
Emerging Role of Protein O-GlcNAcylation in Liver Metabolism: Implications for Diabetes and NAFLD


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Overview of O-GlcNAcylation
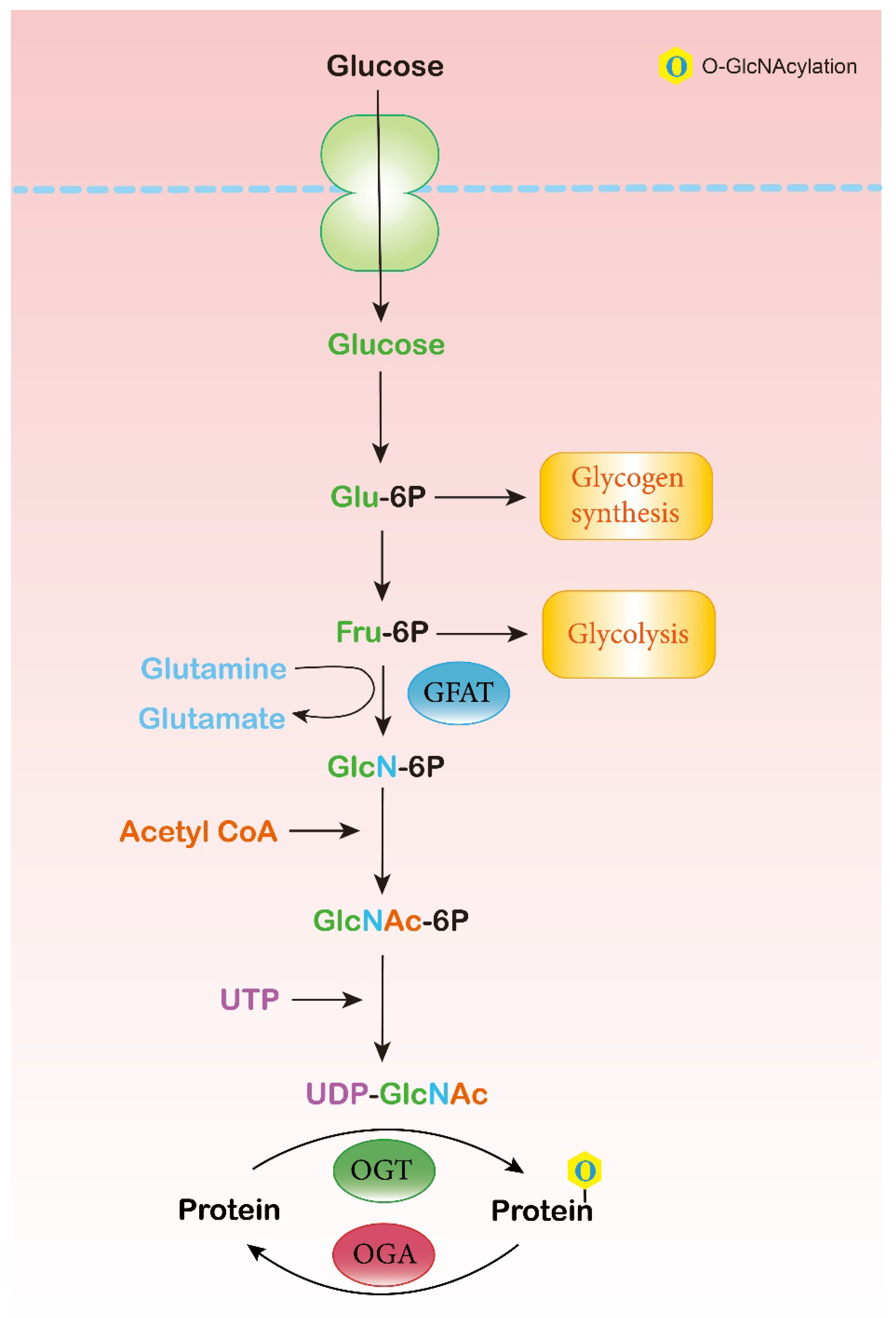
2.1. Hexosamine Biosynthesis Pathway (HBP)
2.2. O-GlcNAc Cycling Enzymes: OGT and OGA
2.3. O-GlcNAc-Modified Proteins
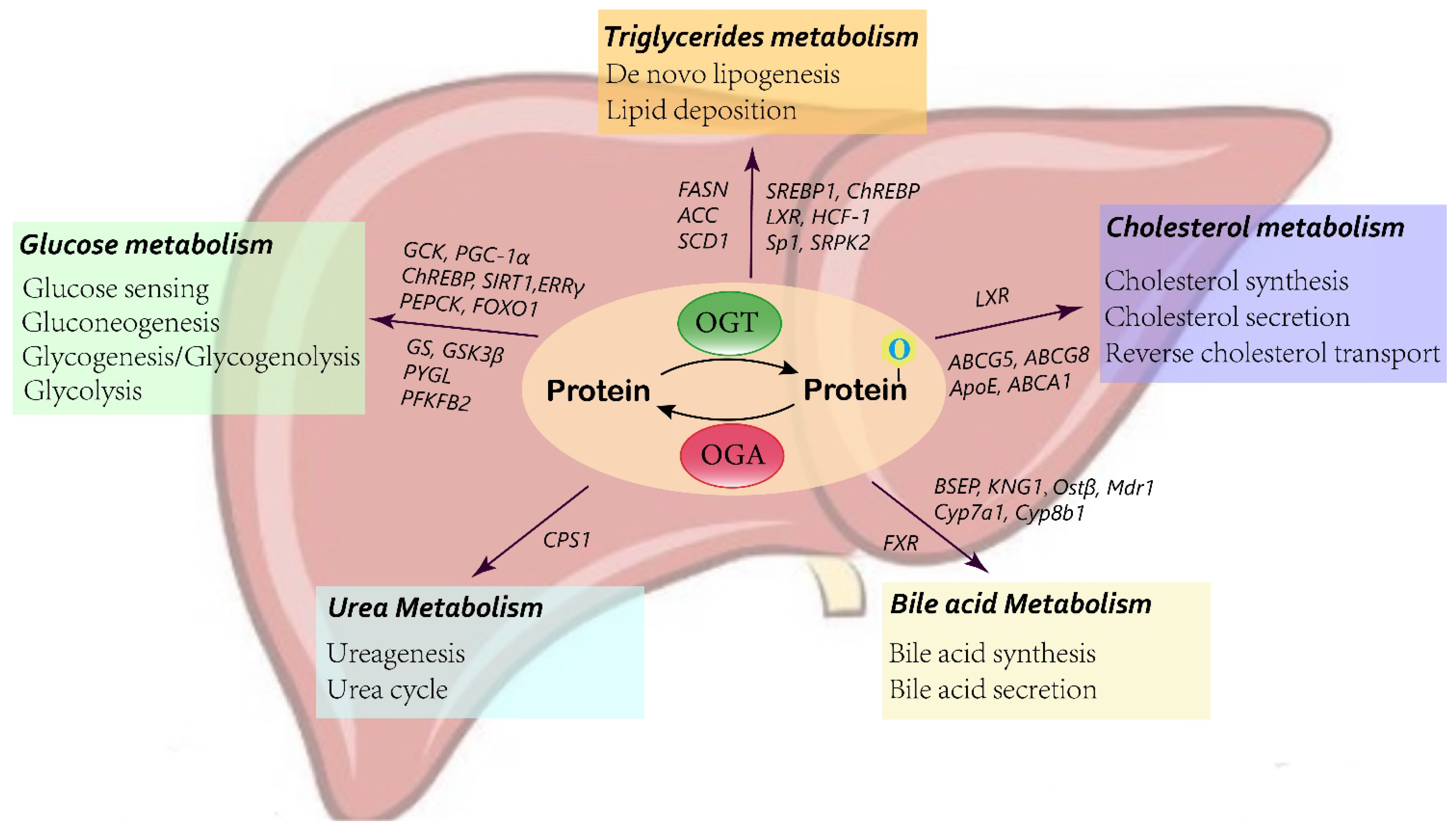
3. O-GlcNAcylation in Liver Metabolism
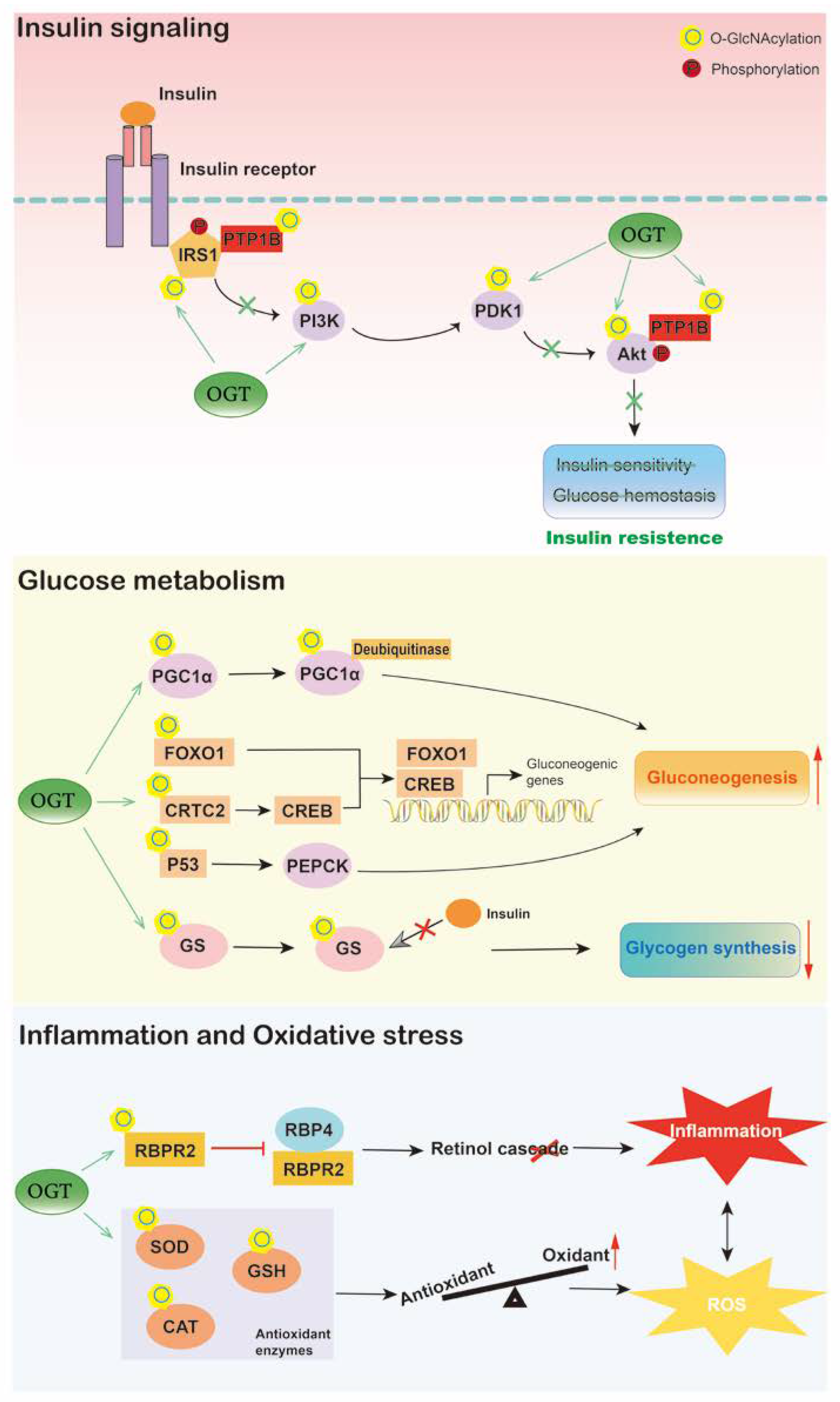
3.1. Glucose Metabolism
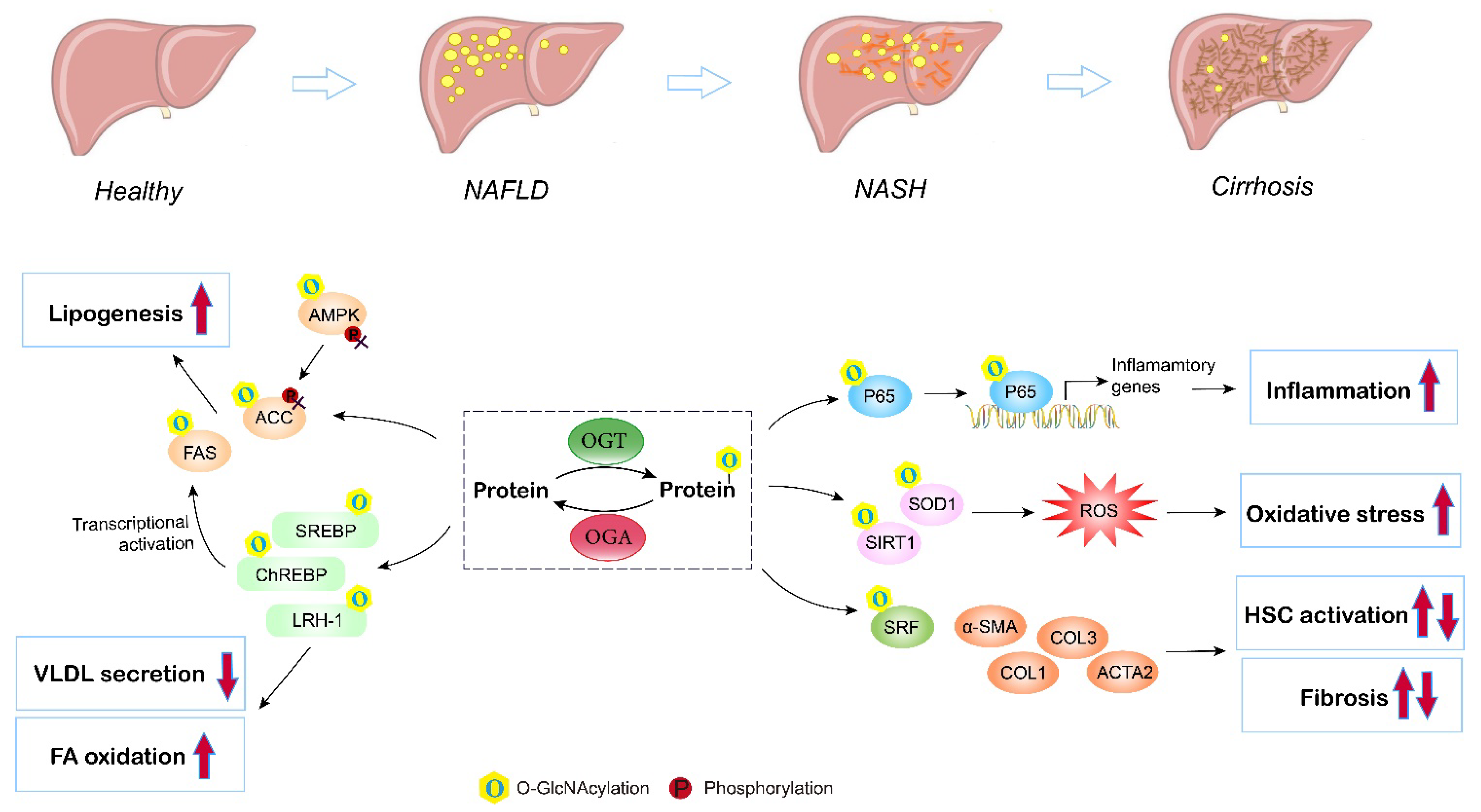
3.2. Lipid Metabolism
3.3. Bile Acid Metabolism
3.4. Urea Metabolism
4. O-GlcNAcylation in Diabetes
5. O-GlcNAcylation in Nonalcoholic Fatty Liver Disease (NAFLD)
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef]
- Vilstrup, H. Synthesis of urea after stimulation with amino acids: Relation to liver function. Gut 1980, 21, 990–995. [Google Scholar] [CrossRef]
- Xiao, J.; Wang, F.; Wong, N.K.; He, J.; Zhang, R.; Sun, R.; Xu, Y.; Liu, Y.; Li, W.; Koike, K.; et al. Global liver disease burdens and research trends: Analysis from a Chinese perspective. J. Hepatol. 2019, 71, 212–221. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Bonora, E.; Targher, G. Nonalcoholic Fatty Liver Disease and Risk of Incident Type 2 Diabetes: A Meta-analysis. Diabetes Care 2018, 41, 372–382. [Google Scholar] [CrossRef]
- Han, E.; Lee, Y.H. Non-Alcoholic Fatty Liver Disease: The Emerging Burden in Cardiometabolic and Renal Diseases. Diabetes Metab. J. 2017, 41, 430–437. [Google Scholar] [CrossRef]
- Bril, F.; Cusi, K. Management of Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes: A Call to Action. Diabetes Care 2017, 40, 419–430. [Google Scholar] [CrossRef]
- Wild, S.H.; Morling, J.R.; McAllister, D.A.; Kerssens, J.; Fischbacher, C.; Parkes, J.; Roderick, P.J.; Sattar, N.; Byrne, C.D. Type 2 diabetes and risk of hospital admission or death for chronic liver diseases. J. Hepatol. 2016, 64, 1358–1364. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef]
- Chatham, J.C.; Zhang, J.; Wende, A.R. Role of O-Linked N-Acetylglucosamine Protein Modification in Cellular (Patho)Physiology. Physiol. Rev. 2021, 101, 427–493. [Google Scholar] [CrossRef]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.S.; Hart, G.W.; Cho, J.W. Chemical approaches to study O-GlcNAcylation. Chem. Soc. Rev. 2013, 42, 4345–4357. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Chen, C.; Liu, R.; Gou, D.; Chang, L.; Deng, H.; Gao, Q.; Zhang, W.; Tuo, L.; Pan, X.; et al. Gluconeogenic enzyme PCK1 deficiency promotes CHK2 O-GlcNAcylation and hepatocellular carcinoma growth upon glucose deprivation. J. Clin. Investig. 2021, 131, e144703. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Murshed, A.; Li, H.; Ma, J.; Zhen, N.; Ding, M.; Zhu, J.; Mao, S.; Tang, X.; Liu, L.; et al. O-GlcNAcylation enhances sensitivity to RSL3-induced ferroptosis via the YAP/TFRC pathway in liver cancer. Cell Death Discov. 2021, 7, 83. [Google Scholar] [CrossRef]
- Zhang, J.; Xun, M.; Li, C.; Chen, Y. The O-GlcNAcylation and its promotion to hepatocellular carcinoma. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188806. [Google Scholar] [CrossRef]
- Lee, D.E.; Lee, S.J.; Kim, S.J.; Lee, H.-S.; Kwon, O.-S. Curcumin Ameliorates Nonalcoholic Fatty Liver Disease through Inhibition of -GlcNAcylation. Nutrients 2019, 11, 2702. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Z.; Xu, M.; Zhang, D.; Ling, J.; Yu, P.; Shen, Y. O-GlycNacylation Remission Retards the Progression of Non-Alcoholic Fatty Liver Disease. Cells 2022, 11, 3637. [Google Scholar] [CrossRef]
