Am. Heart Assoc. 2019, 8, e013267. [Google Scholar] [CrossRef] [PubMed]
- Lofman, I.; Szummer, K.; Olsson, H.; Carrero, J.J.; Lund, L.H.; Jernberg, T. Association Between Mineralocorticoid Receptor Antagonist Use and Outcome in Myocardial Infarction Patients With Heart Failure. J. Am. Heart Assoc. 2018, 7, e009359. [Google Scholar] [CrossRef]
- Nagata, K.; Obata, K.; Xu, J.; Ichihara, S.; Noda, A.; Kimata, H.; Kato, T.; Izawa, H.; Murohara, T.; Yokota, M. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and failure in low-aldosterone hypertensive rats. Hypertension 2006, 47, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Ikejima, H.; Tsujioka, H.; Kuroi, A.; Kobayashi, K.; Muragaki, Y.; Mochizuki, S.; Goto, M.; Yoshida, K.; Akasaka, T. Addition of eplerenone to an angiotensin-converting enzyme inhibitor effectively improves nitric oxide bioavailability. Hypertension 2008, 51, 734–741. [Google Scholar] [CrossRef]
- Sartorio, C.L.; Fraccarollo, D.; Galuppo, P.; Leutke, M.; Ertl, G.; Stefanon, I.; Bauersachs, J. Mineralocorticoid receptor blockade improves vasomotor dysfunction and vascular oxidative stress early after myocardial infarction. Hypertension 2007, 50, 919–925. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Duquaine, D.; King, S.; Pitt, B.; Patel, P. Mineralocorticoid receptor antagonism in experimental atherosclerosis. Circulation 2002, 105, 2212–2216. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Silverio, D.; Sourlas, A.; Montan, P.D.; Guzman, E. Role of spironolactone in the treatment of heart failure with preserved ejection fraction. Ann. Transl. Med. 2018, 6, 461. [Google Scholar] [CrossRef]
- Young, M.J. Targeting the mineralocorticoid receptor in cardiovascular disease. Expert Opin. Ther. Targets 2013, 17, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.A. Aldosterone inhibition and cardiovascular protection: More important than it once appeared. Cardiovasc. Drugs Ther 2010, 24, 345–350. [Google Scholar] [CrossRef]
- Moore, T.D.; Nawarskas, J.J.; Anderson, J.R. Eplerenone: A selective aldosterone receptor antagonist for hypertension and heart failure. Heart Dis. 2003, 5, 354–363. [Google Scholar] [CrossRef]
- Loh, H.H.; Sukor, N. Primary aldosteronism and obstructive sleep apnea: What do we know thus far? Front. Endocrinol. 2022, 13, 976979. [Google Scholar] [CrossRef]
- Fiori, C.Z.; Martinez, D.; Goncalves, S.C.; Montanari, C.C.; Fuchs, F.D. Effect of diuretics and sodium-restricted diet on sleep apnea severity: Study protocol for a randomized controlled trial. Trials 2015, 16, 188. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, H.; Cai, M.; Zou, Y.; Jiang, X.; Song, L.; Liang, E.; Bian, J.; Wu, H.; Hui, R. Effect of spironolactone on patients with resistant hypertension and obstructive sleep apnea. Clin. Exp. Hypertens. 2016, 38, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Goodfriend, T.L.; Calhoun, D.A. Resistant hypertension, obesity, sleep apnea, and aldosterone: Theory and therapy. Hypertension 2004, 43, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Yeghiazarians, Y.; Jneid, H.; Tietjens, J.R.; Redline, S.; Brown, D.L.; El-Sherif, N.; Mehra, R.; Bozkurt, B.; Ndumele, C.E.; Somers, V.K. Obstructive Sleep Apnea and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 144, e56–e67. [Google Scholar] [CrossRef]
- Kim, R.D.; Kapur, V.K.; Redline-Bruch, J.; Rueschman, M.; Auckley, D.H.; Benca, R.M.; Foldvary-Schafer, N.R.; Iber, C.; Zee, P.C.; Rosen, C.L.; et al. An Economic Evaluation of Home Versus Laboratory-Based Diagnosis of Obstructive Sleep Apnea. Sleep 2015, 38, 1027–1037. [Google Scholar] [CrossRef]
- Thornton, C.S.; Tsai, W.H.; Santana, M.J.; Penz, E.D.; Flemons, W.W.; Fraser, K.L.; Hanly, P.J.; Pendharkar, S.R. Effects of Wait Times on Treatment Adherence and Clinical Outcomes in Patients With Severe Sleep-Disordered Breathing: A Secondary Analysis of a Noninferiority Randomized Clinical Trial. JAMA Netw. Open. 2020, 3, e203088. [Google Scholar] [CrossRef]
- Khawaja, I.S.; Olson, E.J.; van der Walt, C.; Bukartyk, J.; Somers, V.; Dierkhising, R.; Morgenthaler, T.I. Diagnostic accuracy of split-night polysomnograms. J. Clin. Sleep Med. 2010, 6, 357–362. [Google Scholar] [CrossRef]
- Chiu, H.Y.; Chen, P.Y.; Chuang, L.P.; Chen, N.H.; Tu, Y.K.; Hsieh, Y.J.; Wang, Y.C.; Guilleminault, C. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: A bivariate meta-analysis. Sleep Med. Rev. 2017, 36, 57–70. [Google Scholar] [CrossRef]
- Rosen, I.M.; Kirsch, D.B.; Chervin, R.D.; Carden, K.A.; Ramar, K.; Aurora, R.N.; Kristo, D.A.; Malhotra, R.K.; Martin, J.L.; Olson, E.J.; et al. Clinical Use of a Home Sleep Apnea Test: An American Academy of Sleep Medicine Position Statement. J. Clin. Sleep Med. 2017, 13, 1205–1207. [Google Scholar] [CrossRef]
- Kump, K.; Whalen, C.; Tishler, P.V.; Browner, I.; Ferrette, V.; Strohl, K.P.; Rosenberg, C.; Redline, S. Assessment of the validity and utility of a sleep-symptom questionnaire. Am. J. Respir. Crit. Care Med. 1994, 150, 735–741. [Google Scholar] [CrossRef]
- Silverberg, D.S.; Oksenberg, A.; Iaina, A. Sleep-related breathing disorders as a major cause of essential hypertension: Fact or fiction? Curr. Opin. Nephrol. Hypertens. 1998, 7, 353–357. [Google Scholar] [CrossRef]
- Fletcher, E.C.; DeBehnke, R.D.; Lovoi, M.S.; Gorin, A.B. Undiagnosed sleep apnea in patients with essential hypertension. Ann. Int. Med. 1985, 103, 190–195. [Google Scholar] [CrossRef]
- Lavie, P.; Ben-Yosef, R.; Rubin, A.E. Prevalence of sleep apnea syndrome among patients with essential hypertension. Am. Heart J. 1984, 108, 373–376. [Google Scholar] [CrossRef]
- Peppard, P.E.; Young, T.; Palta, M.; Skatrud, J. Prospective study of the association between sleep-disordered breathing and hypertension. N. Engl. J. Med. 2000, 342, 1378–1384. [Google Scholar] [CrossRef]
- Logan, A.G.; Perlikowski, S.M.; Mente, A.; Tisler, A.; Tkacova, R.; Niroumand, M.; Leung, R.S.; Bradley, T.D. High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J. Hypertens. 2001, 19, 2271–2277. [Google Scholar] [CrossRef]
- Grote, L.; Hedner, J.; Peter, J.H. Sleep-related breathing disorder is an independent risk factor for uncontrolled hypertension. J. Hypertens. 2000, 18, 679–685. [Google Scholar] [CrossRef]
- Portaluppi, F.; Provini, F.; Cortelli, P.; Plazzi, G.; Bertozzi, N.; Manfredini, R.; Fersini, C.; Lugaresi, E. Undiagnosed sleep-disordered breathing among male nondippers with essential hypertension. J. Hypertens. 1997, 15, 1227–1233. [Google Scholar] [CrossRef]
- Arzt, M.; Woehrle, H.; Oldenburg, O.; Graml, A.; Suling, A.; Erdmann, E.; Teschler, H.; Wegscheider, K.; Schla, H.F.I. Prevalence and Predictors of Sleep-Disordered Breathing in Patients With Stable Chronic Heart Failure: The SchlaHF Registry. JACC Heart Fail 2016, 4, 116–125. [Google Scholar] [CrossRef]
- Chan, J.; Sanderson, J.; Chan, W.; Lai, C.; Choy, D.; Ho, A.; Leung, R. Prevalence of sleep-disordered breathing in diastolic heart failure. Chest 1997, 111, 1488–1493. [Google Scholar] [CrossRef]
- Oldenburg, O.; Wellmann, B.; Buchholz, A.; Bitter, T.; Fox, H.; Thiem, U.; Horstkotte, D.; Wegscheider, K. Nocturnal hypoxaemia is associated with increased mortality in stable heart failure patients. Eur. Heart J. 2016, 37, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Liu, H.; Scherlag, B.J.; Sun, L.; Xing, S.; Xu, J.; Luo, M.; Guo, Y.; Cao, G.; Jiang, H. Atrial fibrillation in obstructive sleep apnea: Neural mechanisms and emerging therapies. Trends Cardiovasc. Med. 2021, 31, 127–132. [Google Scholar] [CrossRef]
- Otto, M.E.; Belohlavek, M.; Romero-Corral, A.; Gami, A.S.; Gilman, G.; Svatikova, A.; Amin, R.S.; Lopez-Jimenez, F.; Khandheria, B.K.; Somers, V.K. Comparison of cardiac structural and functional changes in obese otherwise healthy adults with versus without obstructive sleep apnea. Am. J. Cardiol. 2007, 99, 1298–1302. [Google Scholar] [CrossRef]
- Zaqqa, M.; Afshar, H.; Rasekh, A.; Khoshnevis, R.; Vaughn, W.K.; Massumi, A. Predictors of conversion to sinus rhythm using ibutilide for atrial fibrillation or flutter. Am. J. Cardiol. 2000, 85, 112–114, A119. [Google Scholar] [CrossRef] [PubMed]
- Gami, A.S.; Hodge, D.O.; Herges, R.M.; Olson, E.J.; Nykodym, J.; Kara, T.; Somers, V.K. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J. Am. Coll. Cardiol. 2007, 49, 565–571. [Google Scholar] [CrossRef]
- Defaye, P.; de la Cruz, I.; Marti-Almor, J.; Villuendas, R.; Bru, P.; Senechal, J.; Tamisier, R.; Pepin, J.L. A pacemaker transthoracic impedance sensor with an advanced algorithm to identify severe sleep apnea: The DREAM European study. Heart Rhythm. 2014, 11, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Simantirakis, E.N.; Schiza, S.I.; Marketou, M.E.; Chrysostomakis, S.I.; Chlouverakis, G.I.; Klapsinos, N.C.; Siafakas, N.S.; Vardas, P.E. Severe bradyarrhythmias in patients with sleep apnoea: The effect of continuous positive airway pressure treatment: A long-term evaluation using an insertable loop recorder. Eur. Heart J. 2004, 25, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.A.; Yaggi, H.K.; Concato, J.; Mohsenin, V. Obstructive sleep apnea as a risk factor for coronary events or cardiovascular death. Sleep Breath 2010, 14, 131–136. [Google Scholar] [CrossRef]
