


In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part III


Abstract
1. Introduction
2. Results
2.1. PBPK Models (with and without PK Interaction)
2.2. PD (TGI) Models
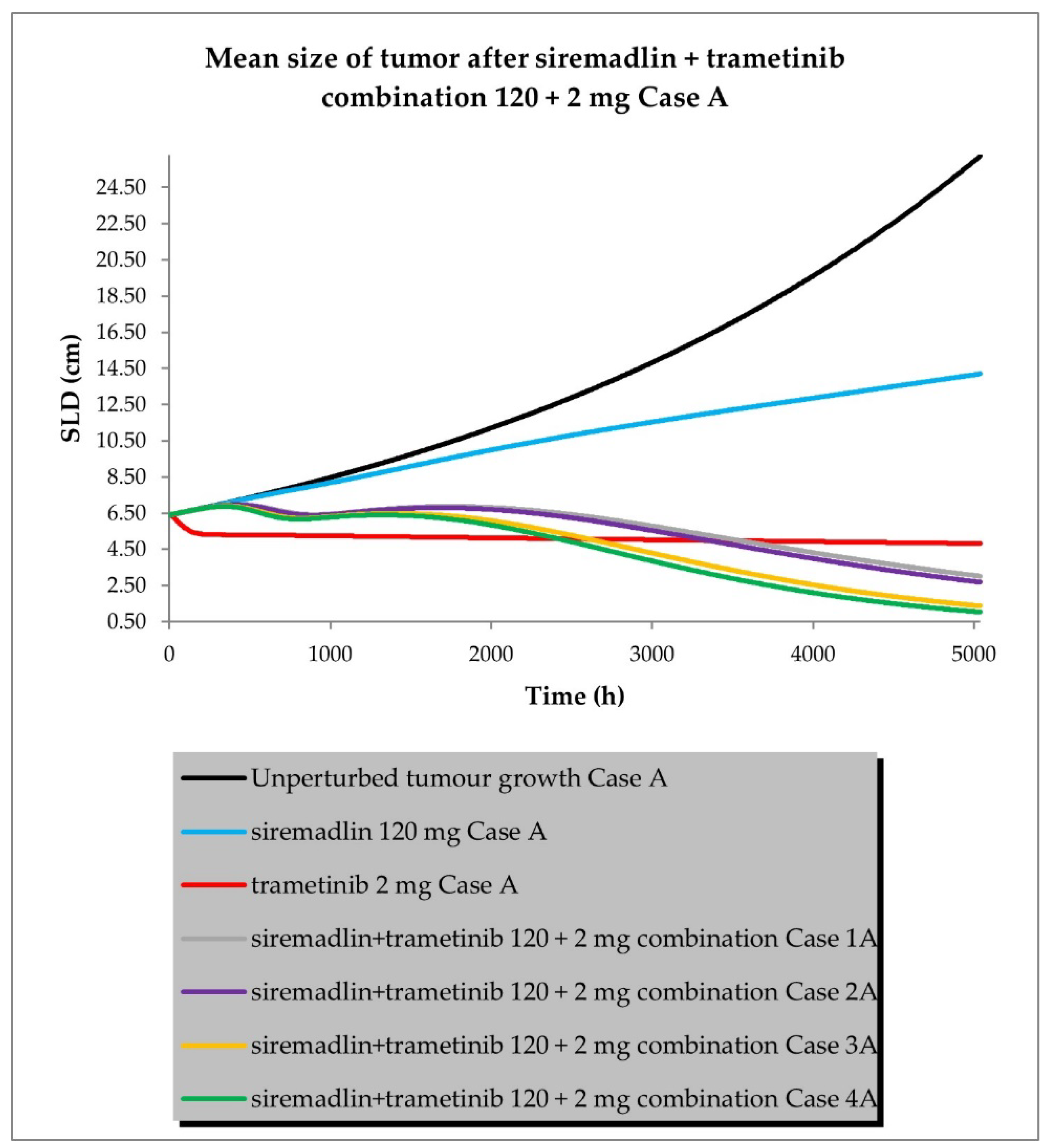
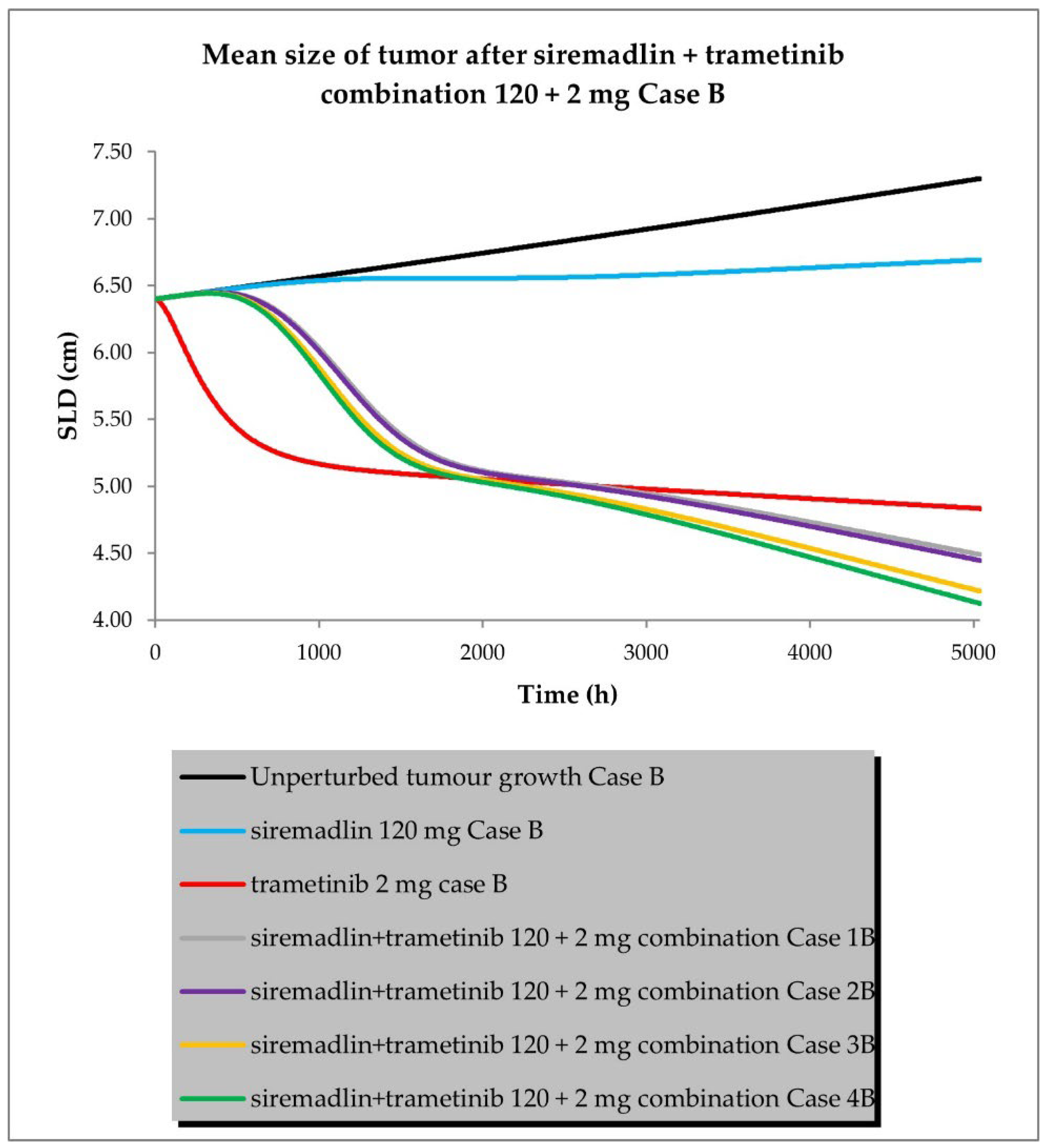
2.3. PBPK/PD Simulations for Drug Combination
- Case A assumed high tumour growth (kgh), high initial tumour size (SLD0), and a high fraction of sensitive cells.
- Case B assumed a similar approach to that described above, but assumed low tumour growth (kgh).
- Case C assumed high tumour growth (kgh), high initial tumour size (SLD0), and a low fraction of sensitive cells.
- Case D assumed high tumour growth (kgh), low initial tumour size (SLD0), and a high fraction of sensitive cells.
- Scenario 1—without PK and PD drug interactions
- Scenario 2—without PK but with PD drug interactions
- Scenario 3—with PK and without PD drug interactions
- Scenario 4—with PK and PD drug interactions
3. Discussion
4. Materials and Methods
4.1. Clinical Studies Used
4.2. Software
4.3. Statistical Methods
4.4. Resimulation of Clinical PK and PD Data for Siremadlin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose (mg) | Regimen | Dosing Schedule | No. of Patients * | No. of Trials | Notes |
|---|---|---|---|---|---|
| 1 | 2A | qdx14 in 28-day cycle | 1 | 10 | |
| 2 | 2A | qdx14 in 28-day cycle | 2 | 10 | |
| 4 | 2A | qdx14 in 28-day cycle | 4 | 10 | |
| 7.5 | 2A | qdx14 in 28-day cycle | 4 | 10 | |
| 15 | 2A | qdx14 in 28-day cycle | 4 | 10 | |
| 20 | 2A | qdx14 in 28-day cycle | 5 | 10 | |
| 15 | 2C | qdx7 in 28-day cycle | 8 | 10 | |
| 20 | 2C | qdx7 in 28-day cycle | 6 | 10 | |
| 25 | 2C | qdx7 in 28-day cycle | 5 | 10 | |
| 12.5 | 1A | qdx1 in 21-day cycle | 1 | 10 | |
| 25 | 1A | qdx1 in 21-day cycle | 1 | 10 | |
| 50 | 1A | qdx1 in 21-day cycle | 4 | 10 | |
| 100 | 1A | qdx1 in 21-day cycle | 4 | 10 | |
| 200 | 1A | qdx1 in 21-day cycle | 5 | 10 | |
| 250 | 1A | qdx1 in 21-day cycle | 9 | 10 | Including patients from eltrombopag group (n = 3) |
| 350 | 1A | qdx1 in 21-day cycle | 5 | 10 | |
| 120 | 1B | qwx2 (day 1/8) in 28-day cycle | 29 | 10 | |
| 150 | 1B | qwx2 (day 1/8) in 28-day cycle | 15 | 10 | Including patients from eltrombopag group (n = 7) |
| 200 | 1B | qwx2 (day 1/8) in 28-day cycle | 3 | 10 |
4.5. Physiologically Based Pharmacokinetic Models
4.5.1. General PBPK Modelling Strategy
4.5.2. Virtual Population Characteristics (System Data)/Patient Population
4.5.3. PBPK Model Verification
4.6. Pharmacodynamic Modelling
4.6.1. General PD (TGI) Modelling Strategy
4.6.2. PD (TGI) Model Development and Verification
4.6.3. Tumour Size Simulation for the Drug Combination