- Raab, S.; Gadault, A.; Very, N.; Decourcelle, A.; Baldini, S.; Schulz, C.; Mortuaire, M.; Lemaire, Q.; Hardiville, S.; Dehennaut, V.; et al. Dual regulation of fatty acid synthase (FASN) expression by O-GlcNAc transferase (OGT) and mTOR pathway in proliferating liver cancer cells. Cell Mol. Life Sci. 2021, 78, 5397–5413. [Google Scholar] [CrossRef] [PubMed]
- Bolanle, I.O.; Palmer, T.M. Targeting Protein -GlcNAcylation, a Link between Type 2 Diabetes Mellitus and Inflammatory Disease. Cells 2022, 11, 705. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hart, G.W. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev. Proteom. 2013, 10, 365–380. [Google Scholar] [CrossRef]
- Chatham, J.C.; Young, M.E.; Zhang, J. Role of O-linked N-acetylglucosamine (O-GlcNAc) modification of proteins in diabetic cardiovascular complications. Curr. Opin. Pharmacol. 2021, 57, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Remigante, A.; Civello, D.A.; Bernardinelli, E.; Szabó, Z.; Morabito, R.; Marino, A.; Sarikas, A.; Patsch, W.; Paulmichl, M.; et al. -GlcNAcylation Suppresses the Ion Current IClswell by Preventing the Binding of the Protein ICln to α-Integrin. Front. Cell Dev. Biol. 2020, 8, 607080. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.; Low, J.-Y.; Tran, P.T.; Wang, H. The hexosamine biosynthetic pathway and cancer: Current knowledge and future therapeutic strategies. Cancer Lett. 2021, 503, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W. Nutrient regulation of signaling and transcription. J. Biol. Chem. 2019, 294, 2211–2231. [Google Scholar] [CrossRef]
- Lecoutre, S.; Maqdasy, S.; Petrus, P.; Ludzki, A.; Couchet, M.; Mejhert, N.; Ryden, M. Glutamine metabolism in adipocytes: A bona fide epigenetic modulator of inflammation. Adipocyte 2020, 9, 620–625. [Google Scholar] [CrossRef]
- Milewski, S. Glucosamine-6-phosphate synthase--the multi-facets enzyme. Biochim. Biophys. Acta 2002, 1597, 173–192. [Google Scholar] [CrossRef]
- Oki, T.; Yamazaki, K.; Kuromitsu, J.; Okada, M.; Tanaka, I. cDNA cloning and mapping of a novel subtype of glutamine:fructose-6-phosphate amidotransferase (GFAT2) in human and mouse. Genomics 1999, 57, 227–234. [Google Scholar] [CrossRef]
- Wang, Z.V.; Deng, Y.; Gao, N.; Pedrozo, Z.; Li, D.L.; Morales, C.R.; Criollo, A.; Luo, X.; Tan, W.; Jiang, N.; et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 2014, 156, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Sayeski, P.P.; Wang, D.; Su, K.; Han, I.O.; Kudlow, J.E. Cloning and partial characterization of the mouse glutamine:fructose-6-phosphate amidotransferase (GFAT) gene promoter. Nucleic Acids Res. 1997, 25, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Su, K.; Baker, J.R.; Yang, X.; Paterson, A.J.; Kudlow, J.E. Phosphorylation of human glutamine:fructose-6-phosphate amidotransferase by cAMP-dependent protein kinase at serine 205 blocks the enzyme activity. J. Biol. Chem. 2000, 275, 21981–21987. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Riesland, L.; Paterson, A.J.; Kudlow, J.E. Phosphorylation of mouse glutamine-fructose-6-phosphate amidotransferase 2 (GFAT2) by cAMP-dependent protein kinase increases the enzyme activity. J. Biol. Chem. 2004, 279, 29988–29993. [Google Scholar] [CrossRef]
- Eguchi, S.; Oshiro, N.; Miyamoto, T.; Yoshino, K.; Okamoto, S.; Ono, T.; Kikkawa, U.; Yonezawa, K. AMP-activated protein kinase phosphorylates glutamine: Fructose-6-phosphate amidotransferase 1 at Ser243 to modulate its enzymatic activity. Genes Cells 2009, 14, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Levine, Z.G.; Walker, S. The Biochemistry of O-GlcNAc Transferase: Which Functions Make It Essential in Mammalian Cells? Annu. Rev. Biochem. 2016, 85, 631–657. [Google Scholar] [CrossRef]
