
Enhanced IRE1α Phosphorylation/Oligomerization-Triggered XBP1 Splicing Contributes to Parkin-Mediated Prevention of SH-SY5Y Cell Death under Nitrosative Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
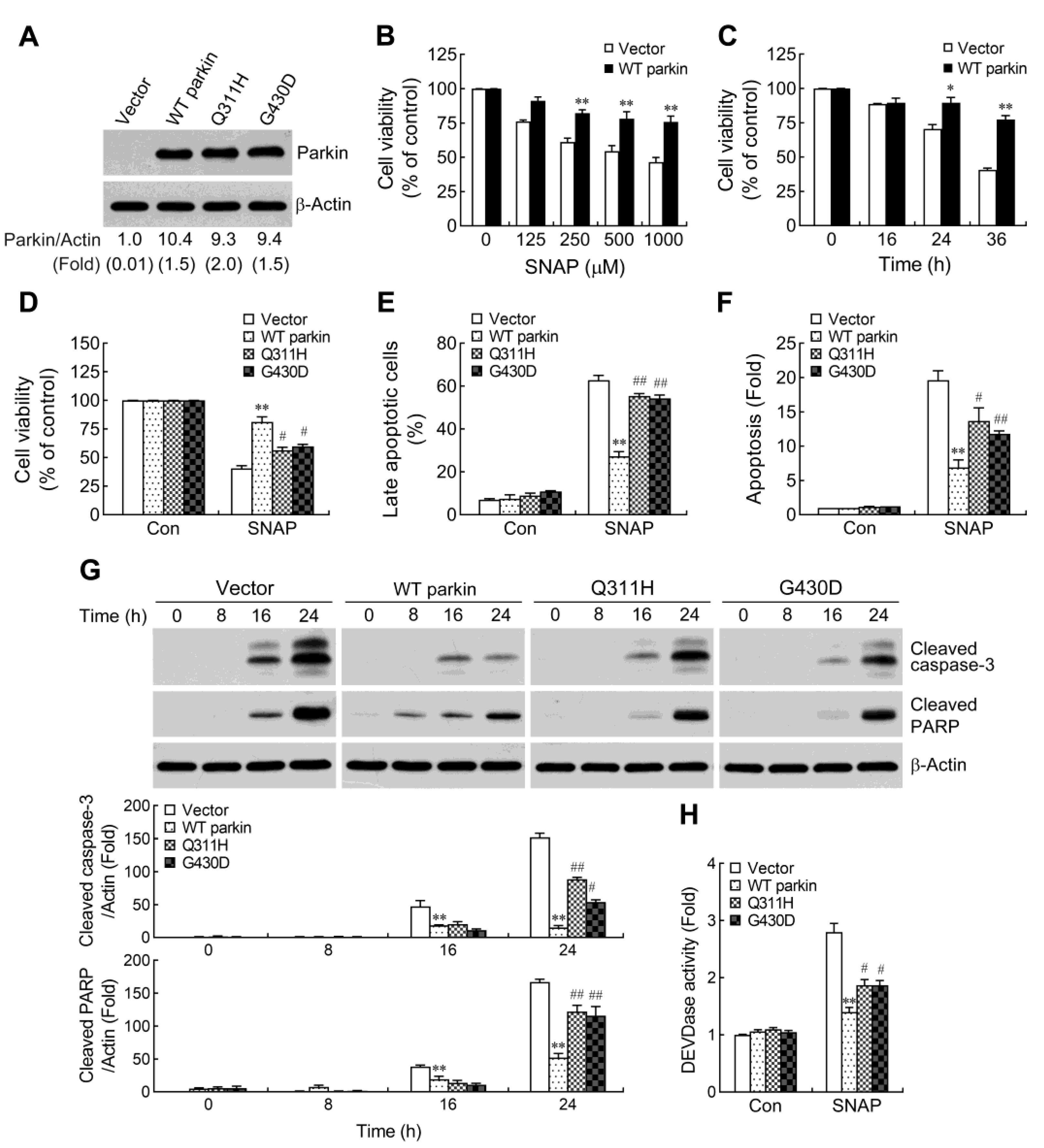
2.1. Pathogenic Mutation of Parkin Impairs the Protective Capacity against NO-Induced Apoptosis
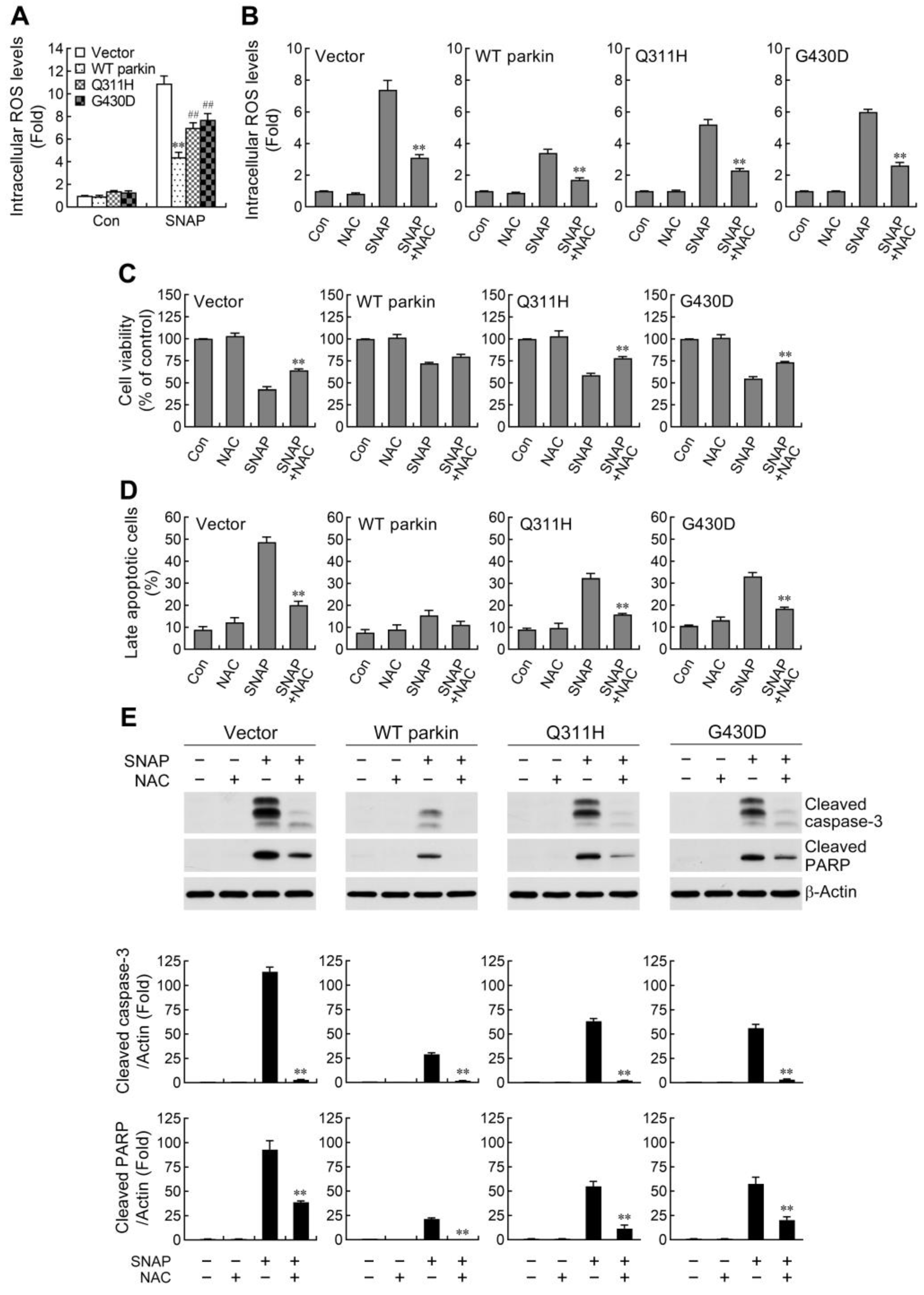
2.2. ROS Reduction Contributes to Parkin-Mediated Abatement of Apoptosis by NO
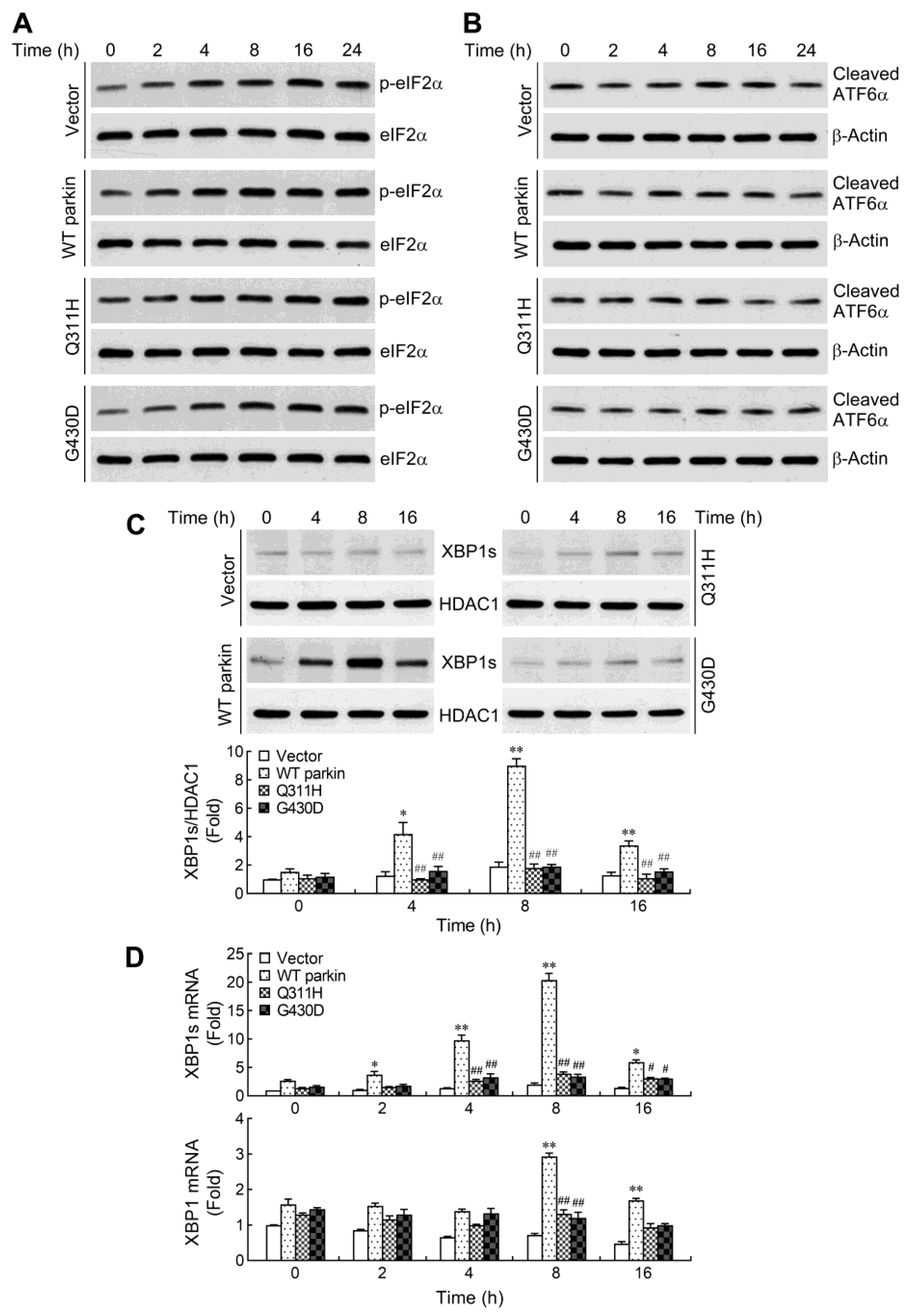
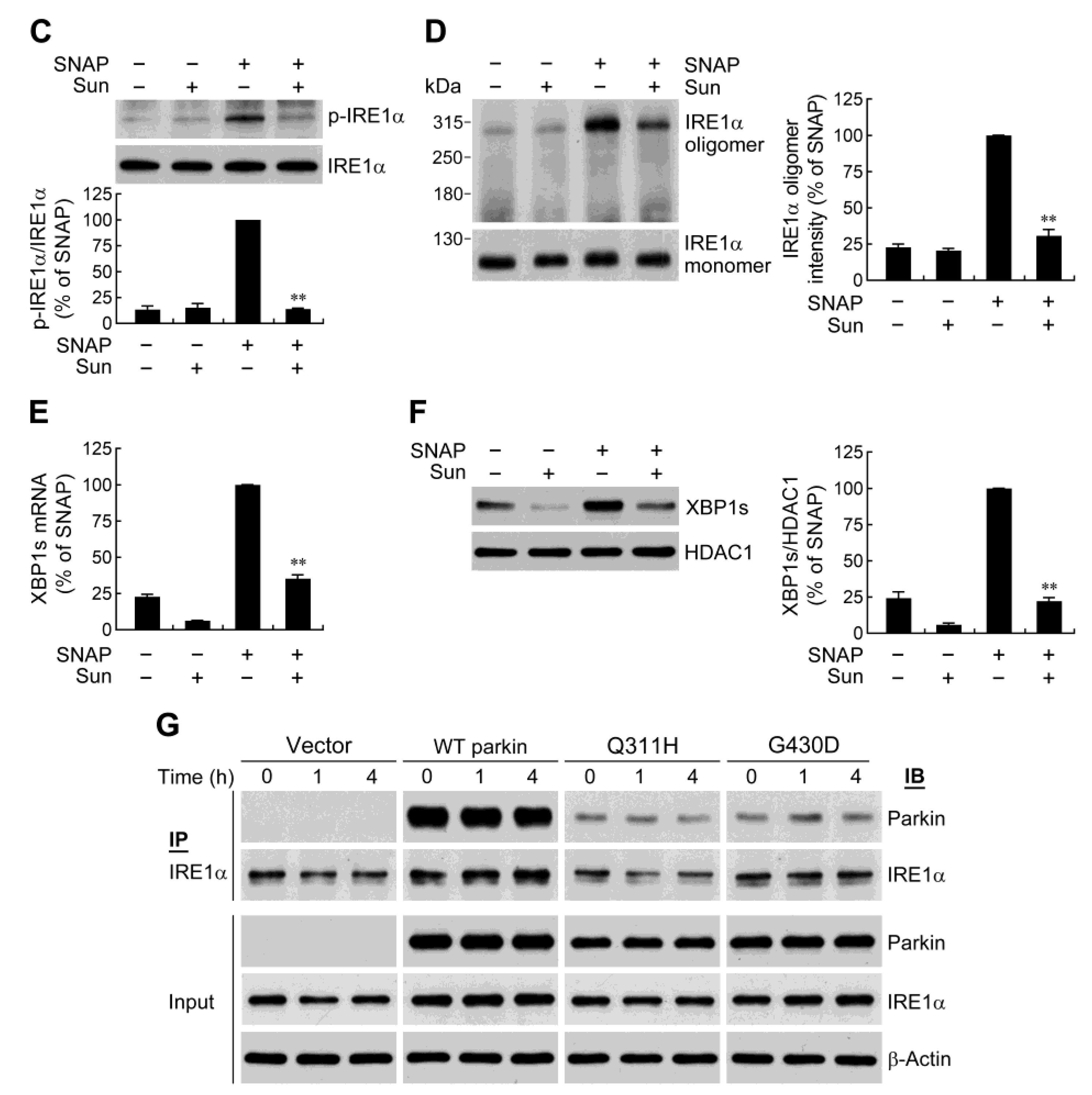
2.3. Parkin Selectively Upregulates IRE1α/XBP1 Signaling in Response to NO
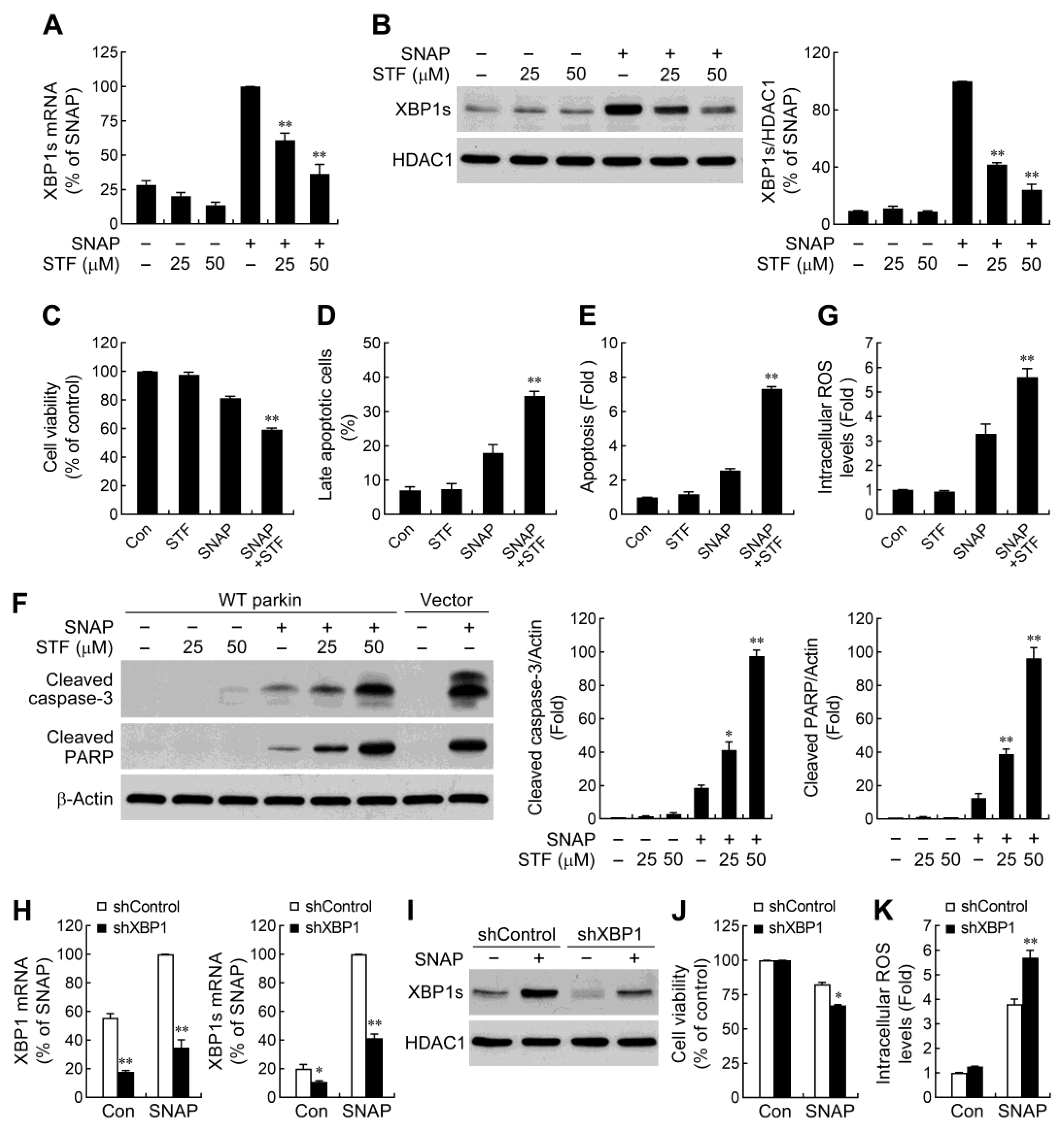
2.4. IRE1α/XBP1 Signaling Conduces to Parkin-Mediated Cell Protection