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yadav, J.; El Hassani, M.; Sodhi, J.; Lauschke, V.M.; Hartman, J.H.; Russell, L.E. Recent Developments in in Vitro and in Vivo Models for Improved Translation of Preclinical Pharmacokinetics and Pharmacodynamics Data. Drug Metab. Rev. 2021, 53, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Jamei, M. Recent Advances in Development and Application of Physiologically-Based Pharmacokinetic (PBPK) Models: A Transition from Academic Curiosity to Regulatory Acceptance. Curr. Pharmacol. Rep. 2016, 2, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Lu, C. PBPK Modeling and Simulation in Drug Research and Development. Acta Pharm. Sin. B 2016, 6, 430–440. [Google Scholar] [CrossRef]
- Jones, H.M.; Dickins, M.; Youdim, K.; Gosset, J.R.; Attkins, N.J.; Hay, T.L.; Gurrell, I.K.; Logan, Y.R.; Bungay, P.J.; Jones, B.C.; et al. Application of PBPK Modelling in Drug Discovery and Development at Pfizer. Xenobiotica 2012, 42, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; He, Q.; Zhang, S.; Li, M.; Tang, Z.; Zhu, X.; Jiao, Z.; Cai, W.; Xiang, X. Application of Physiologically Based Pharmacokinetic Modeling in Preclinical Studies: A Feasible Strategy to Practice the Principles of 3Rs. Front. Pharmacol. 2022, 13, 895556. [Google Scholar] [CrossRef] [PubMed]
- Mattes, W.B. In Vitro to In Vivo Translation. Curr. Opin. Toxicol. 2020, 23–24, 114–118. [Google Scholar] [CrossRef]
- Ireson, C.R.; Alavijeh, M.S.; Palmer, A.M.; Fowler, E.R.; Jones, H.J. The Role of Mouse Tumour Models in the Discovery and Development of Anticancer Drugs. Br. J. Cancer 2019, 121, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, P.S.; Sterk, P.J. Translation of In Vitro Findings to Patients with Asthma: A Timely and Compelling Challenge. Eur. Respir. J. 2019, 54, 1901759. [Google Scholar] [CrossRef]
- Jones, H.; Rowland-Yeo, K. Basic Concepts in Physiologically Based Pharmacokinetic Modeling in Drug Discovery and Development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e63. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhu, J.; Jiang, M.; Sang, L.; Hao, K.; He, H. The Combination of Cell Cultured Technology and in Silico Model to Inform the Drug Development. Pharmaceutics 2021, 13, 704. [Google Scholar] [CrossRef]
- Imaoka, T.; Huang, W.; Shum, S.; Hailey, D.W.; Chang, S.-Y.; Chapron, A.; Yeung, C.K.; Himmelfarb, J.; Isoherranen, N.; Kelly, E.J. Bridging the Gap between in Silico and in Vivo by Modeling Opioid Disposition in a Kidney Proximal Tubule Microphysiological System. Sci. Rep. 2021, 11, 21356. [Google Scholar] [CrossRef] [PubMed]
- Susanto, B.O.; Wicha, S.G.; Hu, Y.; Coates, A.R.M.; Simonsson, U.S.H. Translational Model-Informed Approach for Selection of Tuberculosis Drug Combination Regimens in Early Clinical Development. Clin. Pharmacol. Ther. 2020, 108, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, B.; Lezeau, J.; Werkman, C.; Jacobs, B.; Ait-Oudhia, S. In Vitro to Clinical Translation of Combinatorial Effects of Doxorubicin and Abemaciclib in Rb-Positive Triple Negative Breast Cancer: A Systems-Based Pharmacokinetic/Pharmacodynamic Modeling Approach. Breast Cancer (Dove Med Press). 2021, 13, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Cardilin, T.; Almquist, J.; Jirstrand, M.; Sostelly, A.; Amendt, C.; El Bawab, S.; Gabrielsson, J. Tumor Static Concentration Curves in Combination Therapy. AAPS J. 2017, 19, 456–467. [Google Scholar] [CrossRef]
