
MD Investigation on the Interaction between Carbamazepine and Two CYP Isoforms, CYP3A4 and CYP3A5
Abstract
1. Introduction
2. Computational Details
2.1. Construction of the Simulation Systems
2.2. Molecular Dynamics Simulations
2.3. Analysis of Hydrogen Bonds
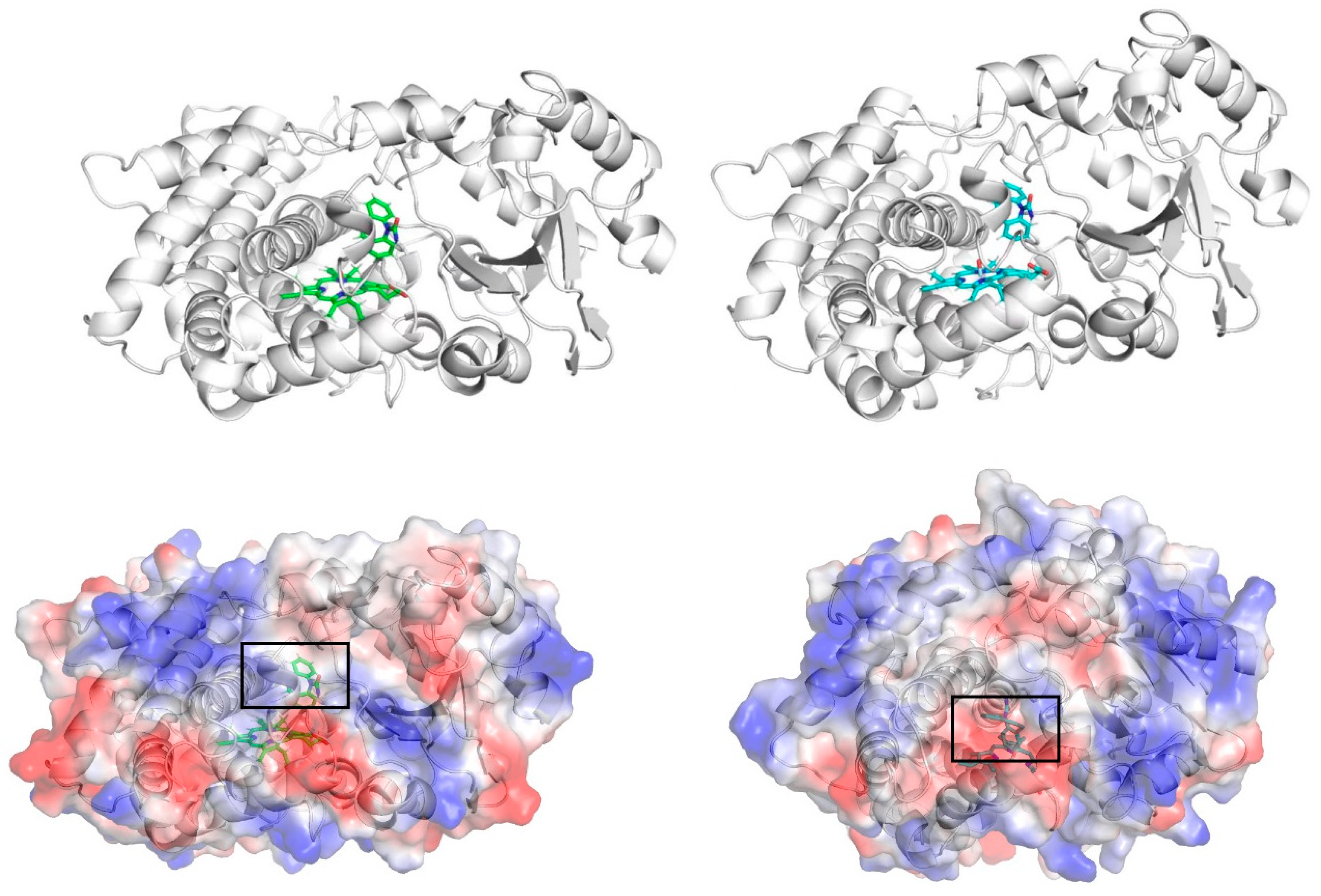
3. Results and Discussion
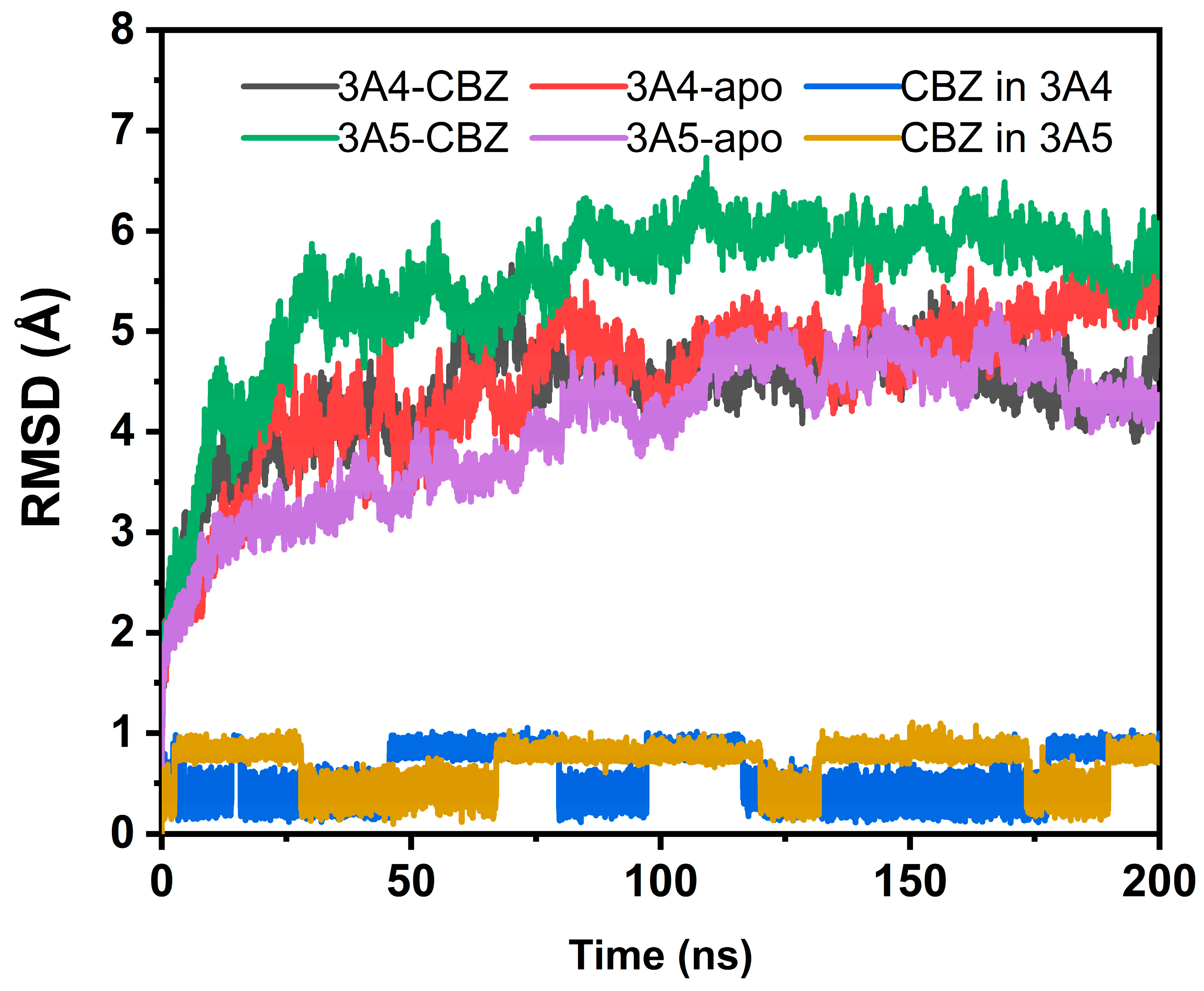
3.1. Stability of Simulation Systems
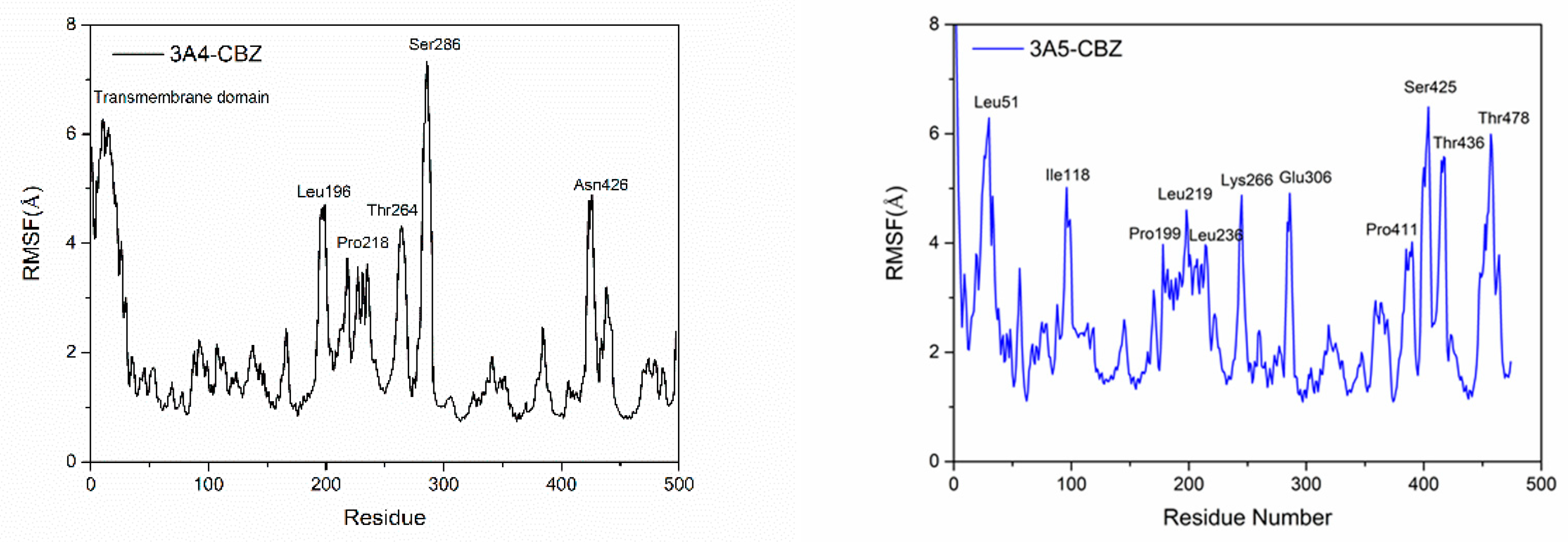
3.2. Flexibility of the Protein Residues in the Four Systems
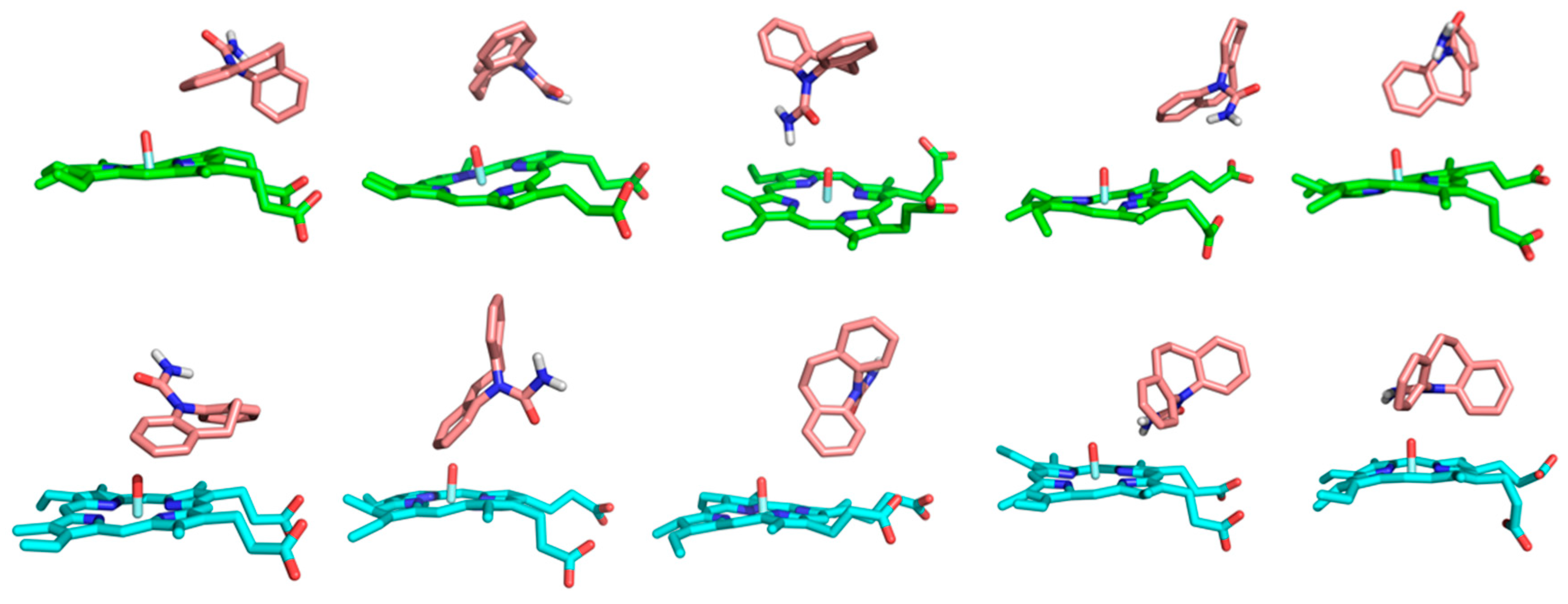
3.3. Dynamic Binding of CBZ to CYP3A4 and CYP3A5
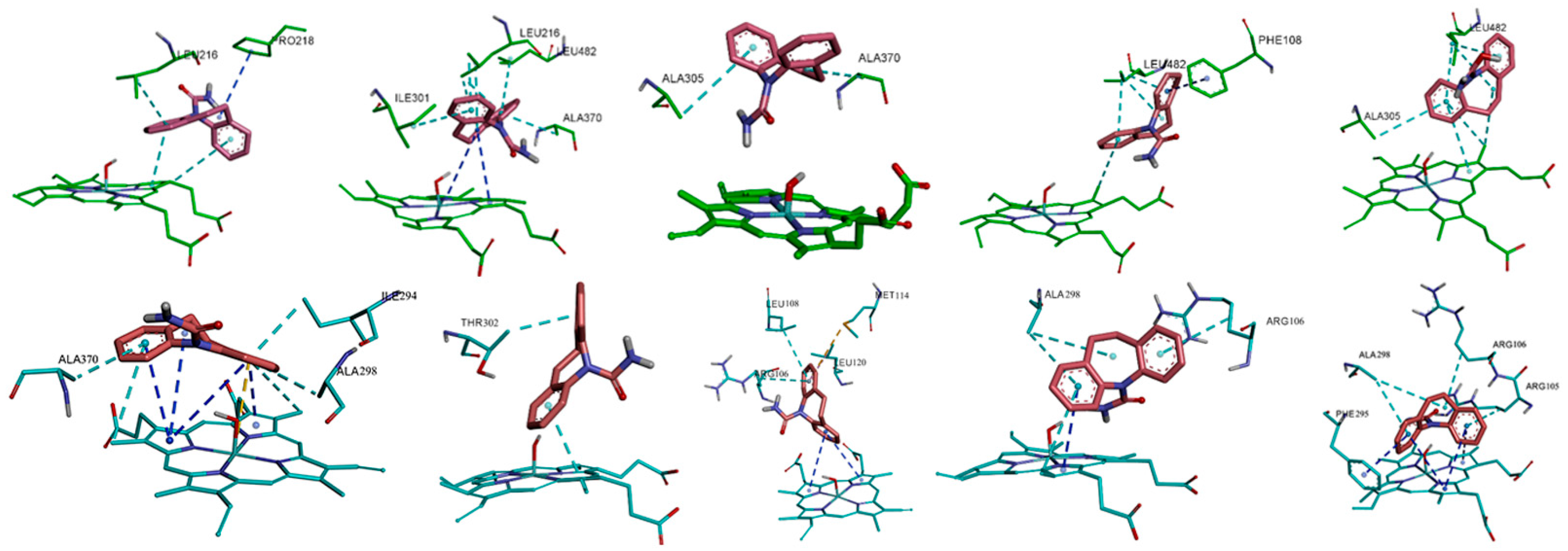
3.4. Hydrogen Bonds and Salt Bridges in the Complex Systems
3.5. Hydrophobic and π-π Interactions Involving the Substrate in the Two Complexes
3.6. Analysis of Electrostatic Surface Potential
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nevitt, S.J.; Marson, A.G.; Weston, J.; Smith, T. Sodium valproate versus phenytoin monotherapy for epilepsy: An individual participant data review. Cochrane Database Syst. Rev. 2018, 8, CD001769. