
Insight into Drug Resistance in Status Epilepticus: Evidence from Animal Models


Abstract
1. Introduction
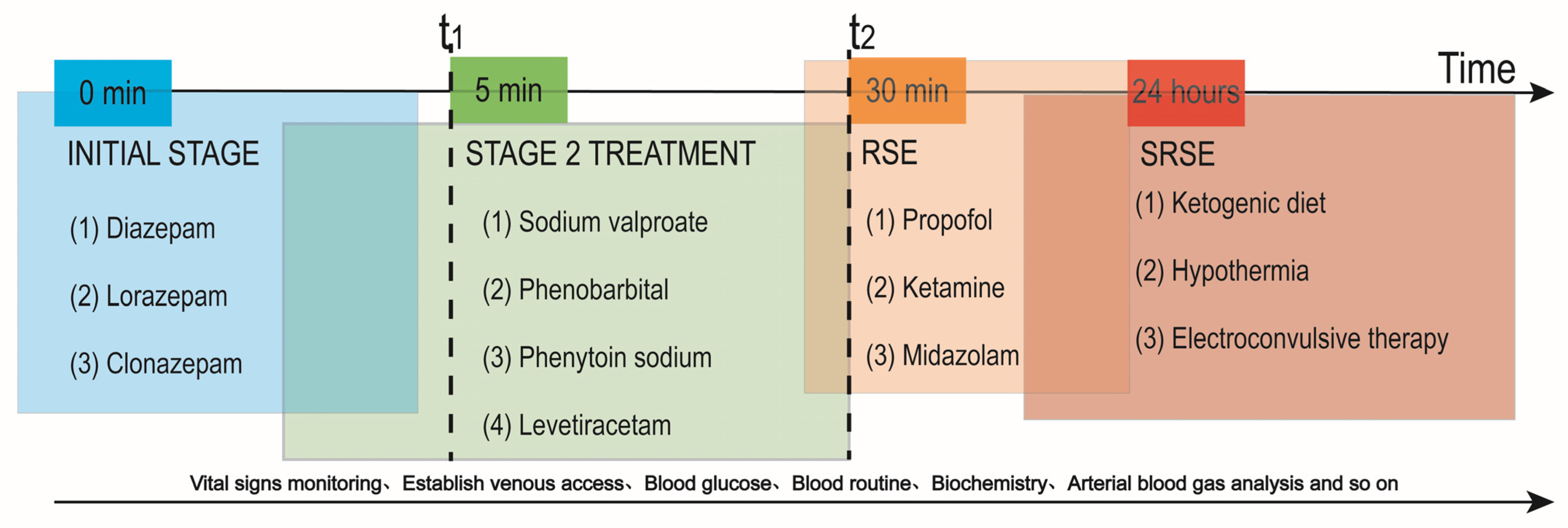
2. Current Dilemmas in Pharmacological Treatment
3. Animal Models of SE
3.1. KA-Induced SE Model
3.2. Pilocarpine-Induced SE Model
3.3. Kindling-Induced SE Model
3.4. Prolonged FS-Induced SE Model
3.5. Other SE Models
4. Common Hypotheses of Drug-Resistant SE
4.1. Degraded GABAergic Transmission Hypothesis
4.2. Augment of AMPA Receptor Function Hypothesis
4.3. Overactivation of Neuroinflammation Hypothesis
4.4. Upregulated P-glycoprotein (P-gp) Hypothesis
4.5. Neurotrophic Factors (NTFs) Hypothesis
5. Summary and Outlook
- (1)
- New Mechanism of SE in both molecular and circuit levels
- (2)
- New smart therapeutics with safe and effective features
- (3)
- Prediction of drug resistance in SE
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SE | Status epilepticus |
| ILAE | International League Against Epilepsy |
| AED | Antiepileptic drug |
| EEG | Electroencephalogram |
| KA | Kainic acid |
| FS | Febrile seizure |
| CNS | Central nervous system |
| RSE | Refractory status epilepticus |
| SRSE | Super-refractory status epilepticus |
| NMDA | N-methyl-D-aspartic acid |
| GABA | γ-aminobutyric acid |
| LPS | Lipopolysaccharide |
| AMPAR | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| IL-1β | Interleukin-1β |
| HMGB1 | High mobility group box 1 |
| TLR4 | Toll-like receptor 4 |
| NF-κB | Nuclear factor kappa-B |
| P-gp | P-glycoprotein |
| COX-2 | Cyclooxygenase-2 |
| NTFs | Neurotrophic factors |
| BDNF | Brain-derived neurotrophic factor |
| FGF2 | Fibroblast growth factor 2 |
| TrkB | Tropomyosin-related kinase B |
References
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus–Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Leitinger, M.; Trinka, E.; Giovannini, G.; Zimmermann, G.; Florea, C.; Rohracher, A.; Kalss, G.; Neuray, C.; Kreidenhuber, R.; Höfler, J.; et al. Epidemiology of status epilepticus in adults: A population-based study on incidence, causes, and outcomes. Epilepsia 2019, 60, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Nazerian, P.; Lazzeretti, D.; Vanni, S.; Donnarumma, E.; Magazzini, S.; Ruggiano, G.; Giannasi, G.; Grifoni, S.; Zaccara, G. Incidence, management and short-term prognosis of status epilepticus in the emergency department: A population survey. Eur. J. Emerg. Med. 2019, 26, 228–230. [Google Scholar] [CrossRef]
- Tiamkao, S.; Pranboon, S.; Thepsuthammarat, K.; Sawanyawisuth, K. Incidences and outcomes of status epilepticus: A 9-year longitudinal national study. Epilepsy Behav. 2015, 49, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Sutter, R.; Dittrich, T.; Semmlack, S.; Rüegg, S.; Marsch, S.; Kaplan, P.W. Acute Systemic Complications of Convulsive Status Epilepticus—A Systematic Review. Crit. Care Med. 2018, 46, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, M.A.; Hocker, S.E. Systemic Complications Following Status Epilepticus. Curr. Neurol. Neurosci. Rep. 2018, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Alldredge, B.K.; Gelb, A.M.; Isaacs, S.M.; Corry, M.D.; Allen, F.; Ulrich, S.; Gottwald, M.D.; O’Neil, N.; Neuhaus, J.M.; Segal, M.R.; et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N. Engl. J. Med. 2001, 345, 631–637. [Google Scholar] [CrossRef]
- McTague, A.; Martland, T.; Appleton, R. Drug management for acute tonic-clonic convulsions including convulsive status epilepticus in children. Cochrane Database Syst. Rev. 2018, 1, Cd001905. [Google Scholar] [CrossRef]
- Silbergleit, R.; Durkalski, V.; Lowenstein, D.; Conwit, R.; Pancioli, A.; Palesch, Y.; Barsan, W. Intramuscular versus intravenous therapy for prehospital status epilepticus. N. Engl. J. Med. 2012, 366, 591–600. [Google Scholar] [CrossRef]
- Alvarez, V.; Rossetti, A.O. Monotherapy or Polytherapy for First-Line Treatment of SE? J. Clin. Neurophysiol. 2016, 33, 14–17. [Google Scholar] [CrossRef]
- Kapur, J.; Elm, J.; Chamberlain, J.M.; Barsan, W.; Cloyd, J.; Lowenstein, D.; Shinnar, S.; Conwit, R.; Meinzer, C.; Cock, H.; et al. Randomized Trial of Three Anticonvulsant Medications for Status Epilepticus. N. Engl. J. Med. 2019, 381, 2103–2113. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.M.; Kapur, J.; Shinnar, S.; Elm, J.; Holsti, M.; Babcock, L.; Rogers, A.; Barsan, W.; Cloyd, J.; Lowenstein, D.; et al. Efficacy of levetiracetam, fosphenytoin, and valproate for established status epilepticus by age group (ESETT): A double-blind, responsive-adaptive, randomised controlled trial. Lancet 2020, 395, 1217–1224. [Google Scholar] [CrossRef]