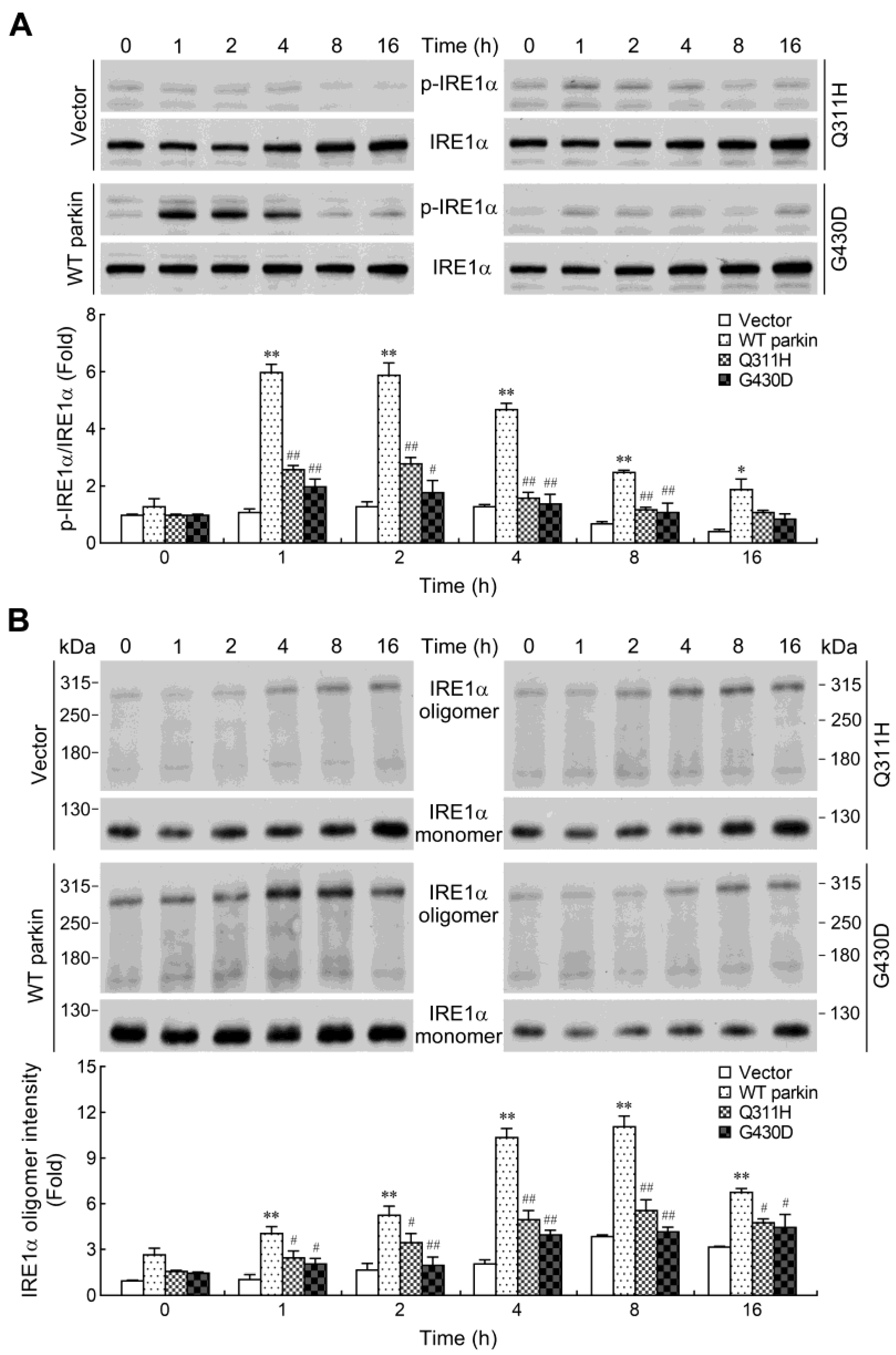
2.5. Parkin Enhances IRE1α Phosphorylation and Oligomerization
2.6. Parkin Forms a Protein Complex with IRE1α
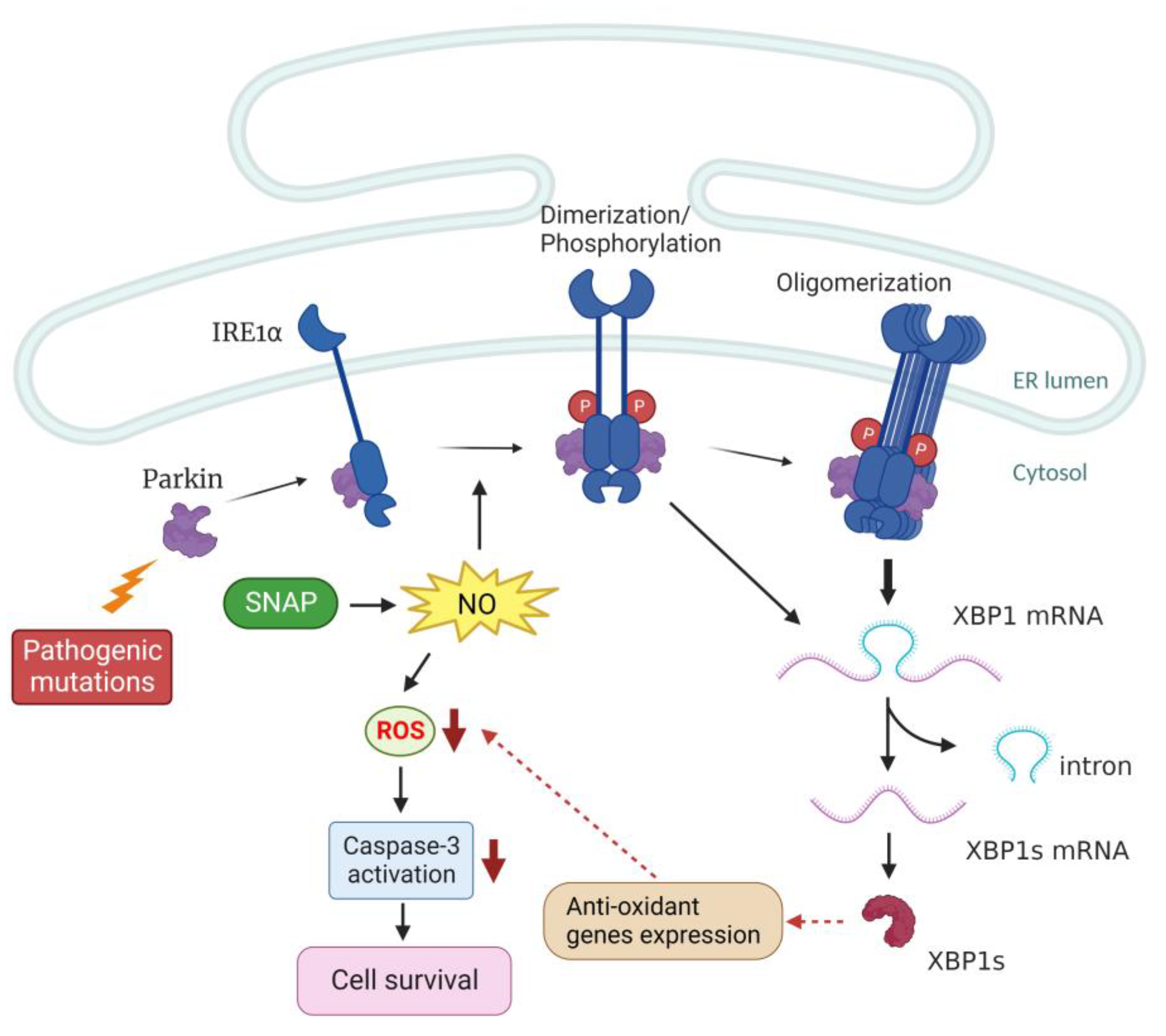
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Generation of Stable Cell Lines
4.3. Cell Cultures and Drug Treatment
4.4. Lentiviral Transduction
4.5. Cell Viability Assay
4.6. Apoptosis Assay
4.7. Measurement of Intracellular ROS
4.8. Real-Time RT-PCR Analysis
4.9. Western Blotting
4.10. Immunoprecipitation
4.11. Caspase Activity Assay
4.12. NO Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; de Jager, P.L.; Feany, M.B. Parkinson’s disease: Genetics and pathogenesis. Annu. Rev. Pathol. 2011, 6, 193–222. [Google Scholar] [CrossRef]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef] [PubMed]