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.L.M.; Goldberg, J.F.M. Antidepressant properties of anticonvulsant drugs for bipolar disorder. J. Clin. Psychopharm. 2003, 23, 182–192. [Google Scholar] [CrossRef]
- Hart, R.G.; Easton, J.D. Carbamazepine and hematological monitoring. Ann. Neurol. 1982, 11, 309–312. [Google Scholar] [CrossRef]
- Shear, N.H.; Spielberg, S.P. Anticonvulsant hypersensitivity syndrome. In vitro assessment of risk. J. Clin. Investig. 1988, 82, 1826–1832. [Google Scholar] [CrossRef]
- Leeder, J.S. Mechanisms of idiosyncratic hypersensitivity reactions to antiepileptic drugs. Epilepsia 1998, 39 (Suppl. 7), S8–S16. [Google Scholar] [CrossRef]
- Kalapos, M.P. Carbamazepine-provoked hepatotoxicity and possible aetiopathological role of glutathione in the events. Retrospective review of old data and call for new investigation. Advers. Drug React. Toxicol. Rev. 2002, 21, 123–141. [Google Scholar]
- Maldonado, N.R.; Tello, J.S.; Garcia-Baquero, E.R.; Castano, A.H. Anticonvulsant hypersensitivity syndrome with fatal outcome. Eur. J. Dermatol. 2002, 12, 503–505. [Google Scholar]