- Lyttle, M.D.; Rainford, N.E.A.; Gamble, C.; Messahel, S.; Humphreys, A.; Hickey, H.; Woolfall, K.; Roper, L.; Noblet, J.; Lee, E.D.; et al. Levetiracetam versus phenytoin for second-line treatment of paediatric convulsive status epilepticus (EcLiPSE): A multicentre, open-label, randomised trial. Lancet 2019, 393, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Dalziel, S.R.; Borland, M.L.; Furyk, J.; Bonisch, M.; Neutze, J.; Donath, S.; Francis, K.L.; Sharpe, C.; Harvey, A.S.; Davidson, A.; et al. Levetiracetam versus phenytoin for second-line treatment of convulsive status epilepticus in children (ConSEPT): An open-label, multicentre, randomised controlled trial. Lancet 2019, 393, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Alolayan, Y.S.; McKinley, K.; Bhatia, R.; Alkhachroum, A. Review and Updates on the Treatment of Refractory and Super Refractory Status Epilepticus. J. Clin. Med. 2021, 10, 3028. [Google Scholar] [CrossRef] [PubMed]
- Vossler, D.G.; Bainbridge, J.L.; Boggs, J.G.; Novotny, E.J.; Loddenkemper, T.; Faught, E.; Amengual-Gual, M.; Fischer, S.N.; Gloss, D.S.; Olson, D.M.; et al. Treatment of Refractory Convulsive Status Epilepticus: A Comprehensive Review by the American Epilepsy Society Treatments Committee. Epilepsy Curr. 2020, 20, 245–264. [Google Scholar] [CrossRef]
- Rosati, A.; De Masi, S.; Guerrini, R. Ketamine for Refractory Status Epilepticus: A Systematic Review. CNS Drugs 2018, 32, 997–1009. [Google Scholar] [CrossRef]
- Stetefeld, H.R.; Schaal, A.; Scheibe, F.; Nichtweiß, J.; Lehmann, F.; Müller, M.; Gerner, S.T.; Huttner, H.B.; Luger, S.; Fuhrer, H.; et al. Isoflurane in (Super-) Refractory Status Epilepticus: A Multicenter Evaluation. Neurocrit. Care 2021, 35, 631–639. [Google Scholar] [CrossRef]
- Shorvon, S. Super-refractory status epilepticus: An approach to therapy in this difficult clinical situation. Epilepsia 2011, 52 (Suppl. 8), 53–56. [Google Scholar] [CrossRef]
- Hocker, S.; Tatum, W.O.; LaRoche, S.; Freeman, W.D. Refractory and super-refractory status epilepticus—An update. Curr. Neurol. Neurosci. Rep. 2014, 14, 452. [Google Scholar] [CrossRef]
- Owens, J. Medical management of refractory status epilepticus. Semin. Pediatr. Neurol. 2010, 17, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, N.; Foreman, B.P.; Alvarez, V.; Cabrera Kang, C.; Probasco, J.C.; Jongeling, A.C.; Meyers, E.; Espinera, A.; Haas, K.F.; Schmitt, S.E.; et al. New-onset refractory status epilepticus: Etiology, clinical features, and outcome. Neurology 2015, 85, 1604–1613. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, D.H.; Alldredge, B.K. Status epilepticus. N. Engl. J. Med. 1998, 338, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, A.O.; Alvarez, V. Update on the management of status epilepticus. Curr. Opin. Neurol. 2021, 34, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Jehi, L. Consequences of status epilepticus in the intensive care unit: What we know and what we need to know. Epilepsy Curr. 2014, 14, 337–338. [Google Scholar] [CrossRef]
- Leppik, I.E. Status epilepticus in the elderly. Epilepsia 2018, 59 (Suppl. 2), 140–143. [Google Scholar] [CrossRef]
- Gasparini, S.; Ferlazzo, E.; Gigli, G.; Pauletto, G.; Nilo, A.; Lettieri, C.; Bilo, L.; Labate, A.; Fortunato, F.; Varrasi, C.; et al. Predictive factors of Status Epilepticus and its recurrence in patients with adult-onset seizures: A multicenter, long follow-up cohort study. Seizure 2021, 91, 397–401. [Google Scholar] [CrossRef]
- Orlandi, N.; Gozzi, A.; Giovannini, G.; Turchi, G.; Cioclu, M.C.; Vaudano, A.E.; Meletti, S. Recurrent status epilepticus: Clinical features and recurrence risk in an adult population. Seizure 2022, 97, 1–7. [Google Scholar] [CrossRef]
- Bateman, D.E. Pseudostatus epilepticus. Lancet 1989, 2, 1278–1279. [Google Scholar] [CrossRef]
- Appleton, R.E. Treatment of childhood epilepsy. Pharmacol. Ther. 1995, 67, 419–431. [Google Scholar] [CrossRef]
- Simonato, M.; Brooks-Kayal, A.R.; Engel, J., Jr.; Galanopoulou, A.S.; Jensen, F.E.; Moshé, S.L.; O’Brien, T.J.; Pitkanen, A.; Wilcox, K.S.; French, J.A. The challenge and promise of anti-epileptic therapy development in animal models. Lancet Neurol. 2014, 13, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Luo, Y.; Hou, G.L.; He, R.J.; Zhang, H.Y.; Yi, Y.L.; Zhang, Y.; Cui, Z.Q. Pretreatment with Methylene Blue Protects Against Acute Seizure and Oxidative Stress in a Kainic Acid-Induced Status Epilepticus Model. Med. Sci. Monit. 2021, 27, e933469. [Google Scholar] [CrossRef] [PubMed]