- Punjabi, N.M.; Caffo, B.S.; Goodwin, J.L.; Gottlieb, D.J.; Newman, A.B.; O’Connor, G.T.; Rapoport, D.M.; Redline, S.; Resnick, H.E.; Robbins, J.A.; et al. Sleep-disordered breathing and mortality: A prospective cohort study. PLoS Med. 2009, 6, e1000132. [Google Scholar] [CrossRef]
- Yeboah, J.; Redline, S.; Johnson, C.; Tracy, R.; Ouyang, P.; Blumenthal, R.S.; Burke, G.L.; Herrington, D.M. Association between sleep apnea, snoring, incident cardiovascular events and all-cause mortality in an adult population: MESA. Atherosclerosis 2011, 219, 963–968. [Google Scholar] [CrossRef]
- Gunta, S.P.; Jakulla, R.S.; Ubaid, A.; Mohamed, K.; Bhat, A.; Lopez-Candales, A.; Norgard, N. Obstructive Sleep Apnea and Cardiovascular Diseases: Sad Realities and Untold Truths regarding Care of Patients in 2022. Cardiovasc. Ther. 2022, 2022, 6006127. [Google Scholar] [CrossRef] [PubMed]
- Imani, M.M.; Sadeghi, M.; Khazaie, H.; Emami, M.; Sadeghi Bahmani, D.; Brand, S. Evaluation of Serum and Plasma Interleukin-6 Levels in Obstructive Sleep Apnea Syndrome: A Meta-Analysis and Meta-Regression. Front. Immunol. 2020, 11, 1343. [Google Scholar] [CrossRef]
- Schulz, R.; Mahmoudi, S.; Hattar, K.; Sibelius, U.; Olschewski, H.; Mayer, K.; Seeger, W.; Grimminger, F. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am. J. Respir. Crit. Care Med. 2000, 162, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Kizawa, T.; Nakamura, Y.; Takahashi, S.; Sakurai, S.; Yamauchi, K.; Inoue, H. Pathogenic role of angiotensin II and oxidised LDL in obstructive sleep apnoea. Eur. Respir. J. 2009, 34, 1390–1398. [Google Scholar] [CrossRef]
- Vatansever, E.; Surmen-Gur, E.; Ursavas, A.; Karadag, M. Obstructive sleep apnea causes oxidative damage to plasma lipids and proteins and decreases adiponectin levels. Sleep Breath 2011, 15, 275–282. [Google Scholar] [CrossRef]
- Yamauchi, M.; Nakano, H.; Maekawa, J.; Okamoto, Y.; Ohnishi, Y.; Suzuki, T.; Kimura, H. Oxidative stress in obstructive sleep apnea. Chest 2005, 127, 1674–1679. [Google Scholar] [CrossRef]
- Rosa, D.P.; Martinez, D.; Picada, J.N.; Semedo, J.G.; Marroni, N.P. Hepatic oxidative stress in an animal model of sleep apnoea: Effects of different duration of exposure. Comp. Hepatol. 2011, 10, 1. [Google Scholar] [CrossRef]
- Castro-Grattoni, A.L.; Suarez-Giron, M.; Benitez, I.; Torres, M.; Almendros, I.; Farre, R.; Montserrat, J.M.; Dalmases, M.; Gozal, D.; Sanchez-de-la-Torre, M.; et al. Effect of age on the cardiovascular remodelling induced by chronic intermittent hypoxia as a murine model of sleep apnoea. Respirology 2020, 25, 312–320. [Google Scholar] [CrossRef]
- Farre, N.; Otero, J.; Falcones, B.; Torres, M.; Jorba, I.; Gozal, D.; Almendros, I.; Farre, R.; Navajas, D. Intermittent Hypoxia Mimicking Sleep Apnea Increases Passive Stiffness of Myocardial Extracellular Matrix. A Multiscale Study. Front. Physiol. 2018, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Bourdier, G.; Detrait, M.; Bouyon, S.; Lemarie, E.; Brasseur, S.; Doutreleau, S.; Pepin, J.L.; Godin-Ribuot, D.; Belaidi, E.; Arnaud, C. Intermittent Hypoxia Triggers Early Cardiac Remodeling and Contractile Dysfunction in the Time-Course of Ischemic Cardiomyopathy in Rats. J. Am. Heart Assoc. 2020, 9, e016369. [Google Scholar] [CrossRef]
- Bao, Q.; Zhang, B.; Suo, Y.; Liu, C.; Yang, Q.; Zhang, K.; Yuan, M.; Yuan, M.; Zhang, Y.; Li, G. Intermittent hypoxia mediated by TSP1 dependent on STAT3 induces cardiac fibroblast activation and cardiac fibrosis. eLife 2020, 9, e49923. [Google Scholar] [CrossRef] [PubMed]
- Abuyassin, B.; Badran, M.; Ayas, N.T.; Laher, I. Intermittent hypoxia causes histological kidney damage and increases growth factor expression in a mouse model of obstructive sleep apnea. PLoS ONE 2018, 13, e0192084. [Google Scholar] [CrossRef]
- Ding, W.X.; Dong, Y.B.; Ding, N.; Zhang, X.F.; Zhang, S.J.; Zhang, X.L.; Liu, J.N.; Lu, G. Adiponectin protects rat heart from left ventricular remodeling induced by chronic intermittent hypoxia via inhibition of TGF-beta/smad2/3 pathway. J. Thorac. Dis. 2014, 6, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.P.; Lin, Y.N.; Li, N.; Sun, X.W.; Ding, Y.J.; Yan, Y.R.; Zhang, L.; Li, Q.Y. Angiotensin-(1-7) Rescues Chronic Intermittent Hypoxia-Aggravated Transforming Growth Factor-beta-Mediated Airway Remodeling in Murine and Cellular Models of Asthma. J. Pharmacol. Exp. Ther. 2020, 375, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Song, Y.; Ning, P.; Zhang, L.; Wu, S.; Quan, J.; Li, Q. Association between tumor necrosis factor alpha and obstructive sleep apnea in adults: A meta-analysis update. BMC Pulm. Med. 2020, 20, 215. [Google Scholar] [CrossRef] [PubMed]
- Htoo, A.K.; Greenberg, H.; Tongia, S.; Chen, G.; Henderson, T.; Wilson, D.; Liu, S.F. Activation of nuclear factor kappaB in obstructive sleep apnea: A pathway leading to systemic inflammation. Sleep Breath 2006, 10, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, D.; Jun, J.; Polotsky, V. Inflammation in sleep apnea: An update. Rev. Endocr. Metab. Disord. 2015, 16, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zheng, X. Tumor necrosis factor alpha is a promising circulating biomarker for the development of obstructive sleep apnea syndrome: A meta-analysis. Oncotarget 2017, 8, 27616–27626. [Google Scholar] [CrossRef]
- Barros, D.; Garcia-Rio, F. Obstructive sleep apnea and dyslipidemia: From animal models to clinical evidence. Sleep 2019, 42, zsy236. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Dong, Y.; Somers, V.K.; Peterson, T.E.; Zhang, Y.; Wang, S.; Li, G.; Singh, P. Intermittent hypoxia regulates vasoactive molecules and alters insulin-signaling in vascular endothelial cells. Sci. Rep. 2018, 8, 14110. [Google Scholar] [CrossRef]
- Thomas, A.; Belaidi, E.; Moulin, S.; Horman, S.; van der Zon, G.C.; Viollet, B.; Levy, P.; Bertrand, L.; Pepin, J.L.; Godin-Ribuot, D.; et al. Chronic Intermittent Hypoxia Impairs Insulin Sensitivity but Improves Whole-Body Glucose Tolerance by Activating Skeletal Muscle AMPK. Diabetes 2017, 66, 2942–2951. [Google Scholar] [CrossRef]
- Tkacova, R.; Dorkova, Z.; Molcanyiova, A.; Radikova, Z.; Klimes, I.; Tkac, I. Cardiovascular risk and insulin resistance in patients with obstructive sleep apnea. Med. Sci. Monit. 2008, 14, CR438–CR444. [Google Scholar]
- Bhushan, B.; Maddalozzo, J.; Sheldon, S.H.; Haymond, S.; Rychlik, K.; Lales, G.C.; Billings, K.R. Metabolic alterations in children with obstructive sleep apnea. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Parish, J.M.; Adam, T.; Facchiano, L. Relationship of metabolic syndrome and obstructive sleep apnea. J. Clin. Sleep Med. 2007, 3, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.P.; Ayappa, I.A.; Caples, S.M.; Kimoff, R.J.; Patel, S.R.; Harrod, C.G. Treatment of Adult Obstructive Sleep Apnea With Positive Airway Pressure: An American Academy of Sleep Medicine Systematic Review, Meta-Analysis, and GRADE Assessment. J. Clin. Sleep Med. 2019, 15, 301–334. [Google Scholar] [CrossRef] [PubMed]
- da Silva Paulitsch, F.; Zhang, L. Continuous positive airway pressure for adults with obstructive sleep apnea and cardiovascular disease: A meta-analysis of randomized trials. Sleep Med. 2019, 54, 28–34. [Google Scholar] [CrossRef]
- Mokhlesi, B.; Finn, L.A.; Hagen, E.W.; Young, T.; Hla, K.M.; Van Cauter, E.; Peppard, P.E. Obstructive sleep apnea during REM sleep and hypertension. results of the Wisconsin Sleep Cohort. Am. J. Respir. Crit. Care Med. 2014, 190, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Cortese, R.; Gileles-Hillel, A.; Khalyfa, A.; Almendros, I.; Akbarpour, M.; Khalyfa, A.A.; Qiao, Z.; Garcia, T.; Andrade, J.; Gozal, D. Aorta macrophage inflammatory and epigenetic changes in a murine model of obstructive sleep apnea: Potential role of CD36. Sci. Rep. 2017, 7, 43648. [Google Scholar] [CrossRef]
- Arriza, J.L.; Weinberger, C.; Cerelli, G.; Glaser, T.M.; Handelin, B.L.; Housman, D.E.; Evans, R.M. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 1987, 237, 268–275. [Google Scholar] [CrossRef]
- Huyet, J.; Pinon, G.M.; Fay, M.R.; Rafestin-Oblin, M.E.; Fagart, J. Structural determinants of ligand binding to the mineralocorticoid receptor. Mol. Cell Endocrinol. 2012, 350, 187–195. [Google Scholar] [CrossRef]
- Rogerson, F.M.; Brennan, F.E.; Fuller, P.J. Mineralocorticoid receptor binding, structure and function. Mol. Cell Endocrinol. 2004, 217, 203–212. [Google Scholar] [CrossRef]
- Galigniana, M.D.; Echeverria, P.C.; Erlejman, A.G.; Piwien-Pilipuk, G. Role of molecular chaperones and TPR-domain proteins in the cytoplasmic transport of steroid receptors and their passage through the nuclear pore. Nucleus 2010, 1, 299–308. [Google Scholar] [CrossRef]