- Leeder, J.S.; Gaedigk, A.; Lu, X.; Cook, V.A. Epitope mapping studies with human anti-cytochrome P450 3A antibodies. Mol. Pharmacol. 1996, 49, 234–243. [Google Scholar]
- He, K.; Bornheim, L.M.; Falick, A.M.; Maltby, D.; Yin, H.; Correia, M.A. Identification of the heme-modified peptides from cumene hydroperoxide-inactivated cytochrome P450 3A4. Biochemistry-US 1998, 37, 17448–17457. [Google Scholar] [CrossRef]
- Kang, P.; Liao, M.; Wester, M.R.; Leeder, J.S.; Pearce, R.E.; Correia, M.A. CYP3A4-mediated carbamazepine (CBZ) metabolism: Formation of a covalent CBZ-CYP3A4 adduct and alteration of the enzyme kinetic profile. Drug Metab. Dispos. 2008, 36, 490–499. [Google Scholar] [CrossRef]
- Park, B.K.; Pirmohamed, M.; Kitteringham, N.R. Role of drug disposition in drug hypersensitivity: A chemical, molecular, and clinical perspective. Chem. Res. Toxicol. 1998, 11, 969–988. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Tomson, T.; Tybring, G.; Bertilsson, L. Carbamazepine metabolism in man. Induction and pharmacogenetic aspects. Clin. Pharmacokinet. 