- Rusina, E.; Bernard, C.; Williamson, A. The Kainic Acid Models of Temporal Lobe Epilepsy. eNeuro 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Sperk, G.; Lassmann, H.; Baran, H.; Seitelberger, F.; Hornykiewicz, O. Kainic acid-induced seizures: Dose-relationship of behavioural, neurochemical and histopathological changes. Brain Res. 1985, 338, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.Q.; Koh, S.; Torgerson, T.; Cole, A.J. Neuronal stress and injury in C57/BL mice after systemic kainic acid administration. Brain Res. 1998, 810, 229–240. [Google Scholar] [CrossRef]
- McKhann, G.M., 2nd; Wenzel, H.J.; Robbins, C.A.; Sosunov, A.A.; Schwartzkroin, P.A. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience 2003, 122, 551–561. [Google Scholar] [CrossRef]
- Cui, H.S.; Kim, M.R.; Sok, D.E. Protection by petaslignolide A, a major neuroprotective compound in the butanol extract of Petasites japonicus leaves, against oxidative damage in the brains of mice challenged with kainic acid. J. Agric. Food Chem. 2005, 53, 8526–8532. [Google Scholar] [CrossRef]
- Terrone, G.; Pauletti, A.; Salamone, A.; Rizzi, M.; Villa, B.R.; Porcu, L.; Sheehan, M.J.; Guilmette, E.; Butler, C.R.; Piro, J.R.; et al. Inhibition of monoacylglycerol lipase terminates diazepam-resistant status epilepticus in mice and its effects are potentiated by a ketogenic diet. Epilepsia 2018, 59, 79–91. [Google Scholar] [CrossRef]
- West, P.J.; Thomson, K.; Billingsley, P.; Pruess, T.; Rueda, C.; Saunders, G.W.; Smith, M.D.; Metcalf, C.S.; Wilcox, K.S. Spontaneous recurrent seizures in an intra-amygdala kainate microinjection model of temporal lobe epilepsy are differentially sensitive to antiseizure drugs. Exp. Neurol. 2022, 349, 113954. [Google Scholar] [CrossRef]
- Lu, M.O.; Zhang, X.M.; Mix, E.; Quezada, H.C.; Jin, T.; Zhu, J.; Adem, A. TNF-α receptor 1 deficiency enhances kainic acid-induced hippocampal injury in mice. J. Neurosci. Res. 2008, 86, 1608–1614. [Google Scholar] [CrossRef]
- Sabilallah, M.; Fontanaud, P.; Linck, N.; Boussadia, B.; Peyroutou, R.; Lasgouzes, T.; Rassendren, F.A.; Marchi, N.; Hirbec, H.E. Evidence for Status Epilepticus and Pro-Inflammatory Changes after Intranasal Kainic Acid Administration in Mice. PLoS ONE 2016, 11, e0150793. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, B.; Stott, J.J.; Donofrio, J.J.; Rogawski, M.A. Treatment of early and late kainic acid-induced status epilepticus with the noncompetitive AMPA receptor antagonist GYKI 52466. Epilepsia 2010, 51, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zheng, Y.; Liu, K.; Chen, J.; Lai, N.; Fei, F.; Shi, J.; Xu, C.; Wang, S.; Nishibori, M.; et al. HMGB1 Is a Therapeutic Target and Biomarker in Diazepam-Refractory Status Epilepticus with Wide Time Window. Neurotherapeutics 2020, 17, 710–721. [Google Scholar] [CrossRef]
- Xu, Z.H.; Wang, Y.; Tao, A.F.; Yu, J.; Wang, X.Y.; Zu, Y.Y.; Zhang, S.H.; Chen, Z. Interleukin-1 receptor is a target for adjunctive control of diazepam-refractory status epilepticus in mice. Neuroscience 2016, 328, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Turski, L.; Ikonomidou, C.; Turski, W.A.; Bortolotto, Z.A.; Cavalheiro, E.A. Review: Cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: A novel experimental model of intractable epilepsy. Synapse 1989, 3, 154–171. [Google Scholar] [CrossRef]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef]
- Turski, W.A.; Cavalheiro, E.A.; Schwarz, M.; Czuczwar, S.J.; Kleinrok, Z.; Turski, L. Limbic seizures produced by pilocarpine in rats: Behavioural, electroencephalographic and neuropathological study. Behav. Brain Res. 1983, 9, 315–335. [Google Scholar] [CrossRef]
- Liu, Z.; Gatt, A.; Werner, S.J.; Mikati, M.A.; Holmes, G.L. Long-term behavioral deficits following pilocarpine seizures in immature rats. Epilepsy Res. 1994, 19, 191–204. [Google Scholar] [CrossRef]
- Maia, O.A.C.; Malheiros-Lima, M.R.; Oliveira, M.A.; Castro, C.L.; Moriya, H.T.; Tavares-de-Lima, W.; Takakura, A.C.; Moreira, T.S. Pilocarpine-induced status epilepticus reduces chemosensory control of breathing. Brain Res. Bull. 2020, 161, 98–105. [Google Scholar] [CrossRef]
- de Aquino, P.E.A.; Rabelo Bezerra, J.; de Souza Nascimento, T.; Tavares, J.; Lustosa, Í.R.; Filho, A.J.M.C.; Mottin, M.; Gaspar, D.M.; Andrade, G.M.; Neves, K.R.T.; et al. A Proline Derivative-Enriched Fraction from Sideroxylon obtusifolium Protects the Hippocampus from Intracerebroventricular Pilocarpine-Induced Injury Associated with Status Epilepticus in Mice. Int. J. Mol. Sci. 2020, 21, 4188. [Google Scholar] [CrossRef]
- Martin, B.S.; Kapur, J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia 2008, 49, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Zaayman, M.; Kuruba, R.; Wu, X. Comparative profile of refractory status epilepticus models following exposure of cholinergic agents pilocarpine, DFP, and soman. Neuropharmacology 2021, 191, 108571. [Google Scholar] [CrossRef]