- Cardilin, T.; Almquist, J.; Jirstrand, M.; Gabrielsson, J. Evaluation and Translation of Combination Therapies in Oncology—A Quantitative Approach. Eur. J. Pharmacol. 2018, 834, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Baaz, M.; Cardilin, T.; Lignet, F.; Jirstrand, M. Optimized Scaling of Translational Factors in Oncology: From Xenografts to RECIST. Cancer Chemother. Pharmacol. 2022, 90, 239–250. [Google Scholar] [CrossRef]
- Witkowski, J.; Polak, S.; Rogulski, Z.; Pawelec, D. In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part I. Int. J. Mol. Sci. 2022, 23, 12984. [Google Scholar] [CrossRef]
- Witkowski, J.; Polak, S.; Rogulski, Z.; Pawelec, D. In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part II. Int. J. Mol. Sci. 2022, 23, 11939. [Google Scholar] [CrossRef]
- Wu, C.-E.; Koay, T.S.; Esfandiari, A.; Ho, Y.-H.; Lovat, P.; Lunec, J. ATM Dependent DUSP6 Modulation of P53 Involved in Synergistic Targeting of MAPK and P53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers 2018, 11, 3. [Google Scholar] [CrossRef]
- Caenepeel, S.; Canon, J.; Hughes, P.; Oliner, J.D.; Rickles, R.J.; Saiki, A.Y. Combination Therapy Including an MDM2 Inhibitor and One or More Additional Pharmaceutically Active Agents for the Treatment of Cancers. U.S. Patent 10,881,648, 5 January 2021. [Google Scholar]
- Decaudin, D.; Frisch Dit Leitz, E.; Nemati, F.; Tarin, M.; Naguez, A.; Zerara, M.; Marande, B.; Vivet-Noguer, R.; Halilovic, E.; Fabre, C.; et al. Preclinical Evaluation of Drug Combinations Identifies Co-Inhibition of Bcl-2/XL/W and MDM2 as a Potential Therapy in Uveal Melanoma. Eur. J. Cancer 2020, 126, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.-M.; VanBrocklin, M.; McWilliams, M.J.; Leppla, S.H.; Duesbery, N.S.; Woude, G.F.V. Apoptosis and Melanogenesis in Human Melanoma Cells Induced by Anthrax Lethal Factor Inactivation of Mitogen-Activated Protein Kinase Kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Alesiani, D.; Cicconi, R.; Mattei, M.; Bei, R.; Canini, A. Inhibition of Mek 1/2 Kinase Activity and Stimulation of Melanogenesis by 5,7-Dimethoxycoumarin Treatment of Melanoma Cells. Int. J. Oncol. 2009, 34, 1727–1735. [Google Scholar] [CrossRef]
- Saud, A.; Sagineedu, S.R.; Ng, H.-S.; Stanslas, J.; Lim, J.C.W. Melanoma Metastasis: What Role Does Melanin Play? (Review). Oncol. Rep. 2022, 48, 217. [Google Scholar] [CrossRef]
- Sarna, M.; Krzykawska-Serda, M.; Jakubowska, M.; Zadlo, A.; Urbanska, K. Melanin Presence Inhibits Melanoma Cell Spread in Mice in a Unique Mechanical Fashion. Sci. Rep. 2019, 9, 9280. [Google Scholar] [CrossRef]