- Grossmann, C.; Ruhs, S.; Langenbruch, L.; Mildenberger, S.; Stratz, N.; Schumann, K.; Gekle, M. Nuclear shuttling precedes dimerization in mineralocorticoid receptor signaling. Chem. Biol. 2012, 19, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Le Billan, F.; Khan, J.A.; Lamribet, K.; Viengchareun, S.; Bouligand, J.; Fagart, J.; Lombes, M. Cistrome of the aldosterone-activated mineralocorticoid receptor in human renal cells. FASEB J. 2015, 29, 3977–3989. [Google Scholar] [CrossRef]
- Cannavo, A.; Bencivenga, L.; Liccardo, D.; Elia, A.; Marzano, F.; Gambino, G.; D’Amico, M.L.; Perna, C.; Ferrara, N.; Rengo, G.; et al. Aldosterone and Mineralocorticoid Receptor System in Cardiovascular Physiology and Pathophysiology. Oxid. Med. Cell Longev. 2018, 2018, 1204598. [Google Scholar] [CrossRef] [PubMed]
- Loffing, J.; Korbmacher, C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflugers. Arch. 2009, 458, 111–135. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The multifaceted mineralocorticoid receptor. Compr. Physiol. 2014, 4, 965–994. [Google Scholar] [CrossRef] [PubMed]
- Funder, J.W.; Pearce, P.T.; Smith, R.; Smith, A.I. Mineralocorticoid action: Target tissue specificity is enzyme, not receptor, mediated. Science 1988, 242, 583–585. [Google Scholar] [CrossRef]
- Mullins, L.J.; Kenyon, C.J.; Bailey, M.A.; Conway, B.R.; Diaz, M.E.; Mullins, J.J. Mineralocorticoid Excess or Glucocorticoid Insufficiency: Renal and Metabolic Phenotypes in a Rat Hsd11b2 Knockout Model. Hypertension 2015, 66, 667–673. [Google Scholar] [CrossRef]
- Mihailidou, A.S.; Loan Le, T.Y.; Mardini, M.; Funder, J.W. Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension 2009, 54, 1306–1312. [Google Scholar] [CrossRef]
- Rossier, M.F.; Lenglet, S.; Vetterli, L.; Python, M.; Maturana, A. Corticosteroids and redox potential modulate spontaneous contractions in isolated rat ventricular cardiomyocytes. Hypertension 2008, 52, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Newfell, B.G.; Iyer, L.K.; Mohammad, N.N.; McGraw, A.P.; Ehsan, A.; Rosano, G.; Huang, P.L.; Mendelsohn, M.E.; Jaffe, I.Z. Aldosterone regulates vascular gene transcription via oxidative stress-dependent and -independent pathways. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1871–1880. [Google Scholar] [CrossRef]
- Turchin, A.; Guo, C.Z.; Adler, G.K.; Ricchiuti, V.; Kohane, I.S.; Williams, G.H. Effect of acute aldosterone administration on gene expression profile in the heart. Endocrinology 2006, 147, 3183–3189. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gekle, M.; Freudinger, R.; Mildenberger, S.; Schenk, K.; Marschitz, I.; Schramek, H. Rapid activation of Na+/H+-exchange in MDCK cells by aldosterone involves MAP-kinase ERK1/2. Pflugers. Arch. 2001, 441, 781–786. [Google Scholar] [CrossRef]
- Blazer-Yost, B.L.; Paunescu, T.G.; Helman, S.I.; Lee, K.D.; Vlahos, C.J. Phosphoinositide 3-kinase is required for aldosterone-regulated sodium reabsorption. Am. J. Physiol. 1999, 277, C531–C536. [Google Scholar] [CrossRef]
- McEneaney, V.; Harvey, B.J.; Thomas, W. Aldosterone regulates rapid trafficking of epithelial sodium channel subunits in renal cortical collecting duct cells via protein kinase D activation. Mol. Endocrinol. 2008, 22, 881–892. [Google Scholar] [CrossRef]
- Mihailidou, A.S.; Mardini, M.; Funder, J.W. Rapid, nongenomic effects of aldosterone in the heart mediated by epsilon protein kinase C. Endocrinology 2004, 145, 773–780. [Google Scholar] [CrossRef]
- Ruhs, S.; Nolze, A.; Hubschmann, R.; Grossmann, C. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: Nongenomic effects via the mineralocorticoid receptor. J. Endocrinol. 2017, 234, T107–T124. [Google Scholar] [CrossRef]
- Ashton, A.W.; Le, T.Y.; Gomez-Sanchez, C.E.; Morel-Kopp, M.C.; McWhinney, B.; Hudson, A.; Mihailidou, A.S. Role of Nongenomic Signaling Pathways Activated by Aldosterone During Cardiac Reperfusion Injury. Mol. Endocrinol. 2015, 29, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, P.; Vega, C.; Pojoga, L.H.; Rivera, A.; Prado, G.N.; Yao, T.M.; Adler, G.; Torres-Grajales, M.; Maldonado, E.R.; Ramos-Rivera, A.; et al. Aldosterone’s rapid, nongenomic effects are mediated by striatin: A modulator of aldosterone’s effect on estrogen action. Endocrinology 2014, 155, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Wuttke, M.; Ruhs, S.; Seiferth, A.; Mildenberger, S.; Rabe, S.; Schwerdt, G.; Gekle, M. Mineralocorticoid receptor inhibits CREB signaling by calcineurin activation. FASEB J. 2010, 24, 2010–2019. [Google Scholar] [CrossRef]
- Grossmann, C.; Ruhs, S.; Seiferth, A.; Gekle, M. Interaction between mineralocorticoid receptor and cAMP/CREB signaling. Steroids 2010, 75, 539–543. [Google Scholar] [CrossRef]
- Ozbaki-Yagan, N.; Liu, X.; Bodnar, A.J.; Ho, J.; Butterworth, M.B. Aldosterone-induced microRNAs act as feedback regulators of mineralocorticoid receptor signaling in kidney epithelia. FASEB J. 2020, 34, 11714–11728. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Kong, Q.; Kone, B.C. Aldosterone reprograms promoter methylation to regulate alphaENaC transcription in the collecting duct. Am. J. Physiol. Renal. Physiol. 2013, 305, F1006–F1013. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Santillo, M.; Colantuoni, A.; Mondola, P.; Guida, B.; Damiano, S. NOX signaling in molecular cardiovascular mechanisms involved in the blood pressure homeostasis. Front. Physiol. 2015, 6, 194. [Google Scholar] [CrossRef]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal 2008, 10, 179–206. [Google Scholar] [CrossRef]
- Fiebeler, A.; Schmidt, F.; Muller, D.N.; Park, J.K.; Dechend, R.; Bieringer, M.; Shagdarsuren, E.; Breu, V.; Haller, H.; Luft, F.C. Mineralocorticoid receptor affects AP-1 and nuclear factor-kappab activation in angiotensin II-induced cardiac injury. Hypertension 2001, 37, 787–793. [Google Scholar] [CrossRef]
- Queisser, N.; Oteiza, P.I.; Stopper, H.; Oli, R.G.; Schupp, N. Aldosterone induces oxidative stress, oxidative DNA damage and NF-kappaB-activation in kidney tubule cells. Mol. Carcinog. 2011, 50, 123–135. [Google Scholar] [CrossRef]
- Hayashi, H.; Kobara, M.; Abe, M.; Tanaka, N.; Gouda, E.; Toba, H.; Yamada, H.; Tatsumi, T.; Nakata, T.; Matsubara, H. Aldosterone nongenomically produces NADPH oxidase-dependent reactive oxygen species and induces myocyte apoptosis. Hypertens. Res. 2008, 31, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, A.; Liccardo, D.; Eguchi, A.; Elliott, K.J.; Traynham, C.J.; Ibetti, J.; Eguchi, S.; Leosco, D.; Ferrara, N.; Rengo, G.; et al. Myocardial pathology induced by aldosterone is dependent on non-canonical activities of G protein-coupled receptor kinases. Nat. Commun. 2016, 7, 10877. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Callera, G.E.; Yogi, A.; He, Y.; Tostes, R.C.; He, G.; Schiffrin, E.L.; Touyz, R.M. Aldosterone and angiotensin II synergistically stimulate migration in vascular smooth muscle cells through c-Src-regulated redox-sensitive RhoA pathways. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1511–1518. [Google Scholar] [CrossRef]
- Nagase, M.; Ayuzawa, N.; Kawarazaki, W.; Ishizawa, K.; Ueda, K.; Yoshida, S.; Fujita, T. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: Role of small GTPase Rac1. Hypertension 2012, 59, 500–506. [Google Scholar] [CrossRef]
- Stas, S.; Whaley-Connell, A.; Habibi, J.; Appesh, L.; Hayden, M.R.; Karuparthi, P.R.; Qazi, M.; Morris, E.M.; Cooper, S.A.; Link, C.D.; et al. Mineralocorticoid receptor blockade attenuates chronic overexpression of the renin-angiotensin-aldosterone system stimulation of reduced nicotinamide adenine dinucleotide phosphate oxidase and cardiac remodeling. Endocrinology 2007, 148, 3773–3780. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Beswick, R.A.; Yamamoto, T.; Palmer, T.; Taylor, T.A.; Pollock, J.S.; Pollock, D.M.; Brands, M.W.; Webb, R.C. Increased reactive oxygen species contributes to kidney injury in mineralocorticoid hypertensive rats. J. Physiol. Pharmacol. 2006, 57, 343–357. [Google Scholar] [PubMed]
- Kamalov, G.; Ahokas, R.A.; Zhao, W.; Zhao, T.; Shahbaz, A.U.; Johnson, P.L.; Bhattacharya, S.K.; Sun, Y.; Gerling, I.C.; Weber, K.T. Uncoupling the coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria seen in aldosteronism. J. Cardiovasc. Pharmacol. 2010, 55, 248–254. [Google Scholar] [CrossRef]
- Ndisang, J.F.; Jadhav, A. The heme oxygenase system attenuates pancreatic lesions and improves insulin sensitivity and glucose metabolism in deoxycorticosterone acetate hypertension. Am. J. Physiol. Regul. Integr. Comp Physiol. 2010, 298, R211–R223. [Google Scholar] [CrossRef]
- Park, Y.M.; Lim, B.H.; Touyz, R.M.; Park, J.B. Expression of NAD(P)H oxidase subunits and their contribution to cardiovascular damage in aldosterone/salt-induced hypertensive rat. J. Korean Med. Sci. 2008, 23, 1039–1045. [Google Scholar] [CrossRef][Green Version]
- Schupp, N.; Kolkhof, P.; Queisser, N.; Gartner, S.; Schmid, U.; Kretschmer, A.; Hartmann, E.; Oli, R.G.; Schafer, S.; Stopper, H. Mineralocorticoid receptor-mediated DNA damage in kidneys of DOCA-salt hypertensive rats. FASEB J. 2011, 25, 968–978. [Google Scholar] [CrossRef]