- Jones, D.M.; Esmaeil, N.; Maren, S.; Macdonald, R.L. Characterization of pharmacoresistance to benzodiazepines in the rat Li-pilocarpine model of status epilepticus. Epilepsy Res. 2002, 50, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Kapur, J.; Macdonald, R.L. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J. Neurosci. 1997, 17, 7532–7540. [Google Scholar] [CrossRef]
- Gorter, J.A.; van Vliet, E.A.; Lopes da Silva, F.H. Which insights have we gained from the kindling and post-status epilepticus models? J. Neurosci. Methods 2016, 260, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W. Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res. 2016, 126, 157–184. [Google Scholar] [CrossRef] [PubMed]
- Borris, D.J.; Bertram, E.H.; Kapur, J. Ketamine controls prolonged status epilepticus. Epilepsy Res. 2000, 42, 117–122. [Google Scholar] [CrossRef]
- Brandt, C.; Glien, M.; Potschka, H.; Volk, H.; Löscher, W. Epileptogenesis and neuropathology after different types of status epilepticus induced by prolonged electrical stimulation of the basolateral amygdala in rats. Epilepsy Res. 2003, 55, 83–103. [Google Scholar] [CrossRef]
- Bankstahl, J.P.; Löscher, W. Resistance to antiepileptic drugs and expression of P-glycoprotein in two rat models of status epilepticus. Epilepsy Res. 2008, 82, 70–85. [Google Scholar] [CrossRef]
- Seinfeld, D.S.; Pellock, J.M. Recent Research on Febrile Seizures: A Review. J. Neurol. Neurophysiol 2013, 4, 19519. [Google Scholar] [CrossRef]
- Shinnar, S. Febrile Seizures and Mesial Temporal Sclerosis. Epilepsy Curr. 2003, 3, 115–118. [Google Scholar] [CrossRef]
- Rakgantsho, C.; Mabandla, M.V. Acetylcholine receptor agonist effect on seizure activity and GABAergic mechanisms involved in prolonged febrile seizure development in an animal model. Brain Res. Bull. 2019, 149, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R. Recent advances in febrile seizures. Indian J. Pediatr. 2014, 81, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Shinnar, S.; Pellock, J.M.; Moshé, S.L.; Maytal, J.; O’Dell, C.; Driscoll, S.M.; Alemany, M.; Newstein, D.; DeLorenzo, R.J. In whom does status epilepticus occur: Age-related differences in children. Epilepsia 1997, 38, 907–914. [Google Scholar] [CrossRef]
- Bender, R.A.; Dubé, C.; Baram, T.Z. Febrile seizures and mechanisms of epileptogenesis: Insights from an animal model. Adv. Exp. Med. Biol. 2004, 548, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yang, Z.; Jin, B.; Qin, X.; Zhu, X.; Sun, J.; Huo, L.; Wang, R.; Shi, Y.; Jia, Z.; et al. Cannabidiol inhibits febrile seizure by modulating AMPA receptor kinetics through its interaction with the N-terminal domain of GluA1/GluA2. Pharmacol. Res. 2020, 161, 105128. [Google Scholar] [CrossRef]
- Chen, B.; Feng, B.; Tang, Y.; You, Y.; Wang, Y.; Hou, W.; Hu, W.; Chen, Z. Blocking GluN2B subunits reverses the enhanced seizure susceptibility after prolonged febrile seizures with a wide therapeutic time-window. Exp. Neurol. 2016, 283, 29–38. [Google Scholar] [CrossRef]
- Wang, X.; Yang, F.; Deng, L.; Qiu, D.; Liu, Y.; Kang, Y. Liraglutide Is Protective against Brain Injury in Mice with Febrile Seizures by Inhibiting Inflammatory Factors. Comput. Math. Methods Med. 2022, 2022, 7563281. [Google Scholar] [CrossRef]
- Dutton, S.B.B.; Dutt, K.; Papale, L.A.; Helmers, S.; Goldin, A.L.; Escayg, A. Early-life febrile seizures worsen adult phenotypes in Scn1a mutants. Exp. Neurol. 2017, 293, 159–171. [Google Scholar] [CrossRef]
- Eun, B.L.; Abraham, J.; Mlsna, L.; Kim, M.J.; Koh, S. Lipopolysaccharide potentiates hyperthermia-induced seizures. Brain Behav. 2015, 5, e00348. [Google Scholar] [CrossRef]
- Baram, T.Z.; Gerth, A.; Schultz, L. Febrile seizures: An appropriate-aged model suitable for long-term studies. Brain Res. Dev. Brain Res. 1997, 98, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.S.; Aboul Ezz, H.S.; Sayed, H.M.; Ali, M.A. Electroencephalographic and biochemical long-lasting abnormalities in animal model of febrile seizure. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2120–2125. [Google Scholar] [CrossRef]
- Tang, Y.; Feng, B.; Wang, Y.; Sun, H.; You, Y.; Yu, J.; Chen, B.; Xu, C.; Ruan, Y.; Cui, S.; et al. Structure-based discovery of CZL80, a caspase-1 inhibitor with therapeutic potential for febrile seizures and later enhanced epileptogenic susceptibility. Br. J. Pharmacol. 2020, 177, 3519–3534. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Tang, Y.; Li, W.; You, Y.; Shi, J.; Xu, C.; Du, Y.; Chen, Z.; Wang, Y. Thermo-sensitive micelles extend therapeutic potential for febrile seizures. Signal Transduct. Target. Ther. 2021, 6, 296. [Google Scholar] [CrossRef]