- Nagel, A.K.; Ball, L.E. O-GlcNAc transferase and O-GlcNAcase: Achieving target substrate specificity. Amino Acids 2014, 46, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, P.; Yan, H.; Sun, H.; Liu, X.; Zhou, F.; Li, L.; Chen, Y.; Muthana, M.M.; Chen, X.; et al. Substrate specificity provides insights into the sugar donor recognition mechanism of O-GlcNAc transferase (OGT). PLoS ONE 2013, 8, e63452. [Google Scholar] [CrossRef] [PubMed]
- Joiner, C.M.; Li, H.; Jiang, J.; Walker, S. Structural characterization of the O-GlcNAc cycling enzymes: Insights into substrate recognition and catalytic mechanisms. Curr. Opin. Struct. Biol. 2019, 56, 97–106. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Nam, Y.; Jiang, J.; Sliz, P.; Walker, S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 2011, 469, 564–567. [Google Scholar] [CrossRef]
- Yang, X.; Ongusaha, P.P.; Miles, P.D.; Havstad, J.C.; Zhang, F.; So, W.V.; Kudlow, J.E.; Michell, R.H.; Olefsky, J.M.; Field, S.J.; et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 2008, 451, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Fardini, Y.; Dehennaut, V.; Lefebvre, T.; Issad, T. O-GlcNAcylation: A New Cancer Hallmark? Front. Endocrinol. 2013, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, M.D.; Yin, R.; Liu, Y.; Yang, Y.; Mitchell-Richards, K.A.; Nam, J.H.; Li, R.; Wang, L.; Iwakiri, Y.; et al. O-GlcNAc transferase suppresses necroptosis and liver fibrosis. JCI Insight 2019, 4, e127709. [Google Scholar] [CrossRef] [PubMed]
- Comtesse, N.; Maldener, E.; Meese, E. Identification of a nuclear variant of MGEA5, a cytoplasmic hyaluronidase and a beta-N-acetylglucosaminidase. Biochem. Biophys. Res. Commun. 2001, 283, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Muha, V.; Authier, F.; Szoke-Kovacs, Z.; Johnson, S.; Gallagher, J.; McNeilly, A.; McCrimmon, R.J.; Teboul, L.; van Aalten, D.M.F. Loss of O-GlcNAcase catalytic activity leads to defects in mouse embryogenesis. J. Biol. Chem. 2021, 296, 100439. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kang, D.O.; Love, D.C.; Hanover, J.A. Enzymatic characterization of O-GlcNAcase isoforms using a fluorogenic GlcNAc substrate. Carbohydr. Res. 2006, 341, 971–982. [Google Scholar] [CrossRef]
- Keembiyehetty, C.N.; Krzeslak, A.; Love, D.C.; Hanover, J.A. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J. Cell Sci. 2011, 124, 2851–2860. [Google Scholar] [CrossRef] [PubMed]
- Ong, Q.; Han, W.; Yang, X. O-GlcNAc as an Integrator of Signaling Pathways. Front. Endocrinol. 2018, 9, 599. [Google Scholar] [CrossRef]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Krześlak, A. Role of O-GlcNAc modification of cellular proteins in signal transduction. Postep. Biochem. 2007, 53, 389–399. [Google Scholar]
- Yang, W.H.; Kim, J.E.; Nam, H.W.; Ju, J.W.; Kim, H.S.; Kim, Y.S.; Cho, J.W. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat. Cell Biol. 2006, 8, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rellan, M.J.; Fondevila, M.F.; Fernandez, U.; Rodriguez, A.; Varela-Rey, M.; Veyrat-Durebex, C.; Seoane, S.; Bernardo, G.; Lopitz-Otsoa, F.; Fernandez-Ramos, D.; et al. O-GlcNAcylated p53 in the liver modulates hepatic glucose production. Nat. Commun. 2021, 12, 5068. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Chang, H.I. O-linked N-acetylglucosamine suppresses thermal aggregation of Sp1. FEBS Lett. 2006, 580, 4645–4652. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Duan, X.; Mao, W.; Li, X.; Li, Z.; Li, Q.; Zheng, Z.; Xu, H.; Chen, M.; Wang, P.G.; et al. O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth. Nat. Commun. 2015, 6, 8468. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Z.; Kolwicz, S.C., Jr.; Abell, L.; Roe, N.D.; Kim, M.; Zhou, B.; Cao, Y.; Ritterhoff, J.; Gu, H.; et al. Defective Branched-Chain Amino Acid Catabolism Disrupts Glucose Metabolism and Sensitizes the Heart to Ischemia-Reperfusion Injury. Cell Metab. 2017, 25, 374–385. [Google Scholar] [CrossRef]
- Bindesboll, C.; Fan, Q.; Norgaard, R.C.; MacPherson, L.; Ruan, H.B.; Wu, J.; Pedersen, T.A.; Steffensen, K.R.; Yang, X.; Matthews, J.; et al. Liver X receptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J. Lipid. Res. 2015, 56, 771–785. [Google Scholar] [CrossRef]
- Kaasik, K.; Kivimae, S.; Allen, J.J.; Chalkley, R.J.; Huang, Y.; Baer, K.; Kissel, H.; Burlingame, A.L.; Shokat, K.M.; Ptacek, L.J.; et al. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 2013, 17, 291–302. [Google Scholar] [CrossRef]
- Qiu, H.; Liu, F.; Tao, T.; Zhang, D.; Liu, X.; Zhu, G.; Xu, Z.; Ni, R.; Shen, A. Modification of p27 with O-linked N-acetylglucosamine regulates cell proliferation in hepatocellular carcinoma. Mol. Carcinog. 2017, 56, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zhu, Y.; Zhang, W.; Liao, Q.; Chen, Y.; Zhao, X.; Guo, Q.; Shen, P.; Zhen, B.; Qian, X.; et al. Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol. Cell 2017, 68, 591–604 e5. [Google Scholar] [CrossRef]
- Oosterveer, M.H.; Schoonjans, K. Hepatic glucose sensing and integrative pathways in the liver. Cell Mol. Life Sci. 2014, 71, 1453–1467. [Google Scholar] [CrossRef] [PubMed]
- Baldini, S.F.; Steenackers, A.; Olivier-Van Stichelen, S.; Mir, A.M.; Mortuaire, M.; Lefebvre, T.; Guinez, C. Glucokinase expression is regulated by glucose through O-GlcNAc glycosylation. Biochem. Biophys. Res. Commun. 2016, 478, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Barberà, A.; Télémaque-Potts, S.; Newgard, C.B.; Guinovart, J.J. Glucokinase overexpression restores glucose utilization and storage in cultured hepatocytes from male Zucker diabetic fatty rats. J. Biol. Chem. 1999, 274, 31833–31838. [Google Scholar] [CrossRef] [PubMed]
- Benhamed, F.; Filhoulaud, G.; Caron, S.; Lefebvre, P.; Staels, B.; Postic, C. O-GlcNAcylation Links ChREBP and FXR to Glucose-Sensing. Front. Endocrinol. 2014, 5, 230. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.S.; Baumel-Alterzon, S.; Scott, D.K.; Herman, M.A. Adaptive and maladaptive roles for ChREBP in the liver and pancreatic islets. J. Biol. Chem. 2021, 296, 100623. [Google Scholar] [CrossRef]
- Kuo, M.; Zilberfarb, V.; Gangneux, N.; Christeff, N.; Issad, T. O-glycosylation of FoxO1 increases its transcriptional activity towards the glucose 6-phosphatase gene. FEBS Lett. 2008, 582, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.; Zilberfarb, V.; Gangneux, N.; Christeff, N.; Issad, T. O-GlcNAc modification of FoxO1 increases its transcriptional activity: A role in the glucotoxicity phenomenon? Biochimie 2008, 90, 679–685. [Google Scholar] [CrossRef]
- Ruan, H.B.; Han, X.; Li, M.D.; Singh, J.P.; Qian, K.; Azarhoush, S.; Zhao, L.; Bennett, A.M.; Samuel, V.T.; Wu, J.; et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab. 2012, 16, 226–237. [Google Scholar] [CrossRef]
- Chattopadhyay, T.; Maniyadath, B.; Bagul, H.P.; Chakraborty, A.; Shukla, N.; Budnar, S.; Rajendran, A.; Shukla, A.; Kamat, S.S.; Kolthur-Seetharam, U. Spatiotemporal gating of SIRT1 functions by O-GlcNAcylation is essential for liver metabolic switching and prevents hyperglycemia. Proc. Natl Acad. Sci. USA 2020, 117, 6890–6900. [Google Scholar] [CrossRef]