- Nagata, D.; Takahashi, M.; Sawai, K.; Tagami, T.; Usui, T.; Shimatsu, A.; Hirata, Y.; Naruse, M. Molecular mechanism of the inhibitory effect of aldosterone on endothelial NO synthase activity. Hypertension 2006, 48, 165–171. [Google Scholar] [CrossRef]
- Taye, A.; Morawietz, H. Spironolactone inhibits NADPH oxidase-induced oxidative stress and enhances eNOS in human endothelial cells. Iran J. Pharm. Res. 2011, 10, 329–337. [Google Scholar]
- Virdis, A.; Neves, M.F.; Amiri, F.; Viel, E.; Touyz, R.M.; Schiffrin, E.L. Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertension 2002, 40, 504–510. [Google Scholar] [CrossRef]
- Mayyas, F.A.; Aljohmani, A.I.; Alzoubi, K.H. The Impact of Spironolactone on Markers of Myocardial Oxidative Status, Inflammation and Remodeling in Hyperthyroid Rats. Curr. Mol. Pharmacol. 2020, 13, 206–215. [Google Scholar] [CrossRef]
- Mayyas, F.; Alzoubi, K.H.; Bonyan, R. The role of spironolactone on myocardial oxidative stress in rat model of streptozotocin-induced diabetes. Cardiovasc. Ther. 2017, 35, e12242. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Leopold, J.A. Mineralocorticoid receptor antagonists and endothelial function. Curr. Opin. Investig. Drugs 2008, 9, 963–969. [Google Scholar] [PubMed]
- Stehr, C.B.; Mellado, R.; Ocaranza, M.P.; Carvajal, C.A.; Mosso, L.; Becerra, E.; Solis, M.; Garcia, L.; Lavandero, S.; Jalil, J.; et al. Increased levels of oxidative stress, subclinical inflammation, and myocardial fibrosis markers in primary aldosteronism patients. J. Hypertens. 2010, 28, 2120–2126. [Google Scholar] [CrossRef]
- Laffer, C.L.; Bolterman, R.J.; Romero, J.C.; Elijovich, F. Effect of salt on isoprostanes in salt-sensitive essential hypertension. Hypertension 2006, 47, 434–440. [Google Scholar] [CrossRef]
- Keidar, S.; Gamliel-Lazarovich, A.; Kaplan, M.; Pavlotzky, E.; Hamoud, S.; Hayek, T.; Karry, R.; Abassi, Z. Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ. Res. 2005, 97, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Renke, M.; Tylicki, L.; Knap, N.; Rutkowski, P.; Neuwelt, A.; Larczynski, W.; Wozniak, M.; Rutkowski, B. Spironolactone attenuates oxidative stress in patients with chronic kidney disease. Hypertension 2008, 52, e132–e133. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef]
- Munoz-Durango, N.; Vecchiola, A.; Gonzalez-Gomez, L.M.; Simon, F.; Riedel, C.A.; Fardella, C.E.; Kalergis, A.M. Modulation of Immunity and Inflammation by the Mineralocorticoid Receptor and Aldosterone. Biomed. Res. Int. 2015, 2015, 652738. [Google Scholar] [CrossRef]
- Ferreira, N.S.; Tostes, R.C.; Paradis, P.; Schiffrin, E.L. Aldosterone, Inflammation, Immune System, and Hypertension. Am. J. Hypertens. 2021, 34, 15–27. [Google Scholar] [CrossRef]
- Gilbert, K.C.; Brown, N.J. Aldosterone and inflammation. Curr. Opin. Endocrinol. Diabetes. Obes. 2010, 17, 199–204. [Google Scholar] [CrossRef]
- Callera, G.E.; Yogi, A.; Briones, A.M.; Montezano, A.C.; He, Y.; Tostes, R.C.; Schiffrin, E.L.; Touyz, R.M. Vascular proinflammatory responses by aldosterone are mediated via c-Src trafficking to cholesterol-rich microdomains: Role of PDGFR. Cardiovasc. Res. 2011, 91, 720–731. [Google Scholar] [CrossRef]
- Ding, W.; Yang, L.; Zhang, M.; Gu, Y. Chronic inhibition of nuclear factor kappa B attenuates aldosterone/salt-induced renal injury. Life Sci. 2012, 90, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cui, R.; Cheng, X.; Du, J. Antiapoptotic effect of serum and glucocorticoid-inducible protein kinase is mediated by novel mechanism activating IkappaB kinase. Cancer Res. 2005, 65, 457–464. [Google Scholar] [CrossRef]
- Rickard, A.J.; Morgan, J.; Bienvenu, L.A.; Fletcher, E.K.; Cranston, G.A.; Shen, J.Z.; Reichelt, M.E.; Delbridge, L.M.; Young, M.J. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension 2012, 60, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Usher, M.G.; Duan, S.Z.; Ivaschenko, C.Y.; Frieler, R.A.; Berger, S.; Schutz, G.; Lumeng, C.N.; Mortensen, R.M. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J. Clin. Investig. 2010, 120, 3350–3364. [Google Scholar] [CrossRef]
- McGraw, A.P.; Bagley, J.; Chen, W.S.; Galayda, C.; Nickerson, H.; Armani, A.; Caprio, M.; Carmeliet, P.; Jaffe, I.Z. Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism. J. Am. Heart Assoc. 2013, 2, e000018. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.J.; Morgan, J.; Chrissobolis, S.; Miller, A.A.; Sobey, C.G.; Young, M.J. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension 2014, 63, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Caprio, M.; Newfell, B.G.; la Sala, A.; Baur, W.; Fabbri, A.; Rosano, G.; Mendelsohn, M.E.; Jaffe, I.Z. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ. Res. 2008, 102, 1359–1367. [Google Scholar] [CrossRef]
- Briet, M.; Barhoumi, T.; Mian, M.O.R.; Coelho, S.C.; Ouerd, S.; Rautureau, Y.; Coffman, T.M.; Paradis, P.; Schiffrin, E.L. Aldosterone-Induced Vascular Remodeling and Endothelial Dysfunction Require Functional Angiotensin Type 1a Receptors. Hypertension 2016, 67, 897–905. [Google Scholar] [CrossRef]
- Lemarie, C.A.; Simeone, S.M.; Nikonova, A.; Ebrahimian, T.; Deschenes, M.E.; Coffman, T.M.; Paradis, P.; Schiffrin, E.L. Aldosterone-induced activation of signaling pathways requires activity of angiotensin type 1a receptors. Circ. Res. 2009, 105, 852–859. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.; Pang, X.; Zhao, J.; Wang, S.; Wu, D. Aldosterone induces C-reactive protein expression via MR-ROS-MAPK-NF-kappaB signal pathway in rat vascular smooth muscle cells. Mol. Cell Endocrinol. 2014, 395, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Bruder-Nascimento, T.; Ferreira, N.S.; Zanotto, C.Z.; Ramalho, F.; Pequeno, I.O.; Olivon, V.C.; Neves, K.B.; Alves-Lopes, R.; Campos, E.; Silva, C.A.; et al. NLRP3 Inflammasome Mediates Aldosterone-Induced Vascular Damage. Circulation 2016, 134, 1866–1880. [Google Scholar] [CrossRef]
- Doi, T.; Doi, S.; Nakashima, A.; Ueno, T.; Yokoyama, Y.; Kohno, N.; Masaki, T. Mizoribine ameliorates renal injury and hypertension along with the attenuation of renal caspase-1 expression in aldosterone-salt-treated rats. PLoS ONE 2014, 9, e93513. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, H.; Zhang, J.; Xie, C.; Fan, C.; Zhang, H.; Wu, P.; Wei, Q.; Tan, W.; Xu, L.; et al. Inflammation and Fibrosis in Perirenal Adipose Tissue of Patients With Aldosterone-Producing Adenoma. Endocrinology 2018, 159, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Savoia, C.; Touyz, R.M.; Amiri, F.; Schiffrin, E.L. Selective mineralocorticoid receptor blocker eplerenone reduces resistance artery stiffness in hypertensive patients. Hypertension 2008, 51, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Silvestre, J.S.; Robert, V.; Heymes, C.; Aupetit-Faisant, B.; Mouas, C.; Moalic, J.M.; Swynghedauw, B.; Delcayre, C. Myocardial production of aldosterone and corticosterone in the rat. Physiological regulation. J. Biol. Chem. 1998, 273, 4883–4891. [Google Scholar] [CrossRef]
- Mohamed, D.M.; Shaqura, M.; Li, X.; Shakibaei, M.; Beyer, A.; Treskatsch, S.; Schafer, M.; Mousa, S.A. Aldosterone Synthase in Peripheral Sensory Neurons Contributes to Mechanical Hypersensitivity during Local Inflammation in Rats. Anesthesiology 2020, 132, 867–880. [Google Scholar] [CrossRef]
- Gomez-Sanchez, C.E.; Zhou, M.Y.; Cozza, E.N.; Morita, H.; Foecking, M.F.; Gomez-Sanchez, E.P. Aldosterone biosynthesis in the rat brain. Endocrinology 1997, 138, 3369–3373. [Google Scholar] [CrossRef]
- Malik, M. Heart rate variability: Standards of measurement, physiological interpretation and clinical use. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Circulation 1996, 93, 1043–1065. [Google Scholar] [CrossRef]
- Wang, W. Chronic administration of aldosterone depresses baroreceptor reflex function in the dog. Hypertension 1994, 24, 571–575. [Google Scholar] [CrossRef]
- Struthers, A.D. Evidence for myocardial synthesis of aldosterone producing myocardial fibrosis in man. Clin. Sci. 2002, 102, 387. [Google Scholar] [CrossRef]
- MacFadyen, R.J.; Barr, C.S.; Struthers, A.D. Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc. Res. 1997, 35, 30–34. [Google Scholar] [CrossRef]
- Burke, S.L.; Barzel, B.; Jackson, K.L.; Gueguen, C.; Young, M.J.; Head, G.A. Role of Mineralocorticoid and Angiotensin Type 1 Receptors in the Paraventricular Nucleus in Angiotensin-Induced Hypertension. Front. Physiol. 2021, 12, 640373. [Google Scholar] [CrossRef]
- Huang, B.S.; Ahmadi, S.; Ahmad, M.; White, R.A.; Leenen, F.H. Central neuronal activation and pressor responses induced by circulating ANG II: Role of the brain aldosterone-"ouabain" pathway. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H422–H430. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dehe, L.; Mousa, S.A.; Aboryag, N.; Shaqura, M.; Beyer, A.; Schafer, M.; Treskatsch, S. Identification of Mineralocorticoid Receptors, Aldosterone, and Its Processing Enzyme CYP11B2 on Parasympathetic and Sympathetic Neurons in Rat Intracardiac Ganglia. Front. Neuroanat. 2021, 15, 802359. [Google Scholar] [CrossRef]