- Feng, B.; Tang, Y.S.; Chen, B.; Xu, Z.H.; Wang, Y.; Wu, D.C.; Zhao, H.W.; Zhang, S.H.; Chen, Z. Early hypoactivity of hippocampal rhythms during epileptogenesis after prolonged febrile seizures in freely-moving rats. Neurosci. Bull. 2015, 31, 297–306. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, T.; Joshi, S.; Williamson, J.M.; Kapur, J. Neocortical injury-induced status epilepticus. Epilepsia 2020, 61, 2811–2824. [Google Scholar] [CrossRef]
- Andrade, P.; Banuelos-Cabrera, I.; Lapinlampi, N.; Paananen, T.; Ciszek, R.; Ndode-Ekane, X.E.; Pitkänen, A. Acute Non-Convulsive Status Epilepticus after Experimental Traumatic Brain Injury in Rats. J. Neurotrauma 2019, 36, 1890–1907. [Google Scholar] [CrossRef]
- Hoover, D.B.; Craig, C.R.; Colasanti, B.K. Cholinergic involvement in cobalt-induced epilepsy in the rat. Exp. Brain Res. 1977, 29, 501–513. [Google Scholar] [CrossRef]
- Wang, J.; Wu, C.; Peng, J.; Patel, N.; Huang, Y.; Gao, X.; Aljarallah, S.; Eubanks, J.H.; McDonald, R.; Zhang, L. Early-Onset Convulsive Seizures Induced by Brain Hypoxia-Ischemia in Aging Mice: Effects of Anticonvulsive Treatments. PLoS ONE 2015, 10, e0144113. [Google Scholar] [CrossRef]
- Rubaj, A.; Zgodziński, W.; Sieklucka-Dziuba, M. The epileptogenic effect of seizures induced by hypoxia: The role of NMDA and AMPA/KA antagonists. Pharmacol. Biochem. Behav. 2003, 74, 303–311. [Google Scholar] [CrossRef]
- Walton, N.Y.; Treiman, D.M. Experimental secondarily generalized convulsive status epilepticus induced by D,L-homocysteine thiolactone. Epilepsy Res. 1988, 2, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, L.S.; DeLorenzo, R.J. Novel therapeutics for treating organophosphate-induced status epilepticus co-morbidities, based on changes in calcium homeostasis. Neurobiol. Dis. 2020, 133, 104418. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Ganesh, T.; Wang, W.; Wang, J.; Dingledine, R. A rat model of organophosphate-induced status epilepticus and the beneficial effects of EP2 receptor inhibition. Neurobiol. Dis. 2020, 133, 104399. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Ganesh, T.; Lelutiu, N.; Gueorguieva, P.; Dingledine, R. Inhibition of the prostaglandin EP2 receptor is neuroprotective and accelerates functional recovery in a rat model of organophosphorus induced status epilepticus. Neuropharmacology 2015, 93, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Ganesh, T.; Manji, Z.; O’Neill, T.; Dingledine, R. Inhibition of the prostaglandin E2 receptor EP2 prevents status epilepticus-induced deficits in the novel object recognition task in rats. Neuropharmacology 2016, 110, 419–430. [Google Scholar] [CrossRef]
- Rojas, A.; Wang, W.; Glover, A.; Manji, Z.; Fu, Y.; Dingledine, R. Beneficial Outcome of Urethane Treatment Following Status Epilepticus in a Rat Organophosphorus Toxicity Model. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Reddy, D.S.; Perumal, D.; Golub, V.; Habib, A.; Kuruba, R.; Wu, X. Phenobarbital as alternate anticonvulsant for organophosphate-induced benzodiazepine-refractory status epilepticus and neuronal injury. Epilepsia Open 2020, 5, 198–212. [Google Scholar] [CrossRef]
- Enderlin, J.; Igert, A.; Auvin, S.; Nachon, F.; Bo, G.D.; Dupuis, N. Characterization of organophosphate-induced brain injuries in a convulsive mouse model of diisopropylfluorophosphate exposure. Epilepsia 2020, 61, e54–e59. [Google Scholar] [CrossRef]
- Zhou, H.; Tang, Y.H.; Zheng, Y. A new rat model of acute seizures induced by tutin. Brain Res. 2006, 1092, 207–213. [Google Scholar] [CrossRef]
- Sechi, G.; De Riu, P.; Mameli, O.; Deiana, G.A.; Cocco, G.A.; Rosati, G. Focal and secondarily generalised convulsive status epilepticus induced by thiocolchicoside in the rat. Seizure 2003, 12, 508–515. [Google Scholar] [CrossRef]
- Treiman, D.M. GABAergic mechanisms in epilepsy. Epilepsia 2001, 42 (Suppl. 3), 8–12. [Google Scholar] [CrossRef]
- Roberts, E.; Frankel, S. gamma-Aminobutyric acid in brain: Its formation from glutamic acid. J. Biol. Chem. 1950, 187, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Buzsáki, G.; Kaila, K.; Raichle, M. Inhibition and brain work. Neuron 2007, 56, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, H.P.; Joshi, S.; Mtchedlishvili, Z.; Brar, J.; Kapur, J. Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J. Neurosci. 2008, 28, 2527–2538. [Google Scholar] [CrossRef]
- Naylor, D.E.; Liu, H.; Wasterlain, C.G. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J. Neurosci. 2005, 25, 7724–7733. [Google Scholar] [CrossRef]