- Misra, J.; Kim, D.K.; Jung, Y.S.; Kim, H.B.; Kim, Y.H.; Yoo, E.K.; Kim, B.G.; Kim, S.; Lee, I.K.; Harris, R.A.; et al. O-GlcNAcylation of Orphan Nuclear Receptor Estrogen-Related Receptor gamma Promotes Hepatic Gluconeogenesis. Diabetes 2016, 65, 2835–2848. [Google Scholar] [CrossRef]
- Yao, R.; Yang, Y.; Lian, S.; Shi, H.; Liu, P.; Liu, Y.; Yang, H.; Li, S. Effects of Acute Cold Stress on Liver O-GlcNAcylation and Glycometabolism in Mice. Int. J. Mol. Sci. 2018, 19, 2815. [Google Scholar] [CrossRef]
- Taylor, R.P.; Parker, G.J.; Hazel, M.W.; Soesanto, Y.; Fuller, W.; Yazzie, M.J.; McClain, D.A. Glucose deprivation stimulates O-GlcNAc modification of proteins through up-regulation of O-linked N-acetylglucosaminyltransferase. J. Biol. Chem. 2008, 283, 6050–6057. [Google Scholar] [CrossRef]
- Keembiyehetty, C.; Love, D.C.; Harwood, K.R.; Gavrilova, O.; Comly, M.E.; Hanover, J.A. Conditional knock-out reveals a requirement for O-linked N-Acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J. Biol. Chem. 2015, 290, 7097–7113. [Google Scholar] [CrossRef]
- Anderson, G. Tumour Microenvironment: Roles of the Aryl Hydrocarbon Receptor, O-GlcNAcylation, Acetyl-CoA and Melatonergic Pathway in Regulating Dynamic Metabolic Interactions across Cell Types-Tumour Microenvironment and Metabolism. Int. J. Mol. Sci. 2020, 22, 141. [Google Scholar] [CrossRef]
- Kokubun, E.; Hirabara, S.M.; Fiamoncini, J.; Curi, R.; Haebisch, H. Changes of glycogen content in liver, skeletal muscle, and heart from fasted rats. Cell Biochem. Funct. 2009, 27, 488–495. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, B.; Hu, Y.; Liu, P.; Lian, S.; Lv, H.; Yang, Y.; Ji, H.; Yang, H.; Liu, J.; et al. O-GlcNAc / Akt pathway regulates glucose metabolism and reduces apoptosis in liver of piglets with acute cold stress. Cryobiology 2021, 100, 125–132. [Google Scholar] [CrossRef]
- Chen, Y.F.; Zhu, J.J.; Li, J.; Ye, X.S. O-GlcNAcylation increases PYGL activity by promoting phosphorylation. Glycobiology 2022, 32, 101–109. [Google Scholar] [CrossRef]
- Yu, X.; Ren, L.-P.; Wang, C.; Zhu, Y.-J.; Xing, H.-Y.; Zhao, J.; Song, G.-Y. Role of X-Box Binding Protein-1 in Fructose-Induced Lipogenesis in HepG2 Cells. Chin. Med. J. 2018, 131, 2310–2319. [Google Scholar] [CrossRef]
- Park, J.; Lee, Y.; Jung, E.-H.; Kim, S.-M.; Cho, H.; Han, I.-O. Glucosamine regulates hepatic lipid accumulation by sensing glucose levels or feeding states of normal and excess. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158764. [Google Scholar] [CrossRef]
- Tan, W.; Jiang, P.; Zhang, W.; Hu, Z.; Lin, S.; Chen, L.; Li, Y.; Peng, C.; Li, Z.; Sun, A.; et al. Posttranscriptional regulation of de novo lipogenesis by glucose-induced O-GlcNAcylation. Mol. Cell 2021, 81, 1890–1904.e7. [Google Scholar] [CrossRef]
- Baldini, S.F.; Wavelet, C.; Hainault, I.; Guinez, C.; Lefebvre, T. The Nutrient-Dependent O-GlcNAc Modification Controls the Expression of Liver Fatty Acid Synthase. J. Mol. Biol. 2016, 428, 3295–3304. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-J.; Lin, T.; Hsieh, P.-C.; Liao, M.-C.; Shin, S.-J. Suppression of Glutamine:fructose-6-phosphate amidotransferase-1 inhibits adipogenesis in 3T3-L1 adipocytes. J. Cell Physiol. 2012, 227, 108–115. [Google Scholar] [CrossRef]