- Almansour, H.; Afat, S.; Serna-Higuita, L.M.; Amaral, T.; Schraag, A.; Peisen, F.; Brendlin, A.; Seith, F.; Klumpp, B.; Eigentler, T.K.; et al. Early Tumor Size Reduction of at Least 10% at the First Follow-Up Computed Tomography Can Predict Survival in the Setting of Advanced Melanoma and Immunotherapy. Acad. Radiol. 2022, 29, 514–522. [Google Scholar] [CrossRef]
- Jain, R.K.; Lee, J.J.; Ng, C.; Hong, D.; Gong, J.; Naing, A.; Wheler, J.; Kurzrock, R. Change in Tumor Size by RECIST Correlates Linearly with Overall Survival in Phase I Oncology Studies. J. Clin. Oncol. 2012, 30, 2684–2690. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, N.; Jullion, A.; Ferretti, S.; Fabre, C.; Meille, C. Translational Modeling of Anticancer Efficacy to Predict Clinical Outcomes in a First-in-Human Phase 1 Study of MDM2 Inhibitor HDM201. AAPS J. 2021, 23, 28. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.Y.K.; Morris, M.J.; Pirhalla, J.L.; Bauman, J.W.; Pendry, C.B.; Orford, K.W.; Morrison, R.A.; Cox, D.S. Trametinib, a First-in-Class Oral MEK Inhibitor Mass Balance Study with Limited Enrollment of Two Male Subjects with Advanced Cancers. Xenobiotica 2014, 44, 352–368. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, D.; Kassir, N.; Chiu, J.; Mouksassi, M.-S.; Leonowens, C.; Cox, D.; DeMarini, D.J.; Gardner, O.; Crist, W.; Patel, K. Population Pharmacokinetics and Exposure-Response of Trametinib, a MEK Inhibitor, in Patients with BRAF V600 Mutation-Positive Melanoma. Cancer Chemother. Pharmacol. 2016, 77, 807–817. [Google Scholar] [CrossRef]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, Pharmacokinetic, Pharmacodynamic, and Efficacy Data for the Oral MEK Inhibitor Trametinib: A Phase 1 Dose-Escalation Trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Elassaiss-Schaap, J.; Heisterkamp, S. Variability as Constant Coefficient of Variation: Can We Right Two Decades in Error? Available online: https://www.page-meeting.org/pdf_assets/4964-Elassaiss-Schaap%20-%20Equations%20variability%20reporting%20PK-PD%20-%20Final.pdf (accessed on 15 December 2022).
- Mistry, H.B.; Orrell, D.; Eftimie, R. Model Based Analysis of the Heterogeneity in the Tumour Size Dynamics Differentiates Vemurafenib, Dabrafenib and Trametinib in Metastatic Melanoma. Cancer Chemother. Pharmacol. 2018, 81, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DeAngelo, D.J.; Chromik, J.; Chatterjee, M.; Bauer, S.; Lin, C.-C.; Suarez, C.; de Vos, F.; Steeghs, N.; Cassier, P.A.; et al. Results from a First-in-Human Phase I Study of Siremadlin (HDM201) in Patients with Advanced Wild-Type TP53 Solid Tumors and Acute Leukemia. Clin. Cancer Res. 2022, 28, 870–881. [Google Scholar] [CrossRef]
- Pharmaceuticals and Medical Devices Agency (PMDA). Mekinist Initial Approval. Available online: https://www.pmda.go.jp/files/000233741.pdf (accessed on 15 December 2022).