- Alexander, Y.; Osto, E.; Schmidt-Trucksass, A.; Shechter, M.; Trifunovic, D.; Duncker, D.J.; Aboyans, V.; Back, M.; Badimon, L.; Cosentino, F.; et al. Endothelial function in cardiovascular medicine: A consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc. Res. 2021, 117, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, T.; Beese, M.; Wyss, K.; Klinge, U.; Haller, H.; Haubitz, M.; Fiebeler, A. Aldosterone modulates endothelial permeability and endothelial nitric oxide synthase activity by rearrangement of the actin cytoskeleton. Hypertension 2013, 61, 501–508. [Google Scholar] [CrossRef]
- Fiebeler, A.; Luft, F.C. The mineralocorticoid receptor and oxidative stress. Heart Fail Rev. 2005, 10, 47–52. [Google Scholar] [CrossRef]
- Briet, M.; Schiffrin, E.L. Vascular actions of aldosterone. J. Vasc. Res. 2013, 50, 89–99. [Google Scholar] [CrossRef]
- Maron, B.A.; Zhang, Y.Y.; Handy, D.E.; Beuve, A.; Tang, S.S.; Loscalzo, J.; Leopold, J.A. Aldosterone increases oxidant stress to impair guanylyl cyclase activity by cysteinyl thiol oxidation in vascular smooth muscle cells. J. Biol. Chem. 2009, 284, 7665–7672. [Google Scholar] [CrossRef]
- Chen, L.; Ding, M.L.; Wu, F.; He, W.; Li, J.; Zhang, X.Y.; Xie, W.L.; Duan, S.Z.; Xia, W.H.; Tao, J. Impaired Endothelial Repair Capacity of Early Endothelial Progenitor Cells in Hypertensive Patients With Primary Hyperaldosteronemia: Role of 5,6,7,8-Tetrahydrobiopterin Oxidation and Endothelial Nitric Oxide Synthase Uncoupling. Hypertension 2016, 67, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Eatman, D.; Peagler, K.; Watson, J.; Rollins-Hairston, A.; Bayorh, M.A. The involvement of prostaglandins in the contractile function of the aorta by aldosterone. BMC Res. Notes 2011, 4, 125. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Rivero, J.; Cachofeiro, V.; Lahera, V.; Aras-Lopez, R.; Marquez-Rodas, I.; Salaices, M.; Xavier, F.E.; Ferrer, M.; Balfagon, G. Participation of prostacyclin in endothelial dysfunction induced by aldosterone in normotensive and hypertensive rats. Hypertension 2005, 46, 107–112. [Google Scholar] [CrossRef]
- Feletou, M.; Huang, Y.; Vanhoutte, P.M. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br. J. Pharmacol. 2011, 164, 894–912. [Google Scholar] [CrossRef]
- Leopold, J.A.; Dam, A.; Maron, B.A.; Scribner, A.W.; Liao, R.; Handy, D.E.; Stanton, R.C.; Pitt, B.; Loscalzo, J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat. Med. 2007, 13, 189–197. [Google Scholar] [CrossRef]
- Oberleithner, H.; Riethmuller, C.; Ludwig, T.; Hausberg, M.; Schillers, H. Aldosterone remodels human endothelium. Acta Physiol. 2006, 187, 305–312. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; Aroor, A.R.; Hill, M.A.; Yang, Y.; Whaley-Connell, A.; Jaisser, F.; Sowers, J.R. Epithelial Sodium Channel in Aldosterone-Induced Endothelium Stiffness and Aortic Dysfunction. Hypertension 2018, 72, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Jeggle, P.; Callies, C.; Tarjus, A.; Fassot, C.; Fels, J.; Oberleithner, H.; Jaisser, F.; Kusche-Vihrog, K. Epithelial sodium channel stiffens the vascular endothelium in vitro and in Liddle mice. Hypertension 2013, 61, 1053–1059. [Google Scholar] [CrossRef]
- Bienvenu, L.A.; Bell, J.R.; Weeks, K.L.; Delbridge, L.M.D.; Young, M.J. New Perspectives on Sex Steroid and Mineralocorticoid Receptor Signaling in Cardiac Ischemic Injury. Front. Physiol. 2022, 13, 896425. [Google Scholar] [CrossRef]
- Mueller, K.B.; Bender, S.B.; Hong, K.; Yang, Y.; Aronovitz, M.; Jaisser, F.; Hill, M.A.; Jaffe, I.Z. Endothelial Mineralocorticoid Receptors Differentially Contribute to Coronary and Mesenteric Vascular Function Without Modulating Blood Pressure. Hypertension 2015, 66, 988–997. [Google Scholar] [CrossRef]
- Davel, A.P.; Jaffe, I.Z.; Tostes, R.C.; Jaisser, F.; Belin de Chantemele, E.J. New roles of aldosterone and mineralocorticoid receptors in cardiovascular disease: Translational and sex-specific effects. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H989–H999. [Google Scholar] [CrossRef] [PubMed]
- Davel, A.P.; Lu, Q.; Moss, M.E.; Rao, S.; Anwar, I.J.; DuPont, J.J.; Jaffe, I.Z. Sex-Specific Mechanisms of Resistance Vessel Endothelial Dysfunction Induced by Cardiometabolic Risk Factors. J. Am. Heart Assoc. 2018, 7, e007675. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, A.; Wallaschofski, H.; Ludemann, J.; Volzke, H.; Markus, M.R.; Rettig, R.; Lendeckel, U.; Reincke, M.; Felix, S.B.; Empen, K.; et al. Plasma aldosterone levels and aldosterone-to-renin ratios are associated with endothelial dysfunction in young to middle-aged subjects. Atherosclerosis 2011, 219, 875–879. [Google Scholar] [CrossRef]
- Nishizaka, M.K.; Zaman, M.A.; Green, S.A.; Renfroe, K.Y.; Calhoun, D.A. Impaired endothelium-dependent flow-mediated vasodilation in hypertensive subjects with hyperaldosteronism. Circulation 2004, 109, 2857–2861. [Google Scholar] [CrossRef]
- Thum, T.; Schmitter, K.; Fleissner, F.; Wiebking, V.; Dietrich, B.; Widder, J.D.; Jazbutyte, V.; Hahner, S.; Ertl, G.; Bauersachs, J. Impairment of endothelial progenitor cell function and vascularization capacity by aldosterone in mice and humans. Eur. Heart J. 2011, 32, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.C.; Lo, S.C.; Chen, Y.L.; Huang, P.H.; Tsai, C.T.; Liang, C.J.; Kuo, C.C.; Kuo, Y.S.; Lee, B.C.; Wu, E.L.; et al. Endothelial progenitor cells in primary aldosteronism: A biomarker of severity for aldosterone vasculopathy and prognosis. J. Clin. Endocrinol. Metab. 2011, 96, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Oki, K.; Kajikawa, M.; Nakashima, A.; Maruhashi, T.; Iwamoto, Y.; Iwamoto, A.; Oda, N.; Hidaka, T.; Kihara, Y.; et al. Effect of aldosterone-producing adenoma on endothelial function and Rho-associated kinase activity in patients with primary aldosteronism. Hypertension 2015, 65, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Sakima, A.; Arima, H.; Matayoshi, T.; Ishida, A.; Ohya, Y. Effect of Mineralocorticoid Receptor Blockade on Arterial Stiffness and Endothelial Function: A Meta-Analysis of Randomized Trials. Hypertension 2021, 77, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Stockand, J.D.; Meszaros, J.G. Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H176–H184. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, J.A.; Kim, K.L.; Jang, H.S.; Kim, J.M.; Lee, J.Y.; Shin, I.S.; Lee, J.S.; Suh, W.; Choi, J.H.; et al. Aldosterone upregulates connective tissue growth factor gene expression via p38 MAPK pathway and mineralocorticoid receptor in ventricular myocytes. J. Korean Med. Sci. 2004, 19, 805–811. [Google Scholar] [CrossRef]
- Tsai, C.F.; Yang, S.F.; Chu, H.J.; Ueng, K.C. Cross-talk between mineralocorticoid receptor/angiotensin II type 1 receptor and mitogen-activated protein kinase pathways underlies aldosterone-induced atrial fibrotic responses in HL-1 cardiomyocytes. Int. J. Cardiol. 2013, 169, 17–28. [Google Scholar] [CrossRef] [PubMed]
- De Giusti, V.C.; Nolly, M.B.; Yeves, A.M.; Caldiz, C.I.; Villa-Abrille, M.C.; Chiappe de Cingolani, G.E.; Ennis, I.L.; Cingolani, H.E.; Aiello, E.A. Aldosterone stimulates the cardiac Na(+)/H(+) exchanger via transactivation of the epidermal growth factor receptor. Hypertension 2011, 58, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Wyatt, A.W.; Klingel, K.; Huang, D.Y.; Hussain, A.; Berchtold, S.; Friedrich, B.; Grahammer, F.; Belaiba, R.S.; Gorlach, A.; et al. SGK1-dependent cardiac CTGF formation and fibrosis following DOCA treatment. J. Mol. Med. 2006, 84, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Azibani, F.; Benard, L.; Schlossarek, S.; Merval, R.; Tournoux, F.; Fazal, L.; Polidano, E.; Launay, J.M.; Carrier, L.; Chatziantoniou, C.; et al. Aldosterone inhibits antifibrotic factors in mouse hypertensive heart. Hypertension 2012, 59, 1179–1187. [Google Scholar] [CrossRef]
- Fraccarollo, D.; Berger, S.; Galuppo, P.; Kneitz, S.; Hein, L.; Schutz, G.; Frantz, S.; Ertl, G.; Bauersachs, J. Deletion of cardiomyocyte mineralocorticoid receptor ameliorates adverse remodeling after myocardial infarction. Circulation 2011, 123, 400–408. [Google Scholar] [CrossRef]
- Lacolley, P.; Challande, P.; Osborne-Pellegrin, M.; Regnault, V. Genetics and pathophysiology of arterial stiffness. Cardiovasc. Res. 2009, 81, 637–648. [Google Scholar] [CrossRef]
- McCurley, A.; Pires, P.W.; Bender, S.B.; Aronovitz, M.; Zhao, M.J.; Metzger, D.; Chambon, P.; Hill, M.A.; Dorrance, A.M.; Mendelsohn, M.E.; et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat. Med. 2012, 18, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Miana, M.; Reboul, P.; Cachofeiro, V.; Martinez-Martinez, E.; de Boer, R.A.; Poirier, F.; Lacolley, P.; Zannad, F.; Rossignol, P.; et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 67–75. [Google Scholar] [CrossRef]
- Tarjus, A.; Martinez-Martinez, E.; Amador, C.; Latouche, C.; El Moghrabi, S.; Berger, T.; Mak, T.W.; Fay, R.; Farman, N.; Rossignol, P.; et al. Neutrophil Gelatinase-Associated Lipocalin, a Novel Mineralocorticoid Biotarget, Mediates Vascular Profibrotic Effects of Mineralocorticoids. Hypertension 2015, 66, 158–166. [Google Scholar] [CrossRef]