- Pang, Y.; Xu, X.; Xiang, X.; Li, Y.; Zhao, Z.; Li, J.; Gao, S.; Liu, Q.; Mai, K.; Ai, Q. High Fat Activates O-GlcNAcylation and Affects AMPK/ACC Pathway to Regulate Lipid Metabolism. Nutrients 2021, 13, 1740. [Google Scholar] [CrossRef] [PubMed]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.F.; Yang, X.; Lefebvre, T.; et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 2011, 60, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Lane, E.A.; Choi, D.W.; Garcia-Haro, L.; Levine, Z.G.; Tedoldi, M.; Walker, S.; Danial, N.N. HCF-1 Regulates De Novo Lipogenesis through a Nutrient-Sensitive Complex with ChREBP. Mol. Cell 2019, 75, 357–371.e7. [Google Scholar] [CrossRef] [PubMed]
- Osborne, T.F. Sterol regulatory element-binding proteins (SREBPs): Key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 2000, 275, 32379–32382. [Google Scholar] [CrossRef] [PubMed]
- Sodi, V.L.; Bacigalupa, Z.A.; Ferrer, C.M.; Lee, J.V.; Gocal, W.A.; Mukhopadhyay, D.; Wellen, K.E.; Ivan, M.; Reginato, M.J. Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene 2018, 37, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Jhu, J.W.; Yan, J.B.; Lin, Z.H.; Lin, S.C.; Peng, I.C. SREBP1-Induced Glutamine Synthetase Triggers a Feedforward Loop to Upregulate SREBP1 through Sp1 O-GlcNAcylation and Augments Lipid Droplet Formation in Cancer Cells. Int. J. Mol. Sci. 2021, 22, 9814. [Google Scholar] [CrossRef]
- Fan, Q.; Norgaard, R.C.; Bindesboll, C.; Lucas, C.; Dalen, K.T.; Babaie, E.; Itkonen, H.M.; Matthews, J.; Nebb, H.I.; Gronning-Wang, L.M. LXRalpha Regulates Hepatic ChREBPalpha Activity and Lipogenesis upon Glucose, but Not Fructose Feeding in Mice. Nutrients 2017, 9, 678. [Google Scholar] [CrossRef]
- Anthonisen, E.H.; Berven, L.; Holm, S.; Nygard, M.; Nebb, H.I.; Gronning-Wang, L.M. Nuclear receptor liver X receptor is O-GlcNAc-modified in response to glucose. J. Biol. Chem. 2010, 285, 1607–1615. [Google Scholar] [CrossRef]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Breevoort, S.R.; Angdisen, J.; Fu, M.; Schmidt, D.R.; Holmstrom, S.R.; Kliewer, S.A.; Mangelsdorf, D.J.; Schulman, I.G. Liver LXRα expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J. Clin. Investig. 2012, 122, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Berge, K.E.; Pomajzl, C.; Richardson, J.A.; Hobbs, H.; Mangelsdorf, D.J. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J. Biol. Chem. 2002, 277, 18793–18800. [Google Scholar] [CrossRef]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef]
- Berrabah, W.; Aumercier, P.; Gheeraert, C.; Dehondt, H.; Bouchaert, E.; Alexandre, J.; Ploton, M.; Mazuy, C.; Caron, S.; Tailleux, A.; et al. Glucose sensing O-GlcNAcylation pathway regulates the nuclear bile acid receptor farnesoid X receptor (FXR). Hepatology 2014, 59, 2022–2033. [Google Scholar] [CrossRef]
- Martínez, A.I.; Pérez-Arellano, I.; Pekkala, S.; Barcelona, B.; Cervera, J. Genetic, structural and biochemical basis of carbamoyl phosphate synthetase 1 deficiency. Mol. Genet. Metab. 2010, 101, 311–323. [Google Scholar] [CrossRef]
- Soria, L.R.; Makris, G.; D’Alessio, A.M.; De Angelis, A.; Boffa, I.; Pravata, V.M.; Rufenacht, V.; Attanasio, S.; Nusco, E.; Arena, P.; et al. O-GlcNAcylation enhances CPS1 catalytic efficiency for ammonia and promotes ureagenesis. Nat. Commun. 2022, 13, 5212. [Google Scholar] [CrossRef]
- Wu, J.; Liu, J.; Lapenta, K.; Desrouleaux, R.; Li, M.D.; Yang, X. Regulation of the urea cycle by CPS1 O-GlcNAcylation in response to dietary restriction and aging. J. Mol. Cell Biol. 2022, 14, mjac016. [Google Scholar] [CrossRef]
- Lewis, G.F.; Carpentier, A.C.; Pereira, S.; Hahn, M.; Giacca, A. Direct and indirect control of hepatic glucose production by insulin. Cell Metab. 2021, 33, 709–720. [Google Scholar] [CrossRef]