- Paine, S.W.; Ménochet, K.; Denton, R.; McGinnity, D.F.; Riley, R.J. Prediction of Human Renal Clearance from Preclinical Species for a Diverse Set of Drugs That Exhibit Both Active Secretion and Net Reabsorption. Drug Metab. Dispos. 2011, 39, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G.; et al. Dose and Schedule Determine Distinct Molecular Mechanisms Underlying the Efficacy of the P53-MDM2 Inhibitor HDM201. Cancer Res. 2018, 78, 6257–6267. [Google Scholar] [CrossRef] [PubMed]
- Leonowens, C.; Pendry, C.; Bauman, J.; Young, G.C.; Ho, M.; Henriquez, F.; Fang, L.; Morrison, R.A.; Orford, K.; Ouellet, D. Concomitant Oral and Intravenous Pharmacokinetics of Trametinib, a MEK Inhibitor, in Subjects with Solid Tumours. Br. J. Clin. Pharmacol. 2014, 78, 524–532. [Google Scholar] [CrossRef]
- Tang, C.; Prueksaritanont, T. Use of In Vivo Animal Models to Assess Pharmacokinetic Drug-Drug Interactions. Pharm. Res. 2010, 27, 1772–1787. [Google Scholar] [CrossRef]
- Jaiswal, S.; Sharma, A.; Shukla, M.; Vaghasiya, K.; Rangaraj, N.; Lal, J. Novel Pre-Clinical Methodologies for Pharmacokinetic Drug–Drug Interaction Studies: Spotlight on “Humanized” Animal Models. Drug Metab. Rev. 2014, 46, 475–493. [Google Scholar] [CrossRef]
- Sjöberg, Å.; Lutz, M.; Tannergren, C.; Wingolf, C.; Borde, A.; Ungell, A.-L. Comprehensive Study on Regional Human Intestinal Permeability and Prediction of Fraction Absorbed of Drugs Using the Ussing Chamber Technique. Eur. J. Pharm. Sci. 2013, 48, 166–180. [Google Scholar] [CrossRef]
- Li, M.; de Graaf, I.A.M.; Groothuis, G.M.M. Precision-Cut Intestinal Slices: Alternative Model for Drug Transport, Metabolism, and Toxicology Research. Expert Opin. Drug Metab. Toxicol. 2016, 12, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Honda, K.; Fujita, K.; Miyachi, Y.; Isoda, K.; Misaka, K.; Suga, Y.; Kato, S.; Tsuchiya, H.; Kato, Y.; et al. Effect of Coadministration of Rifampicin on the Pharmacokinetics of Linezolid: Clinical and Animal Studies. J. Pharm. Health Care Sci. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Howard, G.R.; Johnson, K.E.; Rodriguez Ayala, A.; Yankeelov, T.E.; Brock, A. A Multi-State Model of Chemoresistance to Characterize Phenotypic Dynamics in Breast Cancer. Sci. Rep. 2018, 8, 12058. [Google Scholar] [CrossRef] [PubMed]
- Yoon, N.; Vander Velde, R.; Marusyk, A.; Scott, J.G. Optimal Therapy Scheduling Based on a Pair of Collaterally Sensitive Drugs. Bull. Math. Biol. 2018, 80, 1776–1809. [Google Scholar] [CrossRef]
- Liu, W.; Dowling, J.P.; Murray, W.K.; McArthur, G.A.; Thompson, J.F.; Wolfe, R.; Kelly, J.W. Rate of Growth in Melanomas: Characteristics and Associations of Rapidly Growing Melanomas. Arch. Dermatol. 2006, 142, 1551–1558. [Google Scholar] [CrossRef]
- Chatterjee, M.; Elassaiss-Schaap, J.; Lindauer, A.; Turner, D.; Sostelly, A.; Freshwater, T.; Mayawala, K.; Ahamadi, M.; Stone, J.; de Greef, R.; et al. Population Pharmacokinetic/Pharmacodynamic Modeling of Tumor Size Dynamics in Pembrolizumab-Treated Advanced Melanoma. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 29–39. [Google Scholar] [CrossRef]
- Settleman, J.; Neto, J.M.F.; Bernards, R. Thinking Differently about Cancer Treatment Regimens. Cancer Discov. 2021, 11, 1016–1023. [Google Scholar] [CrossRef]
- Mould, D.; Walz, A.-C.; Lave, T.; Gibbs, J.; Frame, B. Developing Exposure/Response Models for Anticancer Drug Treatment: Special Considerations. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, e00016. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, X.; Chen, G.; Du, J. Drug Resistance in Colorectal Cancer: From Mechanism to Clinic. Cancers 2022, 14, 2928. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Hauschild, A.; Larkin, J.; Ribas, A.; Dréno, B.; Flaherty, K.T.; Ascierto, P.A.; Lewis, K.D.; McKenna, E.; Zhu, Q.; Mun, Y.; et al. Modeled Prognostic Subgroups for Survival and Treatment Outcomes in BRAF V600–Mutated Metastatic Melanoma. JAMA Oncol. 2018, 4, 1382–1388. [Google Scholar] [CrossRef]
- Saiag, P.; Robert, C.; Grob, J.-J.; Mortier, L.; Dereure, O.; Lebbe, C.; Mansard, S.; Grange, F.; Neidhardt, E.-M.; Lesimple, T.; et al. Efficacy, Safety and Factors Associated with Disease Progression in Patients with Unresectable (Stage III) or Distant Metastatic (Stage IV) BRAF V600-Mutant Melanoma: An Open Label, Non-Randomized, Phase IIIb Study of Trametinib in Combination with Dabrafenib. Eur. J. Cancer 2021, 154, 57–65. [Google Scholar] [CrossRef]
- Clinical Trials Results (Study 113220/NCT01072175): Novartis Clinical Trial Results Database. Available online: https://www.novctrd.com/ctrdweb/trialresult/trialresults/pdf?trialResultId=17378 (accessed on 15 December 2022).
- Clinical Trials Results (Study 115306/NCT01584648) Novartis Clinical Trial Results Database. Available online: https://www.novctrd.com/ctrdweb/trialresult/trialresults/pdf?trialResultId=17627 (accessed on 15 December 2022).
- Clinical Trials Results (Study BRF117277/NCT02039947) Novartis Clinical Trial Results Database. Available online: https://www.novctrd.com/ctrdweb/trialresult/trialresults/pdf?trialResultId=17326 (accessed on 15 December 2022).