- Stelzer, A. Regulation of GABAA currents by excitatory amino acids. Adv. Exp. Med. Biol. 1990, 268, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Kapur, J.; Lothman, E.W. NMDA receptor activation mediates the loss of GABAergic inhibition induced by recurrent seizures. Epilepsy Res. 1990, 5, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Min, M.Y.; Melyan, Z.; Kullmann, D.M. Synaptically released glutamate reduces gamma-aminobutyric acid (GABA)ergic inhibition in the hippocampus via kainate receptors. Proc. Natl. Acad. Sci. USA 1999, 96, 9932–9937. [Google Scholar] [CrossRef]
- Silayeva, L.; Deeb, T.Z.; Hines, R.M.; Kelley, M.R.; Munoz, M.B.; Lee, H.H.; Brandon, N.J.; Dunlop, J.; Maguire, J.; Davies, P.A.; et al. KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. USA 2015, 112, 3523–3528. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Donevan, S.D. AMPA receptors in epilepsy and as targets for antiepileptic drugs. Adv. Neurol. 1999, 79, 947–963. [Google Scholar]
- Leo, A.; Giovannini, G.; Russo, E.; Meletti, S. The role of AMPA receptors and their antagonists in status epilepticus. Epilepsia 2018, 59, 1098–1108. [Google Scholar] [CrossRef]
- Rogawski, M.A. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr. 2011, 11, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Borges, K.; Dingledine, R. AMPA receptors: Molecular and functional diversity. Prog. Brain Res. 1998, 116, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Lothman, E. The biochemical basis and pathophysiology of status epilepticus. Neurology 1990, 40, 13–23. [Google Scholar] [PubMed]
- Joshi, S.; Kapur, J. Mechanisms of status epilepticus: α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor hypothesis. Epilepsia 2018, 59 (Suppl. 2), 78–81. [Google Scholar] [CrossRef]
- Chen, J.W.; Naylor, D.E.; Wasterlain, C.G. Advances in the pathophysiology of status epilepticus. Acta Neurol. Scand. Suppl. 2007, 186, 7–15. [Google Scholar] [CrossRef]
- Bredt, D.S.; Nicoll, R.A. AMPA receptor trafficking at excitatory synapses. Neuron 2003, 40, 361–379. [Google Scholar] [CrossRef]
- Jefferys, J.G.; Traub, R.D. Electrophysiological substrates for focal epilepsies. Prog. Brain Res. 1998, 116, 351–358. [Google Scholar] [CrossRef]
- Pitkänen, A.; Mathiesen, C.; Rønn, L.C.; Møller, A.; Nissinen, J. Effect of novel AMPA antagonist, NS1209, on status epilepticus. An experimental study in rat. Epilepsy Res. 2007, 74, 45–54. [Google Scholar] [CrossRef]
- Schmidt-Kastner, R.; Tomac, A.; Hoffer, B.; Bektesh, S.; Rosenzweig, B.; Olson, L. Glial cell-line derived neurotrophic factor (GDNF) mRNA upregulation in striatum and cortical areas after pilocarpine-induced status epilepticus in rats. Brain Res. Mol. Brain Res. 1994, 26, 325–330. [Google Scholar] [CrossRef]
- Donevan, S.D.; Rogawski, M.A. GYKI 52466, a 2,3-benzodiazepine, is a highly selective, noncompetitive antagonist of AMPA/kainate receptor responses. Neuron 1993, 10, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Maroso, M.; Balosso, S.; Sanchez, M.A.; Bartfai, T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav. Immun. 2011, 25, 1281–1289. [Google Scholar] [CrossRef]
- De Simoni, M.G.; Perego, C.; Ravizza, T.; Moneta, D.; Conti, M.; Marchesi, F.; De Luigi, A.; Garattini, S.; Vezzani, A. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur. J. Neurosci. 2000, 12, 2623–2633. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Min, H.J.; Shin, J.S. Increased levels of HMGB1 and pro-inflammatory cytokines in children with febrile seizures. J. Neuroinflammation 2011, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, T.; Terrone, G.; Salamone, A.; Frigerio, F.; Balosso, S.; Antoine, D.J.; Vezzani, A. High Mobility Group Box 1 is a novel pathogenic factor and a mechanistic biomarker for epilepsy. Brain Behav. Immun. 2018, 72, 14–21. [Google Scholar] [CrossRef]
- Varvel, N.H.; Neher, J.J.; Bosch, A.; Wang, W.; Ransohoff, R.M.; Miller, R.J.; Dingledine, R. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. USA 2016, 113, E5665–E5674. [Google Scholar] [CrossRef]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef]
- Ravizza, T.; Gagliardi, B.; Noé, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef]
- Ravizza, T.; Boer, K.; Redeker, S.; Spliet, W.G.; van Rijen, P.C.; Troost, D.; Vezzani, A.; Aronica, E. The IL-1β system in epilepsy-associated malformations of cortical development. Neurobiol. Dis. 2006, 24, 128–143. [Google Scholar] [CrossRef]