1985, 10, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Lin, Y.S.; II, D.J.M.; Calamia, J.C.; Totah, R.A.; Isoherranen, N.; Glodowski, M.; Thummel, K.E. Evidence of significant contribution from CYP3A5 to hepatic drug metabolism. Drug Metab. Dispos. 2004, 32, 1434–1445. [Google Scholar] [CrossRef]
- Scazzone, C.; Agnello, L.; Ragonese, P.; Sasso, B.L.; Bellia, C.; Bivona, G.; Schillaci, R.; Salemi, G.; Ciaccio, M. Association of CYP2R1 rs10766197 with MS risk and disease progression. J. Neurosci. Res. 2018, 96, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Leckband, S.G.; Kelsoe, J.; Leeder, J.S.; Muller, D.J.; Klein, T.E.; Altman, R.B. PharmGKB summary: Carbamazepine pathway. Pharmacogenet. Genom. 2011, 21, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Guengrich, F.P. Cytochrome P-450 3A4: Regulation and role in drug metabolism. Annu. Rev. Pharmacol. 1999, 39, 1–17. [Google Scholar] [CrossRef]
- Mitra, R.; Guo, Z.; Milani, M.; Mesaros, C.; Rodriguez, M.; Nguyen, J.; Luo, X.; Clarke, D.; Lamba, J.; Schuetz, E.; et al. CYP3A4 mediates growth of estrogen receptor-positive breast cancer cells in part by inducing nuclear translocation of phospho-Stat3 through biosynthesis of (±) -14,15-epoxyeicosatrienoic acid (EET). J. Biol. Chem. 2011, 286, 17543–17559. [Google Scholar] [CrossRef]