- West, A.B.; Maidment, N.T. Genetics of parkin-linked disease. Hum. Genet. 2004, 114, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Alves da Costa, C.; Duplan, E.; Rouland, L.; Checler, F. The transcription factor function of parkin: Breaking the dogma. Front. Neurosci. 2019, 12, 965. [Google Scholar] [CrossRef]
- Yang, Y.; Nishimura, I.; Imai, Y.; Takahashi, R.; Lu, B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron 2003, 37, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ren, Y.; Zhao, J.; Feng, J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum. Mol. Genet. 2004, 13, 1745–1754. [Google Scholar] [CrossRef]
- Lo Bianco, C.; Schneider, B.L.; Bauer, M.; Sajadi, A.; Brice, A.; Iwatsubo, T.; Aebischer, P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha synuclein rat model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 17510–17515. [Google Scholar] [CrossRef]
- Henn, I.H.; Bouman, L.; Schlehe, J.S.; Schlierf, A.; Schramm, J.E.; Wegener, E.; Nakaso, K.; Culmsee, C.; Berninger, B.; Krappmann, D.; et al. Parkin mediates neuroprotection through activation of IκB kinase/nuclear factor-κB signaling. J. Neurosci. 2007, 27, 1868–1878. [Google Scholar] [CrossRef]
- Da Costa, C.A.; Sunyach, C.; Giaime, E.; West, A.; Corti, O.; Brice, A.; Safe, S.; Abou-Sleiman, P.M.; Wood, N.W.; Takahashi, H.; et al. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat. Cell Biol. 2009, 11, 1370–1375. [Google Scholar] [CrossRef]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Gelders, G.; Baekelandt, V.; van der Perren, A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. J. Neural Transm. Suppl. 2000, 60, 277–290. [Google Scholar]
- Lofrumento, D.D.; Saponaro, C.; Cianciulli, A.; de Nuccio, F.; Mitolo, V.; Nicolardi, G.; Panaro, M.A. MPTP-induced neuroinflammation increases the expression of pro-inflammatory cytokines and their receptors in mouse brain. Neuroimmunomodulation 2011, 18, 79–88. [Google Scholar] [CrossRef]
- Mrak, R.E.; Griffin, W.S. Glia and their cytokines in progression of neurodegeneration. Neurobiol. Aging 2005, 26, 349–354. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Glial reactions in Parkinson’s disease. Mov. Disord. 2008, 23, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dikshit, M. Apoptotic neuronal death in Parkinson’s disease: Involvement of nitric oxide. Brain Res. Rev. 2007, 54, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; Herrero, M.T.; García-Martín, E.; Agúndez, J.A. An update of the role of nitric oxide in the neurodegenerative processes of Parkinson’s disease. Curr. Med. Chem. 2016, 23, 2666–2679. [Google Scholar] [CrossRef]
- Tiwari, S.; Singh, S. Reciprocal upshot of nitric oxide, endoplasmic reticulum stress, and ubiquitin proteasome system in Parkinson’s disease pathology. Neuroscientist 2021, 27, 340–354. [Google Scholar] [CrossRef]