- Long, G.V.; Eroglu, Z.; Infante, J.; Patel, S.; Daud, A.; Johnson, D.B.; Gonzalez, R.; Kefford, R.; Hamid, O.; Schuchter, L.; et al. Long-Term Outcomes in Patients With BRAF V600-Mutant Metastatic Melanoma Who Received Dabrafenib Combined With Trametinib. J. Clin. Oncol. 2018, 36, 667–673. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Pan, B.; Wei, X.; Xu, X. Patient-Derived Xenograft Models in Hepatopancreatobiliary Cancer. Cancer Cell Int. 2022, 22, 41. [Google Scholar] [CrossRef] [PubMed]
- Blomme, A.; Van Simaeys, G.; Doumont, G.; Costanza, B.; Bellier, J.; Otaka, Y.; Sherer, F.; Lovinfosse, P.; Boutry, S.; Palacios, A.P.; et al. Murine Stroma Adopts a Human-like Metabolic Phenotype in the PDX Model of Colorectal Cancer and Liver Metastases. Oncogene 2018, 37, 1237–1250. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef]
- Hirata, E.; Sahai, E. Tumor Microenvironment and Differential Responses to Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026781. [Google Scholar] [CrossRef]
- Ni, Y.; Zhou, X.; Yang, J.; Shi, H.; Li, H.; Zhao, X.; Ma, X. The Role of Tumor-Stroma Interactions in Drug Resistance Within Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 637675. [Google Scholar] [CrossRef]
- McMillin, D.W.; Negri, J.M.; Mitsiades, C.S. The Role of Tumour–Stromal Interactions in Modifying Drug Response: Challenges and Opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [Google Scholar] [CrossRef]
- Wang, H.Q.; Mulford, I.J.; Sharp, F.; Liang, J.; Kurtulus, S.; Trabucco, G.; Quinn, D.S.; Longmire, T.A.; Patel, N.; Patil, R.; et al. Inhibition of MDM2 Promotes Antitumor Responses in P53 Wild-Type Cancer Cells through Their Interaction with the Immune and Stromal Microenvironment. Cancer Res. 2021, 81, 3079–3091. [Google Scholar] [CrossRef]
- Fang, D.D.; Tang, Q.; Kong, Y.; Wang, Q.; Gu, J.; Fang, X.; Zou, P.; Rong, T.; Wang, J.; Yang, D.; et al. MDM2 Inhibitor APG-115 Synergizes with PD-1 Blockade through Enhancing Antitumor Immunity in the Tumor Microenvironment. J. Immunother. Cancer 2019, 7, 327. [Google Scholar] [CrossRef]
- Zhou, J.; Kryczek, I.; Li, S.; Li, X.; Aguilar, A.; Wei, S.; Grove, S.; Vatan, L.; Yu, J.; Yan, Y.; et al. The Ubiquitin Ligase MDM2 Sustains STAT5 Stability to Control T Cell-Mediated Antitumor Immunity. Nat. Immunol. 2021, 22, 460–470. [Google Scholar] [CrossRef]
- Clinical Trials Results (Study 114267/NCT01245062) GlaxoSmithKline Clinical Trial Results Database. Available online: https://www.gsk-studyregister.com/en/trial-details/?id=114267 (accessed on 15 December 2022).
- Clinical Trials Results (Study CTMT212AUS55/NCT05611229) Novartis Clinical Trial Results Database. Available online: https://www.novctrd.com/ctrdweb/trialresult/trialresults/pdf?trialResultId=17991 (accessed on 15 December 2022).
- Ristl, R. Sample Size Calculator. Available online: https://homepage.univie.ac.at/robin.ristl/samplesize.php?test=fishertest (accessed on 24 November 2022).
- Clinical Trial Results (Study CHDM201X2101/NCT02143635) Novartis Clinical Trial Results Database. Available online: https://www.novctrd.com/ctrdweb/trialresult/trialresults/pdf?trialResultId=17828 (accessed on 15 December 2022).
- Tylutki, Z.; Polak, S.; Wiśniowska, B. Top-down, Bottom-up and Middle-out Strategies for Drug Cardiac Safety Assessment via Modeling and Simulations. Curr. Pharmacol. Rep. 2016, 2, 171–177. [Google Scholar] [CrossRef]
- Kallinowski, F.; Schlenger, K.H.; Runkel, S.; Kloes, M.; Stohrer, M.; Okunieff, P.; Vaupel, P. Blood Flow, Metabolism, Cellular Microenvironment, and Growth Rate of Human Tumor Xenografts. Cancer Res. 1989, 49, 3759–3764. [Google Scholar] [PubMed]
- Hofmann, F. Small Molecule HDM201 Inhibitor HDM201. Presented at the AACR Annual Meeting 2016, New Orleans, LA, USA, 16–20 April 2016. [Google Scholar]
- Lei, Y.; Zhenglin, Y.; Heng, L. MDM2 Inhibitors. U.S. Patent 11,339,171, 24 May 2022. [Google Scholar]
- Food and Drug Administration (FDA). Trametinib Pharmacology Review. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204114Orig1s000PharmR.pdf (accessed on 15 December 2022).
- Benjaminsen, I.C.; Graff, B.A.; Brurberg, K.G.; Rofstad, E.K. Assessment of Tumor Blood Perfusion by High-Resolution Dynamic Contrast-Enhanced MRI: A Preclinical Study of Human Melanoma Xenografts. Magn. Reson. Med. 2004, 52, 269–276. [Google Scholar] [CrossRef]
- Graff, B.A.; Benjaminsen, I.C.; Melås, E.A.; Brurberg, K.G.; Rofstad, E.K. Changes in Intratumor Heterogeneity in Blood Perfusion in Intradermal Human Melanoma Xenografts during Tumor Growth Assessed by DCE-MRI. Magn. Reson. Imaging 2005, 23, 961–966. [Google Scholar] [CrossRef]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef]
- Tsakalozou, E.; Alam, K.; Babiskin, A.; Zhao, L. Physiologically-Based Pharmacokinetic Modeling to Support Determination of Bioequivalence for Dermatological Drug Products: Scientific and Regulatory Considerations. Clin. Pharmacol. Ther. 2022, 111, 1036–1049. [Google Scholar] [CrossRef]
- Khoshaein, N.; Ezuruike, U.; Hatley, O.; Gill, K.; Gardner, I. Performance Verification and Application of a Cancer Population for Use in Physiologically Based Pharmacokinetic Modelling. Available online: https://www.certara.com/app/uploads/2019/09/Khoshaein_2019_JRC_cancer.pdf (accessed on 2 December 2022).
- Hatley, O. Predicting Drug Exposure in Cancer Patients Using a PBPK Oncology Population. Available online: https://www.certara.com/blog/predicting-drug-exposure-in-cancer-patients-using-a-pbpk-oncology-population/ (accessed on 2 December 2022).