- Yamakoshi, Y.; Kishimoto, T.; Sugimura, K.; Kawashima, H. Human prostate CYP3A5: Identification of a unique 5′-untranslated sequence and characterization of purified recombinant protein. Biochem. Biophys. Res. Commun. 1999, 260, 676–681. [Google Scholar] [CrossRef]
- Mitra, R.; Goodman, O.B., Jr. CYP3A5 regulates prostate cancer cell growth by facilitating nuclear translocation of AR. Prostate 2015, 75, 527–538. [Google Scholar] [CrossRef]
- Jiang, F.; Chen, L.; Yang, Y.-C.; Wang, X.-M.; Wang, R.-Y.; Li, L.; Wen, W.; Chang, Y.-X.; Chen, C.-Y.; Tang, J.; et al. CYP3A5 Functions as a Tumor Suppressor in Hepatocellular Carcinoma by Regulating mTORC2/Akt Signaling. Cancer Res. 2015, 75, 1470–1481. [Google Scholar] [CrossRef]
- Lolodi, O.; Wang, Y.-M.; Wright, W.C.; Chen, T. Differential Regulation of CYP3A4 and CYP3A5 and Its Implication in Drug Discovery. Curr. Drug Metab. 2017, 18, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Discovery Studio 3.1; Accelrys: San Diego, CA, USA, 2008.