- Tan, C.C.; Zhang, J.G.; Tan, M.S.; Chen, H.; Meng, D.W.; Jiang, T.; Meng, X.F.; Li, Y.; Sun, Z.; Li, M.M.; et al. NLRP1 inflammasome is activated in patients with medial temporal lobe epilepsy and contributes to neuronal pyroptosis in amygdala kindling-induced rat model. J. Neuroinflamm. 2015, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Varvel, N.H.; Jiang, J.; Dingledine, R. Candidate drug targets for prevention or modification of epilepsy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 229–247. [Google Scholar] [CrossRef]
- Jiang, J.; Yang, M.S.; Quan, Y.; Gueorguieva, P.; Ganesh, T.; Dingledine, R. Therapeutic window for cyclooxygenase-2 related anti-inflammatory therapy after status epilepticus. Neurobiol. Dis. 2015, 76, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Kemper, T.; Qiu, J.; Jiang, J. Defining the therapeutic time window for suppressing the inflammatory prostaglandin E2 signaling after status epilepticus. Expert Rev. Neurother. 2016, 16, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Cardo, C.; O’Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.; Melamed, M.R.; Bertino, J.R. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W. How to explain multidrug resistance in epilepsy? Epilepsy Curr. 2005, 5, 107–112. [Google Scholar] [CrossRef]
- Xie, Y.; Yu, N.; Chen, Y.; Zhang, K.; Ma, H.Y.; Di, Q. HMGB1 regulates P-glycoprotein expression in status epilepticus rat brains via the RAGE/NF-κB signaling pathway. Mol. Med. Rep. 2017, 16, 1691–1700. [Google Scholar] [CrossRef]
- Deng, X.; Shao, Y.; Xie, Y.; Feng, Y.; Wu, M.; Wang, M.; Chen, Y. MicroRNA-146a-5p Downregulates the Expression of P-Glycoprotein in Rats with Lithium-Pilocarpine-Induced Status Epilepticus. Biol. Pharm. Bull. 2019, 42, 744–750. [Google Scholar] [CrossRef]
- Ciceri, P.; Zhang, Y.; Shaffer, A.F.; Leahy, K.M.; Woerner, M.B.; Smith, W.G.; Seibert, K.; Isakson, P.C. Pharmacology of celecoxib in rat brain after kainate administration. J. Pharmacol. Exp. Ther. 2002, 302, 846–852. [Google Scholar] [CrossRef]
- Kanter-Schlifke, I.; Georgievska, B.; Kirik, D.; Kokaia, M. Seizure suppression by GDNF gene therapy in animal models of epilepsy. Mol. Ther. 2007, 15, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kastner, R.; Humpel, C.; Wetmore, C.; Olson, L. Cellular hybridization for BDNF, trkB, and NGF mRNAs and BDNF-immunoreactivity in rat forebrain after pilocarpine-induced status epilepticus. Exp. Brain Res. 1996, 107, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Mudò, G.; Jiang, X.H.; Timmusk, T.; Bindoni, M.; Belluardo, N. Change in neurotrophins and their receptor mRNAs in the rat forebrain after status epilepticus induced by pilocarpine. Epilepsia 1996, 37, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, J.N.; Shah, S.K.; McCloskey, D.P.; Goodman, J.H.; Elkady, A.; Atassi, H.; Hylton, D.; Rudge, J.S.; Scharfman, H.E.; Croll, S.D. Vascular endothelial growth factor is up-regulated after status epilepticus and protects against seizure-induced neuronal loss in hippocampus. Neuroscience 2008, 151, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, J.N.; Lenzer, J.; Salerni, E.A.; Shah, S.K.; Elkady, A.; Khalid, S.; Quinteros, D.; Rotella, F.; Betancourth, D.; Croll, S.D. Vascular endothelial growth factor attenuates status epilepticus-induced behavioral impairments in rats. Epilepsy Behav. 2010, 19, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Simonato, M. Neurotrophic factors and status epilepticus. Epilepsia 2018, 59 (Suppl. 2), 87–91. [Google Scholar] [CrossRef]
- Isackson, P.J.; Huntsman, M.M.; Murray, K.D.; Gall, C.M. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: Temporal patterns of induction distinct from NGF. Neuron 1991, 6, 937–948. [Google Scholar] [CrossRef]
- He, X.P.; Pan, E.; Sciarretta, C.; Minichiello, L.; McNamara, J.O. Disruption of TrkB-mediated phospholipase Cgamma signaling inhibits limbic epileptogenesis. J. Neurosci. 2010, 30, 6188–6196. [Google Scholar] [CrossRef]
- Gu, B.; Huang, Y.Z.; He, X.P.; Joshi, R.B.; Jang, W.; McNamara, J.O. A Peptide Uncoupling BDNF Receptor TrkB from Phospholipase Cγ1 Prevents Epilepsy Induced by Status Epilepticus. Neuron 2015, 88, 484–491. [Google Scholar] [CrossRef]
- Obeid, M.; Rosenberg, E.C.; Klein, P.M.; Jensen, F.E. Lestaurtinib (CEP-701) attenuates “second hit” kainic acid-induced seizures following early life hypoxic seizures. Epilepsy Res. 2014, 108, 806–810. [Google Scholar] [CrossRef]
- Liu, X.; Geng, J.; Guo, H.; Zhao, H.; Ai, Y. Propofol inhibited apoptosis of hippocampal neurons in status epilepticus through miR-15a-5p/NR2B/ERK1/2 pathway. Cell Cycle 2020, 19, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Beamer, E.; Fischer, W.; Engel, T. The ATP-Gated P2X7 Receptor As a Target for the Treatment of Drug-Resistant Epilepsy. Front. Neurosci. 2017, 11, 21. [Google Scholar] [CrossRef]