- Reig-López, J.; Maldonado, M.D.M.; Merino-Sanjuan, M.; Cruz-Collazo, A.M.; Ruiz-Calderón, J.F.; Mangas-Sanjuán, V.; Dharmawardhane, S.; Duconge, J. Physiologically-Based Pharmacokinetic/Pharmacodynamic Model of MBQ-167 to Predict Tumor Growth Inhibition in Mice. Pharmaceutics 2020, 12, 975. [Google Scholar] [CrossRef]
- Tejera-Vaquerizo, A.; Cañueto, J.; Toll, A.; Santos-Juanes, J.; Jaka, A.; Ferrandiz, C.; Sanmartín, O.; Ribero, S.; Moreno-Ramírez, D.; Almazán, F.; et al. Estimated Effect of COVID-19 Lockdown on Skin Tumor Size and Survival: An Exponential Growth Model. Actas Dermo-Sifiliográficas (Engl. Ed.) 2020, 111, 629–638. [Google Scholar] [CrossRef]
- Lobo, E.D.; Balthasar, J.P. Pharmacodynamic Modeling of Chemotherapeutic Effects: Application of a Transit Compartment Model to Characterize Methotrexate Effects in Vitro. AAPS J. 2002, 4, 212–222. [Google Scholar] [CrossRef] [PubMed]













| Dose (mg) | Representative AUC0-inf Predicted (nM × h) | Population AUC0-inf Predicted (nM × h) | AUC0-inf Observed (nM × h) | Representative AUC Predicted/ Observed | Population AUC Predicted/ Observed |
|---|---|---|---|---|---|
| 1 | 257.90 | 286.40 (44.8%) | 241.80 (-%) | 1.07 | 1.18 |
| 2 | 515.80 | 537.83 (42.5%) | 304.46 (31.5%) | 1.69 | 1.77 |
| 4 | 1031.60 | 1073.86 (36.5%) | 387.64 (22.0%) | 2.66 | 2.77 |
| 7.5 | 1934.26 | 2013.49 (36.5%) | 1076.86(49.8%) | 1.80 | 1.87 |
| 12.5 | 3223.76 | 3580.00 (44.8%) | 2670.28 (-%) | 1.21 | 1.34 |
| 15 | 3868.52 | 3816.39 (40.0%) | 2343.49 (71.0%) | 1.65 | 1.63 |
| 20 | 5158.02 | 5119.24 (36.9%) | 4122.18 (29.6%) | 1.25 | 1.24 |
| 25 | 6447.53 | 6399.05 (36.9%) | 4803.12 (28.3%) | 1.34 | 1.33 |
| 50 | 12,895.05 | 13,423.28 (36.5%) | 14,455.63 (25.6%) | 0.89 | 0.93 |
| 100 | 25,790.10 | 26,846.56 (36.5%) | 25,723.34 (58.5%) | 1.00 | 1.04 |
| 120 | 30,948.13 | 29,889.60 (38.8%) | 33,275.78 (62.7%) | 0.93 | 0.90 |
| 150 | 38,685.16 | 37,968.46 (41.8%) | 42,719.97 (43.2%) | 0.91 | 0.89 |
| 200 | 51,580.21 | 51,192.34 (36.9%) | 47,271.75 (56.2%) | 1.09 | 1.08 |
| 250 | 64,475.26 | 63,698.83 (43.2%) | 74,579.68 (71.2%) | 0.86 | 0.85 |
| 350 | 90,265.34 | 89,586.57 (36.9%) | 99,211.21 (34.4%) | 0.91 | 0.90 |
| Dose (mg) | Representative Cmax Predicted (nM) | Population Cmax Predicted (nM) | Cmax Observed (nM) | Representative Cmax Predicted/ Observed | Population Cmax Predicted/ Observed |
|---|---|---|---|---|---|
| 1 | 14.37 | 14.05 (33.6%) | 14.22 (-%) | 1.01 | 0.99 |
| 2 | 28.75 | 28.77 (27.5%) | 21.61 (23.7%) | 1.33 | 1.33 |
| 4 | 57.49 | 58.14 (27.0%) | 31.69 (22.8%) | 1.81 | 1.83 |
| 7.5 | 107.79 | 109.01 (27.0%) | 70.22 (43.9%) | 1.54 | 1.55 |
| 12.5 | 179.64 | 175.62 (33.6%) | 212.46 (-%) | 0.85 | 0.83 |
| 15 | 215.60 | 209.79 (26.2%) | 164.74 (56.9%) | 1.31 | 1.27 |
| 20 | 266.36 | 278.51 (28.6%) | 269.17 (20.3%) | 0.99 | 1.03 |
| 25 | 359.32 | 348.15 (28.6%) | 422.57 (27.5%) | 0.85 | 0.82 |
| 50 | 718.63 | 726.77 (27.0%) | 840.82 (13.0%) | 0.85 | 0.86 |
| 100 | 1437.19 | 1453.48 (27.0%) | 1194.25 (30.1%) | 1.20 | 1.22 |
| 120 | 1724.76 | 1596.25 (26.2) | 1871.59 (51.5%) | 0.92 | 0.85 |
| 150 | 2156.00 | 2093.20 (25.4%) | 2600.42 (27.8%) | 0.83 | 0.80 |
| 200 | 2874.55 | 2785.12 (28.6%) | 2104.39 (43.7%) | 1.37 | 1.32 |
| 250 | 3593.23 | 3482.48 (27.4%) | 3629.21 (69.5%) | 0.99 | 0.96 |
| 350 | 5030.18 | 4873.72 (28.6%) | 4066.91 (56.9%) | 1.24 | 1.20 |
| Drug | Trametinib | Trametinib | Trametinib | Trametinib |
|---|---|---|---|---|
| Representative AUC0–24 h (day 1) | 165.39 | - | - | - |
| Population AUC0–24 h (day 1) | 170.70 (29%) | - | 200.76 (20%) * | 109.60 (2%) ** |
| Representative AUC0–24 h (day 15) | 570.91 | - | - | - |
| Population AUC0–24 h (day 15) | 656.45 (50%) | 601.24 (22%) | - | 586.28 (16%) ** |
| Population AUC ratio (day 1) | - | - | 0.85 | 1.56 |
| Population AUC ratio (day 15) | - | 1.09 | - | 1.12 |
| Representative Cmax (day 1) | 18.08 | - | - | - |
| Population Cmax (day 1) | 18.50 (19%) | - | 24.44 (3%) * | 10.45 (0.3%) ** |