- Wu, G.; Robertson, D.H.; Books, C.L., III; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Shahrokh, K.; Orendt, A.; Yost, G.S.; Cheatham, T.E., III. Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J. Comput. Chem. 2012, 33, 119–133. [Google Scholar] [CrossRef]
- Toukmaji, A.Y.; Board, J.A., Jr. Ewald summation techniques in perspective: A survey. Comput. Phys. Commun. 1996, 95, 73–92. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3A4-CBZ | % | 3A5-CBZ | % |
|---|---|---|---|
| 499CBZ@O17-108PHE@H-N | 71.37 | 476CBZ@O17-106ARG@HH11-NH1 | 46.38 |
| 499CBZ@O17-105ARG@HE-NE 499CBZ@O17-105ARG@HH22-NH2 | 32.21 30.38 | ||
| 498HEM@O2A-105ARG@HH22-NH2 498HEM@O1A-105ARG@HH22-NH2 498HEM@O2D-105ARG@HH21-NH2 | 54.83 52.05 36.51 | 475HEM@O1A-105ARG@H-N 475HEM@O1A-105ARG@H-N | 47.88 46.13 |
| 498HEM@O1D-126TRP@HE1-NE1 498HEM@O2D-126TRP@HE1-NE1 | 89.12 72.36 | 475HEM@O1D-126TRP@HE1-NE1 475HEM@O2D-126TRP@HE1-NE1 | 82.79 75.31 |
| 498HEM@O1D-130ARG@HH22-NH2 498HEM@O2D-130ARG@HH22-NH2 498HEM@O2D-130ARG@HE-NE 498HEM@O1D-130ARG@HE-NE | 47.95 45.50 33.74 25.19 | 475HEM@O2D-130ARG@HE-NE 475HEM@O2D-130ARG@HH21-NH2 475HEM@O1D-130ARG@HH21-NH2 | 35.91 28.43 26.18 |
| 498HEM@O2A-440ARG@HH22-NH2 498HEM@O1A-440ARG@H22-NH2 | 45.28 43.29 | 475HEM@O2A-375ARG@HH21-NH2 | 28.68 |
| 3A4-CBZ | % | 3A5-CBZ | % |
|---|---|---|---|
| 123ASP@OD2-105ARG@HH12-NH1 123ASP@OD2-105ARG@HH11-NH1 123ASP@OD1-105ARG@HH12-NH1 123ASP@OD1-105ARG@HH11-NH1 123ASP@OD2-105ARG@HH22-NH2 123ASP@OD2-105ARG@HH21-NH2 | 32.88 32.88 27.31 27.31 21.75 21.75 | 306GLU@OE2-105ARG@HH22-NH2 306GLU@OE2-105ARG@HH21-NH2 306GLU@OE1-105ARG@HH22-NH2 306GLU@OE1-105ARG@HH21-NH2 306GLU@OE1-105ARG@HH12-NH1 306GLU@OE1-105ARG@HH11-NH1 306GLU@OE2-105ARG@HH12-NH1 306GLU@OE2-105ARG@HH11-NH1 | 99.62 99.62 99.31 99.31 98.62 98.62 98.50 98.50 |
| 61ASP@OD2-372ARG@HH22-NH2 61ASP@OD2-372ARG@HH21-NH2 61ASP@OD2-372ARG@HE-NE 61ASP@OD1-372ARG@HH22-NH2 61ASP@OD1-372ARG@HH21-NH2 61ASP@OD1-372ARG@HE-NE | 100.00 100.00 100.00 100.00 100.00 99.81 | 374GLU@OE1-105ARG@HH22-NH2 374GLU@OE1-105ARG@HH21-NH2 374GLU@OE2-105ARG@HH22-NH2 374GLU@OE2-105ARG@HH21-NH2 374GLU@OE1-105ARG@HE-NE 374GLU@OE2-105ARG@HE-NE | 99.19 99.19 97.69 97.69 93.81 91.37 |