- Harvey, A.P.; Montezano, A.C.; Hood, K.Y.; Lopes, R.A.; Rios, F.; Ceravolo, G.; Graham, D.; Touyz, R.M. Vascular dysfunction and fibrosis in stroke-prone spontaneously hypertensive rats: The aldosterone-mineralocorticoid receptor-Nox1 axis. Life Sci. 2017, 179, 110–119. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, K.; Chen, J.; Wang, M.H.; Wang, J.; Liu, P.; Huang, H. Roles of aldosterone in vascular calcification: An update. Eur. J. Pharmacol. 2016, 786, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.W.; He, W.B.; Xie, C.M.; Gao, M.; Feng, L.Y.; Liu, Z.Y.; Wang, J.F.; Huang, H.; Liu, P.M. Aldosterone enhances high phosphate-induced vascular calcification through inhibition of AMPK-mediated autophagy. J. Cell Mol. Med. 2020, 24, 13648–13659. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, J.; Alesutan, I.; Leibrock, C.B.; Quintanilla-Martinez, L.; Kuhn, V.; Feger, M.; Mia, S.; Ahmed, M.S.; Rosenblatt, K.P.; Kuro, O.M.; et al. Spironolactone ameliorates PIT1-dependent vascular osteoinduction in klotho-hypomorphic mice. J. Clin. Investig. 2013, 123, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, I.Z.; Tintut, Y.; Newfell, B.G.; Demer, L.L.; Mendelsohn, M.E. Mineralocorticoid receptor activation promotes vascular cell calcification. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Tatsumoto, N.; Yamada, S.; Tokumoto, M.; Eriguchi, M.; Noguchi, H.; Torisu, K.; Tsuruya, K.; Kitazono, T. Spironolactone ameliorates arterial medial calcification in uremic rats: The role of mineralocorticoid receptor signaling in vascular calcification. Am. J. Physiol. Renal. Physiol. 2015, 309, F967–F979. [Google Scholar] [CrossRef]
- Gkaliagkousi, E.; Anyfanti, P.; Triantafyllou, A.; Gavriilaki, E.; Nikolaidou, B.; Lazaridis, A.; Vamvakis, A.; Douma, S. Aldosterone as a mediator of microvascular and macrovascular damage in a population of normotensive to early-stage hypertensive individuals. J. Am. Soc. Hypertens. 2018, 12, 50–57. [Google Scholar] [CrossRef]
- Mahmud, A.; Feely, J. Aldosterone-to-renin ratio, arterial stiffness, and the response to aldosterone antagonism in essential hypertension. Am. J. Hypertens. 2005, 18, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Lupoli, R.; Tortora, A.; Cacciapuoti, M.; Lupoli, G.A.; Tarantino, P.; Nasto, A.; Di Minno, M.N. Cardiovascular risk markers in patients with primary aldosteronism: A systematic review and meta-analysis of literature studies. Int. J. Cardiol. 2016, 208, 46–55. [Google Scholar] [CrossRef]
- Strauch, B.; Petrak, O.; Zelinka, T.; Wichterle, D.; Holaj, R.; Kasalicky, M.; Safarik, L.; Rosa, J.; Widimsky, J., Jr. Adrenalectomy improves arterial stiffness in primary aldosteronism. Am. J. Hypertens. 2008, 21, 1086–1092. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Ginsberg, H.N. Insulin resistance and cardiovascular disease. J. Clin. Investig. 2000, 106, 453–458. [Google Scholar] [CrossRef] [PubMed]
- DeMarco, V.G.; Habibi, J.; Jia, G.; Aroor, A.R.; Ramirez-Perez, F.I.; Martinez-Lemus, L.A.; Bender, S.B.; Garro, M.; Hayden, M.R.; Sun, Z.; et al. Low-Dose Mineralocorticoid Receptor Blockade Prevents Western Diet-Induced Arterial Stiffening in Female Mice. Hypertension 2015, 66, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Habibi, J.; Chen, D.; Hulse, J.L.; Whaley-Connell, A.; Sowers, J.R.; Jia, G. Targeting mineralocorticoid receptors in diet-induced hepatic steatosis and insulin resistance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2022, 322, R253–R262. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Sowers, J.R. The role of mineralocorticoid receptor signaling in the cross-talk between adipose tissue and the vascular wall. Cardiovasc. Res. 2017, 113, 1055–1063. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; Aroor, A.R.; Martinez-Lemus, L.A.; DeMarco, V.G.; Ramirez-Perez, F.I.; Sun, Z.; Hayden, M.R.; Meininger, G.A.; Mueller, K.B.; et al. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ. Res. 2016, 118, 935–943. [Google Scholar] [CrossRef]
- Igbekele, A.E.; Jia, G.; Hill, M.A.; Sowers, J.R.; Jia, G. Mineralocorticoid Receptor Activation in Vascular Insulin Resistance and Dysfunction. Int. J. Mol. Sci. 2022, 23, 8954. [Google Scholar] [CrossRef]
- Jia, G.; Lockette, W.; Sowers, J.R. Mineralocorticoid receptors in the pathogenesis of insulin resistance and related disorders: From basic studies to clinical disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R276–R286. [Google Scholar] [CrossRef]
- Preiss, D.; Zetterstrand, S.; McMurray, J.J.; Ostergren, J.; Michelson, E.L.; Granger, C.B.; Yusuf, S.; Swedberg, K.; Pfeffer, M.A.; Gerstein, H.C.; et al. Predictors of development of diabetes in patients with chronic heart failure in the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) program. Diabetes Care 2009, 32, 915–920. [Google Scholar] [CrossRef]
- Zhao, J.V.; Xu, L.; Lin, S.L.; Schooling, C.M. Spironolactone and glucose metabolism, a systematic review and meta-analysis of randomized controlled trials. J. Am. Soc. Hypertens. 2016, 10, 671–682. [Google Scholar] [CrossRef]
- Rossing, P.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Chan, J.C.N.; Kooy, A.; McCafferty, K.; Schernthaner, G.; et al. Finerenone in Predominantly Advanced CKD and Type 2 Diabetes With or Without Sodium-Glucose Cotransporter-2 Inhibitor Therapy. Kidney Int. Rep. 2022, 7, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Lipscombe, D. L-type calcium channels: Highs and new lows. Circ. Res. 2002, 90, 933–935. [Google Scholar] [CrossRef]
- Boixel, C.; Gavillet, B.; Rougier, J.S.; Abriel, H. Aldosterone increases voltage-gated sodium current in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2257–H2266. [Google Scholar] [CrossRef]
- Aronsen, J.M.; Swift, F.; Sejersted, O.M. Cardiac sodium transport and excitation-contraction coupling. J. Mol. Cell Cardiol. 2013, 61, 11–19. [Google Scholar] [CrossRef]
- Matsui, S.; Satoh, H.; Kawashima, H.; Nagasaka, S.; Niu, C.F.; Urushida, T.; Katoh, H.; Watanabe, Y.; Hayashi, H. Non-genomic effects of aldosterone on intracellular ion regulation and cell volume in rat ventricular myocytes. Can. J. Physiol. Pharmacol. 2007, 85, 264–273. [Google Scholar] [CrossRef][Green Version]
- Mattiazzi, A. Positive inotropic effect of angiotensin II. Increases in intracellular Ca2+ or changes in myofilament Ca2+ responsiveness? J. Pharmacol. Toxicol. Methods 1997, 37, 205–214. [Google Scholar] [CrossRef]
- Denham, N.C.; Pearman, C.M.; Caldwell, J.L.; Madders, G.W.P.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front. Physiol. 2018, 9, 1380. [Google Scholar] [CrossRef]
- Ouvrard-Pascaud, A.; Sainte-Marie, Y.; Benitah, J.P.; Perrier, R.; Soukaseum, C.; Nguyen Dinh Cat, A.; Royer, A.; Le Quang, K.; Charpentier, F.; Demolombe, S.; et al. Conditional mineralocorticoid receptor expression in the heart leads to life-threatening arrhythmias. Circulation 2005, 111, 3025–3033. [Google Scholar] [CrossRef]
- Diaz, R.G.; Perez, N.G.; Morgan, P.E.; Villa-Abrille, M.C.; Caldiz, C.I.; Nolly, M.B.; Portiansky, E.L.; Ennis, I.L.; Cingolani, H.E. Myocardial mineralocorticoid receptor activation by stretching and its functional consequences. Hypertension 2014, 63, 112–118. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Jaffe, I.Z.; Mendelsohn, M.E. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ. Res. 2005, 96, 643–650. [Google Scholar] [CrossRef]
- Marzolla, V.; Armani, A.; Mammi, C.; Moss, M.E.; Pagliarini, V.; Pontecorvo, L.; Antelmi, A.; Fabbri, A.; Rosano, G.; Jaffe, I.Z.; et al. Essential role of ICAM-1 in aldosterone-induced atherosclerosis. Int. J. Cardiol. 2017, 232, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.; Rudolph, A.E.; Frierdich, G.E.; Nachowiak, D.A.; Kekec, B.K.; Blomme, E.A.; McMahon, E.G.; Delyani, J.A. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1802–H1810. [Google Scholar] [CrossRef]
- Keidar, S.; Hayek, T.; Kaplan, M.; Pavlotzky, E.; Hamoud, S.; Coleman, R.; Aviram, M. Effect of eplerenone, a selective aldosterone blocker, on blood pressure, serum and macrophage oxidative stress, and atherosclerosis in apolipoprotein E-deficient mice. J. Cardiovasc. Pharmacol. 2003, 41, 955–963. [Google Scholar] [CrossRef]
- Suzuki, J.; Iwai, M.; Mogi, M.; Oshita, A.; Yoshii, T.; Higaki, J.; Horiuchi, M. Eplerenone with valsartan effectively reduces atherosclerotic lesion by attenuation of oxidative stress and inflammation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 917–921. [Google Scholar] [CrossRef]
- Gueret, A.; Harouki, N.; Favre, J.; Galmiche, G.; Nicol, L.; Henry, J.P.; Besnier, M.; Thuillez, C.; Richard, V.; Kolkhof, P.; et al. Vascular Smooth Muscle Mineralocorticoid Receptor Contributes to Coronary and Left Ventricular Dysfunction After Myocardial Infarction. Hypertension 2016, 67, 717–723. [Google Scholar] [CrossRef]
- Inoue, K.; Goldwater, D.; Allison, M.; Seeman, T.; Kestenbaum, B.R.; Watson, K.E. Serum Aldosterone Concentration, Blood Pressure, and Coronary Artery Calcium: The Multi-Ethnic Study of Atherosclerosis. Hypertension 2020, 76, 113–120. [Google Scholar] [CrossRef]