- Majumdar, G.W.J.; Markowitz, P.; Martinez-Hernandez, A.; Raghow, R.; Solomon, S.S. Insulin stimulates and diabetes inhibits O-linked N-acetylglucosamine transferase and O-glycosylation of Sp1. Diabetes 2004, 53, 3184–3192. [Google Scholar] [CrossRef] [PubMed]
- McClain, D.A.; Lubas, W.A.; Cooksey, R.C.; Hazel, M.; Parker, G.J.; Love, D.C.; Hanover, J.A. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc. Natl. Acad. Sci. USA 2002, 99, 10695–10699. [Google Scholar] [CrossRef] [PubMed]
- McClain, D.A. Hexosamines as mediators of nutrient sensing and regulation in diabetes. J. Diabetes Complicat. 2002, 16, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tang, Z.; Shen, A.; Tao, T.; Wan, C.; Zhu, X.; Huang, J.; Zhang, W.; Xia, N.; Wang, S.; et al. The Role of PTP1B O-GlcNAcylation in Hepatic Insulin Resistance. Int. J. Mol. Sci. 2015, 16, 22856–22869. [Google Scholar] [CrossRef]
- Dentin, R.H.S.; Xie, J.; Yates, J., 3rd; Montminy, M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science 2008, 319, 1402–1405. [Google Scholar] [CrossRef]
- Parker, G.; Taylor, R.; Jones, D.; McClain, D. Hyperglycemia and inhibition of glycogen synthase in streptozotocin-treated mice: Role of O-linked N-acetylglucosamine. J. Biol. Chem. 2004, 279, 20636–20642. [Google Scholar] [CrossRef]
- Graham, T.E.; Yang, Q.; Blüher, M.; Hammarstedt, A.; Ciaraldi, T.P.; Henry, R.R.; Wason, C.J.; Oberbach, A.; Jansson, P.-A.; Smith, U.; et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med. 2006, 354, 2552–2563. [Google Scholar] [CrossRef]
- Shin, S.J.; Chen, C.H.; Kuo, W.C.; Chan, H.C.; Chan, H.C.; Lin, K.D.; Ke, L.Y. Disruption of retinoid homeostasis induces RBP4 overproduction in diabetes: O-GlcNAcylation involved. Metabolism 2020, 113, 154403. [Google Scholar] [CrossRef]
- Dinic, S.; Arambasic, J.; Mihailovic, M.; Uskokovic, A.; Grdovic, N.; Markovic, J.; Karadzic, B.; Poznanovic, G.; Vidakovic, M. Decreased O-GlcNAcylation of the key proteins in kinase and redox signalling pathways is a novel mechanism of the beneficial effect of alpha-lipoic acid in diabetic liver. Br. J. Nutr. 2013, 110, 401–412. [Google Scholar] [CrossRef]
- McGreal, S.R.; Bhushan, B.; Walesky, C.; McGill, M.R.; Lebofsky, M.; Kandel, S.E.; Winefield, R.D.; Jaeschke, H.; Zachara, N.E.; Zhang, Z.; et al. Modulation of O-GlcNAc Levels in the Liver Impacts Acetaminophen-Induced Liver Injury by Affecting Protein Adduct Formation and Glutathione Synthesis. Toxicol. Sci. 2018, 162, 599–610. [Google Scholar] [CrossRef]
- Duggirala, R.; Blangero, J.; Almasy, L.; Dyer, T.D.; Williams, K.L.; Leach, R.J.; O’Connell, P.; Stern, M.P. Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am. J. Hum. Genet. 1999, 64, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Lehman, D.M.; Fu, D.-J.; Freeman, A.B.; Hunt, K.J.; Leach, R.J.; Johnson-Pais, T.; Hamlington, J.; Dyer, T.D.; Arya, R.; Abboud, H.; et al. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc-selective N-acetyl-beta-D glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes 2005, 54, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Springhorn, C.; Matsha, T.E.; Erasmus, R.T.; Essop, M.F. Exploring leukocyte O-GlcNAcylation as a novel diagnostic tool for the earlier detection of type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, 4640–4649. [Google Scholar] [CrossRef]