| 123ASP@OD2-440ARG@HH12-NH1 123ASP@OD2-440ARG@HH11-NH1 123ASP@OD1-440ARG@HH12-NH1 123ASP@OD1-440ARG@HH11-NH1 123ASP@OD2-440ARG@HH22-NH2 123ASP@OD2-440ARG@HH21-NH2 123ASP@OD1-440ARG@HH22-NH2 123ASP@OD1-440ARG@HH21-NH2 | 65.19 65.19 64.56 64.56 63.81 63.81 63.81 63.81 | 301GLU@OE2-106ARG@HH22-NH2 301GLU@OE2-106ARG@HH21-NH2 301GLU@OE2-106ARG@HH12-NH1 301GLU@OE2-106ARG@HH11-NH1 301GLU@OE1-106ARG@HH22-NH2 301GLU@OE1-106ARG@HH21-NH2 301GLU@OE1-106ARG@HH12-NH1 301GLU@OE1-106ARG@HH11-NH1 | 54.00 54.00 52.44 52.44 49.06 49.06 46.00 46.00 |
| 306GLU@OE2-106ARG@HH22-NH2 306GLU@OE2-106ARG@HH21-NH2 306GLU@OE1-106ARG@HH22-NH2 306GLU@OE1-106ARG@HH21-NH2 306GLU@OE2-106ARG@HH12-NH1 306GLU@OE2-106ARG@HH11-NH1 306GLU@OE1-106ARG@HH12-NH1 306GLU@OE1-106ARG@HH11-NH1 | 66.00 66.00 60.87 60.87 42.12 42.12 38.62 38.62 | ||
| 374GLU@OE2-106ARG@HE-NE 374GLU@OE1-106ARG@HE-NE 374GLU@OE2-106ARG@HH22-NH2 374GLU@OE2-106ARG@HH21-NH2 374GLU@OE1-106ARG@HH22-NH2 374GLU@OE1-106ARG@HH21-NH2 | 29.69 28.44 23.87 23.87 20.56 20.56 | ||
| 123ASP@OD1-130ARG@HH22-NH2 123ASP@OD1-130ARG@HH21-NH2 123ASP@OD2-130ARG@HH22-NH2 123ASP@OD2-130ARG@HH21-NH2 123ASP@OD1-130ARG@HH12-NH1 123ASP@OD1-130ARG@HH11-NH1 123ASP@OD2-130ARG@HH12-NH1 123ASP@OD2-130ARG@HH11-NH1 | 36.56 36.56 34.31 34.31 33.19 33.19 32.25 32.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Xu, Y. MD Investigation on the Interaction between Carbamazepine and Two CYP Isoforms, CYP3A4 and CYP3A5. Int. J. Mol. Sci. 2023, 24, 2188. https://doi.org/10.3390/ijms24032188
Liu S, Xu Y. MD Investigation on the Interaction between Carbamazepine and Two CYP Isoforms, CYP3A4 and CYP3A5. International Journal of Molecular Sciences. 2023; 24(3):2188. https://doi.org/10.3390/ijms24032188
Chicago/Turabian StyleLiu, Shuhui, and Yang Xu. 2023. "MD Investigation on the Interaction between Carbamazepine and Two CYP Isoforms, CYP3A4 and CYP3A5" International Journal of Molecular Sciences 24, no. 3: 2188. https://doi.org/10.3390/ijms24032188
APA StyleLiu, S., & Xu, Y. (2023). MD Investigation on the Interaction between Carbamazepine and Two CYP Isoforms, CYP3A4 and CYP3A5. International Journal of Molecular Sciences, 24(3), 2188. https://doi.org/10.3390/ijms24032188