| Representative Cmax (day 15) | 36.44 | - | - | - |
| Population Cmax (day 15) | 40.93 (36%) | 36.07 (28%) | - | 31.67 (12%) ** |
| Population Cmax ratio (day 1) | - | - | 0.76 | 1.77 |
| Population Cmax ratio (day 15) | - | 1.13 | - | 1.29 |
| Source | Current study | Digitised from Infante et al. [32] | Digitised from Ho et al. [30] | Digitised from Ouellet et al. [31] |
| Drug | Siremadlin | Trametinib |
|---|---|---|
| Dose (mg) | 120 | 2 |
| AUC no PK DDI | 19,240.73 | 174.01 |
| AUC PK DDI | 21,858.34 | 133.15 |
| AUC ratio (PK DDI) | 1.1360 | 0.7652 |
| Cmax no PK DDI | 1724.96 | 19.02 |
| Cmax PK DDI | 2580.39 | 9.61 |
| Cmax ratio (PK DDI) | 1.4959 | 0.5051 |
| Trial | n | No. of Trials | Difference (Drug Combination vs. Trametinib) | %ORR (Mean from 10 Trials) | Statistical Significance (p Value) |
|---|---|---|---|---|---|
| Siremadlin Case A | 29 | 10 | 51.64% | 0.00% | <0.0001 |
| Trametinib Case A | 214 | 10 | 0.00% | 51.64% | <0.0001 |
| Case 1a | 243 | 10 | 26.51% | 78.15% | <0.0001 |
| Case 2a | 243 | 10 | 31.16% | 82.80% | <0.0001 |
| Case 3a | 243 | 10 | 45.81% | 97.45% | <0.0001 |
| Case 4a | 243 | 10 | 47.29% | 98.93% | <0.0001 |
| Siremadlin Case B | 29 | 10 | 25.84% | 0.00% | <0.0001 |
| Trametinib Case B | 214 | 10 | 0.00% | 25.84% | <0.0001 |
| Case 1b | 243 | 10 | 26.67% | 52.51% | <0.0001 |
| Case 2b | 243 | 10 | 30.70% | 56.54% | <0.0001 |
| Case 3b | 243 | 10 | 52.22% | 78.07% | <0.0001 |
| Case 4b | 243 | 10 | 59.14% | 84.98% | <0.0001 |
| Siremadlin Case C | 29 | 10 | 51.64% | 0.00% | <0.0001 |
| Trametinib Case C | 214 | 10 | 0.00% | 51.64% | <0.0001 |
| Case 1c | 243 | 10 | 14.41% | 66.05% | <0.0001 |
| Case 2c | 243 | 10 | 20.83% | 72.47% | <0.0001 |
| Case 3c | 243 | 10 | 42.52% | 94.16% | <0.0001 |
| Case 4c | 243 | 10 | 45.81% | 97.45% | <0.0001 |
| Siremadlin Case D | 29 | 10 | 51.64% | 0.00% | <0.0001 |
| Trametinib Case D | 214 | 10 | 0.00% | 51.64% | <0.0001 |
| Case 1d | 243 | 10 | 26.51% | 78.15% | <0.0001 |
| Case 2d | 243 | 10 | 31.16% | 82.80% | <0.0001 |
| Case 3d | 243 | 10 | 45.81% | 97.45% | <0.0001 |
| Case 4d | 243 | 10 | 47.29% | 98.93% | <0.0001 |
| Drug | Siremadlin | Trametinib | Trametinib | Trametinib | Trametinib |
|---|---|---|---|---|---|
| NCT number | NCT02143635 | NCT00687622 | NCT01387204 | NCT01245062 | NCT00687622/ NCT01037127/ NCT01245062 |
| Phase | 1/2 | 1/2 | 1 | 3 | 1/2/3 |
| Doses (mg) | 1–350 | 0.125–10 | 2 | 2 | 0.125–10/2/2 |
| Administration | Oral | Oral | Oral | Oral | Oral/oral/oral |
| n | 115 | 206 | 2 | 214 | 206/97/214 |
| Women (%) | 44 | 46 | 0 | 44 | 46/30/44 |
| Age (Years) | 18–80 | 19–92 | 54–66 | 23–85 | 19–92/23–79/ 23–85 |
| Dataset purpose | PK/PD training/ verification | PK training | PK training | PD training/ verification | PK verification |
| Reference | Guerreiro et al. [29] Stein et al. [36] Jeay et al. [39] | Infante et al. [32] | Ho et al. [30] | Flaherty et al. [35] Mistry et al. [34] | Ouellet et al. [31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witkowski, J.; Polak, S.; Pawelec, D.; Rogulski, Z. In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part III. Int. J. Mol. Sci. 2023, 24, 2239. https://doi.org/10.3390/ijms24032239
Witkowski J, Polak S, Pawelec D, Rogulski Z. In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part III. International Journal of Molecular Sciences. 2023; 24(3):2239. https://doi.org/10.3390/ijms24032239
Chicago/Turabian StyleWitkowski, Jakub, Sebastian Polak, Dariusz Pawelec, and Zbigniew Rogulski. 2023. "In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part III" International Journal of Molecular Sciences 24, no. 3: 2239. https://doi.org/10.3390/ijms24032239
APA StyleWitkowski, J., Polak, S., Pawelec, D., & Rogulski, Z. (2023). In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part III. International Journal of Molecular Sciences, 24(3), 2239. https://doi.org/10.3390/ijms24032239