- Engel, T.; Gomez-Villafuertes, R.; Tanaka, K.; Mesuret, G.; Sanz-Rodriguez, A.; Garcia-Huerta, P.; Miras-Portugal, M.T.; Henshall, D.C.; Diaz-Hernandez, M. Seizure suppression and neuroprotection by targeting the purinergic P2X7 receptor during status epilepticus in mice. FASEB J. 2012, 26, 1616–1628. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Xu, C.; Wang, S.; Tan, N.; Chen, C.; Chen, L.; Wu, X.; Fei, F.; Cheng, H.; et al. Direct Septum-Hippocampus Cholinergic Circuit Attenuates Seizure Through Driving Somatostatin Inhibition. Biol. Psychiatry 2020, 87, 843–856. [Google Scholar] [CrossRef]
- Fei, F.; Wang, X.; Xu, C.; Shi, J.; Gong, Y.; Cheng, H.; Lai, N.; Ruan, Y.; Ding, Y.; Wang, S.; et al. Discrete subicular circuits control generalization of hippocampal seizures. Nat. Commun. 2022, 13, 5010. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Z. An update for epilepsy research and antiepileptic drug development: Toward precise circuit therapy. Pharmacol. Ther. 2019, 201, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Fei, F.; Zhang, Q.; Wang, X.; Gong, Y.; Chen, X.; Zheng, Y.; Tan, B.; Xu, C.; Xie, H.; et al. Nanoengineered on-demand drug delivery system improves efficacy of pharmacotherapy for epilepsy. Sci. Adv. 2022, 8, eabm3381. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Wang, Y.; Liang, J.; Yue, J.; Xu, C.; Lu, L.; Xu, Z.; Gao, J.; Du, Y.; Chen, Z. Angiopep-conjugated electro-responsive hydrogel nanoparticles: Therapeutic potential for epilepsy. Angew. Chem. Int. Ed. Engl. 2014, 53, 12436–12440. [Google Scholar] [CrossRef]
- Wang, Y.; Ying, X.; Chen, L.; Liu, Y.; Wang, Y.; Liang, J.; Xu, C.; Guo, Y.; Wang, S.; Hu, W.; et al. Electroresponsive Nanoparticles Improve Antiseizure Effect of Phenytoin in Generalized Tonic-Clonic Seizures. Neurotherapeutics 2016, 13, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Hanin, A.; Lambrecq, V.; Denis, J.A.; Imbert-Bismut, F.; Rucheton, B.; Lamari, F.; Bonnefont-Rousselot, D.; Demeret, S.; Navarro, V. Cerebrospinal fluid and blood biomarkers of status epilepticus. Epilepsia 2020, 61, 6–18. [Google Scholar] [CrossRef]
- Kamousi, B.; Karunakaran, S.; Gururangan, K.; Markert, M.; Decker, B.; Khankhanian, P.; Mainardi, L.; Quinn, J.; Woo, R.; Parvizi, J. Monitoring the Burden of Seizures and Highly Epileptiform Patterns in Critical Care with a Novel Machine Learning Method. Neurocritical Care 2021, 34, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Baldassano, S.N.; Hill, C.E.; Shankar, A.; Bernabei, J.; Khankhanian, P.; Litt, B. Big data in status epilepticus. Epilepsy Behav. 2019, 101, 106457. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Model | Mode of Operation | Mechanism | Mortality Rate |
|---|---|---|---|
| KA model | Intraperitoneal injection | KA binds directly to non-NMDA (KA) receptors in the neuronal postsynaptic membrane, producing excitatory postsynaptic potentials that lead to seizures. | 47–75% |
| Intraventricular injection | 8–21% | ||
| Intranasal injection | Lower than intraventricular injection | ||
| Pilocarpine model | Lithium–pilocarpine | Pilocarpine can stimulate not only the M receptor, but also NMDA receptors, metabolic glutamate receptors, resulting in activation of the excitatory system in the brain. | 27.4–40% |
| Intracerebral administration | |||
| Kindling model | Hippocampus | Repeated electrical stimulations cause a gradual change in the excitatory synaptic plasticity and lower seizure threshold. | —— |
| Amygdala | |||
| Prolonged FS | LPS-induced FS | An imbalance between the excitatory neurotransmitter glutamate and the inhibitory neurotransmitter GABA. | About 50% |
| Heat-induced FS | |||
| FS induced by heat combined with LPS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Zhang, Q.; Wang, Y.; Chen, J.; Wang, Y. Insight into Drug Resistance in Status Epilepticus: Evidence from Animal Models. Int. J. Mol. Sci. 2023, 24, 2039. https://doi.org/10.3390/ijms24032039
Wang F, Zhang Q, Wang Y, Chen J, Wang Y. Insight into Drug Resistance in Status Epilepticus: Evidence from Animal Models. International Journal of Molecular Sciences. 2023; 24(3):2039. https://doi.org/10.3390/ijms24032039
Chicago/Turabian StyleWang, Fei, Qingyang Zhang, Yu Wang, Junzi Chen, and Yi Wang. 2023. "Insight into Drug Resistance in Status Epilepticus: Evidence from Animal Models" International Journal of Molecular Sciences 24, no. 3: 2039. https://doi.org/10.3390/ijms24032039
APA StyleWang, F., Zhang, Q., Wang, Y., Chen, J., & Wang, Y. (2023). Insight into Drug Resistance in Status Epilepticus: Evidence from Animal Models. International Journal of Molecular Sciences, 24(3), 2039. https://doi.org/10.3390/ijms24032039