- Myslicki, J.P.; Shearer, J.; Hittel, D.S.; Hughey, C.C.; Belke, D.D. O-GlcNAc modification is associated with insulin sensitivity in the whole blood of healthy young adult males. Diabetol. Metab. Syndr. 2014, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Coomer, M.; Essop, M.F. Differential hexosamine biosynthetic pathway gene expression with type 2 diabetes. Mol. Genet. Metab. Rep. 2014, 1, 158–169. [Google Scholar] [CrossRef]
- Sun, Y.; Demagny, H.; Schoonjans, K. Emerging functions of the nuclear receptor LRH-1 in liver physiology and pathology. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166145. [Google Scholar] [CrossRef] [PubMed]
- Parlati, L.; Régnier, M.; Guillou, H.; Postic, C. New targets for NAFLD. JHEP Rep. 2021, 3, 100346. [Google Scholar] [CrossRef]
- Farzanegi, P.; Dana, A.; Ebrahimpoor, Z.; Asadi, M.; Azarbayjani, M.A. Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): Roles of oxidative stress and inflammation. Eur. J. Sport Sci. 2019, 19, 994–1003. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Lee, S.J.; Nam, M.J.; Lee, D.E.; Park, J.W.; Kang, B.S.; Lee, D.S.; Lee, H.S.; Kwon, O.S. Silibinin Ameliorates O-GlcNAcylation and Inflammation in a Mouse Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2165. [Google Scholar] [CrossRef]
- Sage, A.T.; Walter, L.A.; Shi, Y.; Khan, M.I.; Kaneto, H.; Capretta, A.; Werstuck, G.H. Hexosamine biosynthesis pathway flux promotes endoplasmic reticulum stress, lipid accumulation, and inflammatory gene expression in hepatic cells. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E499–E511. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, X.; Wu, J.L.; Fu, L.; Liu, K.; Liu, D.; Chen, G.G.; Lai, P.B.; Wong, N.; Yu, J. O-GlcNAc transferase promotes fatty liver-associated liver cancer through inducing palmitic acid and activating endoplasmic reticulum stress. J. Hepatol. 2017, 67, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis--A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 373, 96. [Google Scholar] [CrossRef]
- Fan, X.; Chuan, S.; Hongshan, W. Protein O glycosylation regulates activation of hepatic stellate cells. Inflammation 2013, 36, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ong, Q.; Wong, C.C.; Chu, E.S.H.; Sung, J.J.Y.; Yang, X.; Yu, J. O-GlcNAcylation inhibits hepatic stellate cell activation. J. Gastroenterol. Hepatol. 2021, 36, 3477–3486. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.R.; Hart, G.W. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef]
- Housley, M.P.; Udeshi, N.D.; Rodgers, J.T.; Shabanowitz, J.; Puigserver, P.; Hunt, D.F.; Hart, G.W. A PGC-1alpha-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J. Biol. Chem. 2009, 284, 5148–5157. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Z.; Xie, T.; Liu, J.; Zhang, Q.; Xiao, X. Emerging Role of Protein O-GlcNAcylation in Liver Metabolism: Implications for Diabetes and NAFLD. Int. J. Mol. Sci. 2023, 24, 2142. https://doi.org/10.3390/ijms24032142
Xie Z, Xie T, Liu J, Zhang Q, Xiao X. Emerging Role of Protein O-GlcNAcylation in Liver Metabolism: Implications for Diabetes and NAFLD. International Journal of Molecular Sciences. 2023; 24(3):2142. https://doi.org/10.3390/ijms24032142
Chicago/Turabian StyleXie, Ziyan, Ting Xie, Jieying Liu, Qian Zhang, and Xinhua Xiao. 2023. "Emerging Role of Protein O-GlcNAcylation in Liver Metabolism: Implications for Diabetes and NAFLD" International Journal of Molecular Sciences 24, no. 3: 2142. https://doi.org/10.3390/ijms24032142
APA StyleXie, Z., Xie, T., Liu, J., Zhang, Q., & Xiao, X. (2023). Emerging Role of Protein O-GlcNAcylation in Liver Metabolism: Implications for Diabetes and NAFLD. International Journal of Molecular Sciences, 24(3), 2142. https://doi.org/10.3390/ijms24032142