- Ivanes, F.; Susen, S.; Mouquet, F.; Pigny, P.; Cuilleret, F.; Sautiere, K.; Collet, J.P.; Beygui, F.; Hennache, B.; Ennezat, P.V.; et al. Aldosterone, mortality, and acute ischaemic events in coronary artery disease patients outside the setting of acute myocardial infarction or heart failure. Eur. Heart J. 2012, 33, 191–202. [Google Scholar] [CrossRef]
- Beygui, F.; Collet, J.P.; Benoliel, J.J.; Vignolles, N.; Dumaine, R.; Barthelemy, O.; Montalescot, G. High plasma aldosterone levels on admission are associated with death in patients presenting with acute ST-elevation myocardial infarction. Circulation 2006, 114, 2604–2610. [Google Scholar] [CrossRef]
- van der Heijden, C.; Smeets, E.M.M.; Aarntzen, E.; Noz, M.P.; Monajemi, H.; Kersten, S.; Kaffa, C.; Hoischen, A.; Deinum, J.; Joosten, L.A.B.; et al. Arterial Wall Inflammation and Increased Hematopoietic Activity in Patients With Primary Aldosteronism. J. Clin. Endocrinol. Metab. 2020, 105, e1967–e1980. [Google Scholar] [CrossRef]
- Bucerius, J.; Hyafil, F.; Verberne, H.J.; Slart, R.H.; Lindner, O.; Sciagra, R.; Agostini, D.; Ubleis, C.; Gimelli, A.; Hacker, M.; et al. Position paper of the Cardiovascular Committee of the European Association of Nuclear Medicine (EANM) on PET imaging of atherosclerosis. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 780–792. [Google Scholar] [CrossRef]
- Monticone, S.; D’Ascenzo, F.; Moretti, C.; Williams, T.A.; Veglio, F.; Gaita, F.; Mulatero, P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Hundemer, G.L.; Curhan, G.C.; Yozamp, N.; Wang, M.; Vaidya, A. Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: A retrospective cohort study. Lancet Diabetes Endocrinol. 2018, 6, 51–59. [Google Scholar] [CrossRef]
- Montalescot, G.; Pitt, B.; Lopez de Sa, E.; Hamm, C.W.; Flather, M.; Verheugt, F.; Shi, H.; Turgonyi, E.; Orri, M.; Vincent, J.; et al. Early eplerenone treatment in patients with acute ST-elevation myocardial infarction without heart failure: The Randomized Double-Blind Reminder Study. Eur. Heart J. 2014, 35, 2295–2302. [Google Scholar] [CrossRef]
- Beygui, F.; Cayla, G.; Roule, V.; Roubille, F.; Delarche, N.; Silvain, J.; Van Belle, E.; Belle, L.; Galinier, M.; Motreff, P.; et al. Early Aldosterone Blockade in Acute Myocardial Infarction: The ALBATROSS Randomized Clinical Trial. J. Am. Coll Cardiol. 2016, 67, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Filippatos, G.; Anker, S.D.; Agarwal, R.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Schloemer, P.; Tornus, I.; Joseph, A.; et al. Finerenone and Cardiovascular Outcomes in Patients With Chronic Kidney Disease and Type 2 Diabetes. Circulation 2021, 143, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Tsai, C.H.; Pan, C.T.; Chang, Y.Y.; Chen, Z.W.; Wu, V.C.; Hung, C.S.; Lin, Y.H. Left ventricular remodeling and dysfunction in primary aldosteronism. J. Hum. Hypertens. 2021, 35, 131–147. [Google Scholar] [CrossRef]
- He, B.J.; Joiner, M.L.; Singh, M.V.; Luczak, E.D.; Swaminathan, P.D.; Koval, O.M.; Kutschke, W.; Allamargot, C.; Yang, J.; Guan, X.; et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat. Med. 2011, 17, 1610–1618. [Google Scholar] [CrossRef]
- Hung, C.S.; Chou, C.H.; Liao, C.W.; Lin, Y.T.; Wu, X.M.; Chang, Y.Y.; Chen, Y.H.; Wu, V.C.; Su, M.J.; Ho, Y.L.; et al. Aldosterone Induces Tissue Inhibitor of Metalloproteinases-1 Expression and Further Contributes to Collagen Accumulation: From Clinical to Bench Studies. Hypertension 2016, 67, 1309–1320. [Google Scholar] [CrossRef]
- Sakamuri, S.S.; Valente, A.J.; Siddesha, J.M.; Delafontaine, P.; Siebenlist, U.; Gardner, J.D.; Bysani, C. TRAF3IP2 mediates aldosterone/salt-induced cardiac hypertrophy and fibrosis. Mol. Cell Endocrinol. 2016, 429, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Calvier, L.; Fernandez-Celis, A.; Rousseau, E.; Jurado-Lopez, R.; Rossoni, L.V.; Jaisser, F.; Zannad, F.; Rossignol, P.; Cachofeiro, V.; et al. Galectin-3 blockade inhibits cardiac inflammation and fibrosis in experimental hyperaldosteronism and hypertension. Hypertension 2015, 66, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Mummidi, S.; Das, N.A.; Carpenter, A.J.; Kandikattu, H.; Krenz, M.; Siebenlist, U.; Valente, A.J.; Chandrasekar, B. Metformin inhibits aldosterone-induced cardiac fibroblast activation, migration and proliferation in vitro, and reverses aldosterone+salt-induced cardiac fibrosis in vivo. J. Mol. Cell Cardiol. 2016, 98, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Brilla, C.G.; Pick, R.; Tan, L.B.; Janicki, J.S.; Weber, K.T. Remodeling of the rat right and left ventricles in experimental hypertension. Circ. Res. 1990, 67, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.K.; Morgan, J.; Kennaway, D.R.; Bienvenu, L.A.; Rickard, A.J.; Delbridge, L.M.D.; Fuller, P.J.; Clyne, C.D.; Young, M.J. Deoxycorticosterone/Salt-Mediated Cardiac Inflammation and Fibrosis Are Dependent on Functional CLOCK Signaling in Male Mice. Endocrinology 2017, 158, 2906–2917. [Google Scholar] [CrossRef] [PubMed]
- Oestreicher, E.M.; Martinez-Vasquez, D.; Stone, J.R.; Jonasson, L.; Roubsanthisuk, W.; Mukasa, K.; Adler, G.K. Aldosterone and not plasminogen activator inhibitor-1 is a critical mediator of early angiotensin II/NG-nitro-L-arginine methyl ester-induced myocardial injury. Circulation 2003, 108, 2517–2523. [Google Scholar] [CrossRef]
- Lopez-Andres, N.; Martin-Fernandez, B.; Rossignol, P.; Zannad, F.; Lahera, V.; Fortuno, M.A.; Cachofeiro, V.; Diez, J. A role for cardiotrophin-1 in myocardial remodeling induced by aldosterone. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2372–H2382. [Google Scholar] [CrossRef]
- Park, Y.M.; Park, M.Y.; Suh, Y.L.; Park, J.B. NAD(P)H oxidase inhibitor prevents blood pressure elevation and cardiovascular hypertrophy in aldosterone-infused rats. Biochem. Biophys. Res. Commun. 2004, 313, 812–817. [Google Scholar] [CrossRef]
- Freel, E.M.; Mark, P.B.; Weir, R.A.; McQuarrie, E.P.; Allan, K.; Dargie, H.J.; McClure, J.D.; Jardine, A.G.; Davies, E.; Connell, J.M. Demonstration of blood pressure-independent noninfarct myocardial fibrosis in primary aldosteronism: A cardiac magnetic resonance imaging study. Circ. Cardiovasc. Imaging 2012, 5, 740–747. [Google Scholar] [CrossRef]
- Rossi, G.P.; Di Bello, V.; Ganzaroli, C.; Sacchetto, A.; Cesari, M.; Bertini, A.; Giorgi, D.; Scognamiglio, R.; Mariani, M.; Pessina, A.C. Excess aldosterone is associated with alterations of myocardial texture in primary aldosteronism. Hypertension 2002, 40, 23–27. [Google Scholar] [CrossRef]
- Ohno, Y.; Sone, M.; Inagaki, N.; Kawashima, A.; Takeda, Y.; Yoneda, T.; Kurihara, I.; Itoh, H.; Tsuiki, M.; Ichijo, T.; et al. Nadir Aldosterone Levels After Confirmatory Tests Are Correlated With Left Ventricular Hypertrophy in Primary Aldosteronism. Hypertension 2020, 75, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.W.; Huang, K.C.; Lee, J.K.; Lin, L.C.; Chen, C.W.; Chang, Y.Y.; Liao, C.W.; Wu, V.C.; Hung, C.S.; Lin, Y.H.; et al. Aldosterone induces left ventricular subclinical systolic dysfunction: A strain imaging study. J. Hypertens. 2018, 36, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Sacchetto, A.; Visentin, P.; Canali, C.; Graniero, G.R.; Palatini, P.; Pessina, A.C. Changes in left ventricular anatomy and function in hypertension and primary aldosteronism. Hypertension 1996, 27, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Rev. Esp. Cardiol. 2022, 75, 523. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, P.; Cleland, J.G.; Bhandari, S.; Tala, S.; Gustafsson, F.; Fay, R.; Lamiral, Z.; Dobre, D.; Pitt, B.; Zannad, F. Determinants and consequences of renal function variations with aldosterone blocker therapy in heart failure patients after myocardial infarction: Insights from the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study. Circulation 2012, 125, 271–279. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef]
- Filippatos, G.; Anker, S.D.; Bohm, M.; Gheorghiade, M.; Kober, L.; Krum, H.; Maggioni, A.P.; Ponikowski, P.; Voors, A.A.; Zannad, F.; et al. A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease. Eur. Heart J. 2016, 37, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Kosmala, W.; Rojek, A.; Przewlocka-Kosmala, M.; Wright, L.; Mysiak, A.; Marwick, T.H. Effect of Aldosterone Antagonism on Exercise Tolerance in Heart Failure With Preserved Ejection Fraction. J. Am. Coll Cardiol. 2016, 68, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, F.; Wachter, R.; Schmidt, A.G.; Kraigher-Krainer, E.; Colantonio, C.; Kamke, W.; Duvinage, A.; Stahrenberg, R.; Durstewitz, K.; Loffler, M.; et al. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: The Aldo-DHF randomized controlled trial. JAMA 2013, 309, 781–791. [Google Scholar] [CrossRef]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Cleland, J.G.; Girerd, N.; Bozec, E.; Rossignol, P.; Pellicori, P.; Cosmi, F.; Mariottoni, B.; Solomon, S.D.; Pitt, B.; et al. Spironolactone effect on cardiac structure and function of patients with heart failure and preserved ejection fraction: A pooled analysis of three randomized trials. Eur. J. Heart Fail 2022. Online Version of Record. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.M.; Rueda, A.; Sainte-Marie, Y.; Pereira, L.; Zissimopoulos, S.; Zhu, X.; Schaub, R.; Perrier, E.; Perrier, R.; Latouche, C.; et al. Mineralocorticoid modulation of cardiac ryanodine receptor activity is associated with downregulation of FK506-binding proteins. Circulation 2009, 119, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Benitah, J.P.; Perrier, E.; Gomez, A.M.; Vassort, G. Effects of aldosterone on transient outward K+ current density in rat ventricular myocytes. J. Physiol. 2001, 537, 151–160. [Google Scholar] [CrossRef]
- Reil, J.C.; Hohl, M.; Selejan, S.; Lipp, P.; Drautz, F.; Kazakow, A.; Munz, B.M.; Muller, P.; Steendijk, P.; Reil, G.H.; et al. Aldosterone promotes atrial fibrillation. Eur. Heart J. 2012, 33, 2098–2108. [Google Scholar] [CrossRef]
- Lammers, C.; Dartsch, T.; Brandt, M.C.; Rottlander, D.; Halbach, M.; Peinkofer, G.; Ockenpoehler, S.; Weiergraeber, M.; Schneider, T.; Reuter, H.; et al. Spironolactone prevents aldosterone induced increased duration of atrial fibrillation in rat. Cell Physiol. Biochem. 2012, 29, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, Y.; Ramirez, R.J.; Kaur, K.; Salvador-Montanes, O.; Ponce-Balbuena, D.; Ramos-Mondragon, R.; Ennis, S.R.; Guerrero-Serna, G.; Berenfeld, O.; Jalife, J. Eplerenone Reduces Atrial Fibrillation Burden Without Preventing Atrial Electrical Remodeling. J. Am. Coll Cardiol. 2017, 70, 2893–2905. [Google Scholar] [CrossRef]
- Mulatero, P.; Monticone, S.; Deinum, J.; Amar, L.; Prejbisz, A.; Zennaro, M.C.; Beuschlein, F.; Rossi, G.P.; Nishikawa, T.; Morganti, A.; et al. Genetics, prevalence, screening and confirmation of primary aldosteronism: A position statement and consensus of the Working Group on Endocrine Hypertension of The European Society of Hypertension. J. Hypertens. 2020, 38, 1919–1928. [Google Scholar] [CrossRef]
- Seccia, T.M.; Letizia, C.; Muiesan, M.L.; Lerco, S.; Cesari, M.; Bisogni, V.; Petramala, L.; Maiolino, G.; Volpin, R.; Rossi, G.P. Atrial fibrillation as presenting sign of primary aldosteronism: Results of the Prospective Appraisal on the Prevalence of Primary Aldosteronism in Hypertensive (PAPPHY) Study. J. Hypertens. 2020, 38, 332–339. [Google Scholar] [CrossRef]
- Rossi, G.P.; Maiolino, G.; Flego, A.; Belfiore, A.; Bernini, G.; Fabris, B.; Ferri, C.; Giacchetti, G.; Letizia, C.; Maccario, M.; et al. Adrenalectomy Lowers Incident Atrial Fibrillation in Primary Aldosteronism Patients at Long Term. Hypertension 2018, 71, 585–591. [Google Scholar] [CrossRef]
- Swedberg, K.; Zannad, F.; McMurray, J.J.; Krum, H.; van Veldhuisen, D.J.; Shi, H.; Vincent, J.; Pitt, B.; Investigators, E.-H.S. Eplerenone and atrial fibrillation in mild systolic heart failure: Results from the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization And SurvIval Study in Heart Failure) study. J. Am. Coll Cardiol. 2012, 59, 1598–1603. [Google Scholar] [CrossRef]
- Rienstra, M.; Hobbelt, A.H.; Alings, M.; Tijssen, J.G.P.; Smit, M.D.; Brugemann, J.; Geelhoed, B.; Tieleman, R.G.; Hillege, H.L.; Tukkie, R.; et al. Targeted therapy of underlying conditions improves sinus rhythm maintenance in patients with persistent atrial fibrillation: Results of the RACE 3 trial. Eur. Heart J. 2018, 39, 2987–2996. [Google Scholar] [CrossRef] [PubMed]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomstrom-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef] [PubMed]
- Buffolo, F.; Tetti, M.; Mulatero, P.; Monticone, S. Aldosterone as a Mediator of Cardiovascular Damage. Hypertension 2022, 79, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Funder, J.W.; Carey, R.M.; Mantero, F.; Murad, M.H.; Reincke, M.; Shibata, H.; Stowasser, M.; Young, W.F., Jr. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol. Metab. 2016, 101, 1889–1916. [Google Scholar] [CrossRef]
- Barcelo, A.; Pierola, J.; Esquinas, C.; de la Pena, M.; Arque, M.; Alonso-Fernandez, A.; Bauca, J.M.; Robles, J.; Barcelo, B.; Barbe, F. Relationship between aldosterone and the metabolic syndrome in patients with obstructive sleep apnea hypopnea syndrome: Effect of continuous positive airway pressure treatment. PLoS ONE 2014, 9, e84362. [Google Scholar] [CrossRef]
- Marzolla, V.; Armani, A.; Zennaro, M.C.; Cinti, F.; Mammi, C.; Fabbri, A.; Rosano, G.M.; Caprio, M. The role of the mineralocorticoid receptor in adipocyte biology and fat metabolism. Mol. Cell Endocrinol. 2012, 350, 281–288. [Google Scholar] [CrossRef]
- Raff, H.; Roarty, T.P. Renin, ACTH, and aldosterone during acute hypercapnia and hypoxia in conscious rats. Am. J. Physiol. 1988, 254, R431–R435. [Google Scholar] [CrossRef]
- Zhang, J.; Tian, L.; Guo, L. Changes of aldosterone levels in patients with type 2 diabetes complicated by moderate to severe obstructive sleep apnea-hypopnea syndrome before and after treatment with continuous positive airway pressure. J. Int. Med. Res. 2019, 47, 4723–4733. [Google Scholar] [CrossRef]
- Svatikova, A.; Olson, L.J.; Wolk, R.; Phillips, B.G.; Adachi, T.; Schwartz, G.L.; Somers, V.K. Obstructive sleep apnea and aldosterone. Sleep 2009, 32, 1589–1592. [Google Scholar] [CrossRef]
- Wolk, R.; Shamsuzzaman, A.S.; Somers, V.K. Obesity, sleep apnea, and hypertension. Hypertension 2003, 42, 1067–1074. [Google Scholar] [CrossRef]
- Vecchiola, A.; Lagos, C.F.; Carvajal, C.A.; Baudrand, R.; Fardella, C.E. Aldosterone Production and Signaling Dysregulation in Obesity. Curr. Hypertens. Rep. 2016, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Goodfriend, T.L.; Egan, B.M.; Kelley, D.E. Plasma aldosterone, plasma lipoproteins, obesity and insulin resistance in humans. Prostaglandins. Leukot. Essent. Fatty Acids 1999, 60, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga, C.C.; Gaddam, K.K.; Ahmed, M.I.; Pimenta, E.; Thomas, S.J.; Harding, S.M.; Oparil, S.; Cofield, S.S.; Calhoun, D.A. Severity of obstructive sleep apnea is related to aldosterone status in subjects with resistant hypertension. J. Clin. Sleep Med. 2010, 6, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Pratt-Ubunama, M.N.; Nishizaka, M.K.; Boedefeld, R.L.; Cofield, S.S.; Harding, S.M.; Calhoun, D.A. Plasma aldosterone is related to severity of obstructive sleep apnea in subjects with resistant hypertension. Chest 2007, 131, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Dudenbostel, T.; Calhoun, D.A. Resistant hypertension, obstructive sleep apnoea and aldosterone. J. Hum. Hypertens. 2012, 26, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Yu, Y.; Kang, Y.M.; Wei, S.G.; Felder, R.B. Aldosterone acts centrally to increase brain renin-angiotensin system activity and oxidative stress in normal rats. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1067–H1074. [Google Scholar] [CrossRef] [PubMed]
- Krasinska, B.; Cofta, S.; Szczepaniak-Chichel, L.; Rzymski, P.; Trafas, T.; Paluszkiewicz, L.; Tykarski, A.; Krasinski, Z. The Effects of Eplerenone on the Circadian Blood Pressure Pattern and Left Ventricular Hypertrophy in Patients with Obstructive Sleep Apnea and Resistant Hypertension-A Randomized, Controlled Trial. J. Clin. Med. 2019, 8, 1671. [Google Scholar] [CrossRef] [PubMed]
- Krasinska, B.; Miazga, A.; Cofta, S.; Szczepaniak-Chichel, L.; Trafas, T.; Krasinski, Z.; Pawlaczyk-Gabriel, K.; Tykarski, A. Effect of eplerenone on the severity of obstructive sleep apnea and arterial stiffness in patients with resistant arterial hypertension. Pol. Arch. Med. Wewn 2016, 126, 330–339. [Google Scholar] [CrossRef]
- Rodenstein, D.O.; D’Odemont, J.P.; Pieters, T.; Aubert-Tulkens, G. Diurnal and nocturnal diuresis and natriuresis in obstructive sleep apnea. Effects of nasal continuous positive airway pressure therapy. Am. Rev. Respir. Dis. 1992, 145, 1367–1371. [Google Scholar] [CrossRef]
- Pedrosa, R.P.; Drager, L.F.; de Paula, L.K.G.; Amaro, A.C.S.; Bortolotto, L.A.; Lorenzi-Filho, G. Effects of OSA treatment on BP in patients with resistant hypertension: A randomized trial. Chest 2013, 144, 1487–1494. [Google Scholar] [CrossRef]
- Moller, D.S.; Lind, P.; Strunge, B.; Pedersen, E.B. Abnormal vasoactive hormones and 24-hour blood pressure in obstructive sleep apnea. Am. J. Hypertens. 2003, 16, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Meston, N.; Davies, R.J.; Mullins, R.; Jenkinson, C.; Wass, J.A.; Stradling, J.R. Endocrine effects of nasal continuous positive airway pressure in male patients with obstructive sleep apnoea. J. Intern. Med. 2003, 254, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Joyeux-Faure, M.; Baguet, J.P.; Barone-Rochette, G.; Faure, P.; Sosner, P.; Mounier-Vehier, C.; Levy, P.; Tamisier, R.; Pepin, J.L. Continuous Positive Airway Pressure Reduces Night-Time Blood Pressure and Heart Rate in Patients With Obstructive Sleep Apnea and Resistant Hypertension: The RHOOSAS Randomized Controlled Trial. Front. Neurol. 2018, 9, 318. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).