- Li, X.; Ye, X.; Li, X.; Sun, X.; Liang, Q.; Tao, L.; Kang, X.; Chen, J. Salidroside protects against MPP(+)-induced apoptosis in PC12 cells by inhibiting the NO pathway. Brain Res. 2011, 1382, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.A.; Delattre, A.M.; Carabelli, B.; Pudell, C.; Bortolanza, M.; Staziaki, P.V.; Visentainer, J.V.; Montanher, P.F.; del Bel, E.A.; Ferraz, A.C. Neuroprotective effect of omega-3 polyunsaturated fatty acids in the 6-OHDA model of Parkinson’s disease is mediated by a reduction of inducible nitric oxide synthase. Nutr. Neurosci. 2018, 21, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Sunico, C.R.; Nakamura, T.; Rockenstein, E.; Mante, M.; Adame, A.; Chan, S.F.; Newmeyer, T.F.; Masliah, E.; Nakanishi, N.; Lipton, S.A. S-Nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson’s disease. Mol. Neurodegener. 2013, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Kotzbauer, P.T.; Uryu, K.; Leight, S.; Trojanowski, J.Q.; Lee, V.M. Neuroinflammation and oxidation/nitration of α-synuclein linked to dopaminergic neurodegeneration. J. Neurosci. 2008, 28, 7687–7698. [Google Scholar] [CrossRef]
- Gao, H.M.; Zhang, F.; Zhou, H.; Kam, W.; Wilson, B.; Hong, J.S. Neuroinflammation and α-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ. Health Perspect. 2011, 119, 807–814. [Google Scholar] [CrossRef]
- Vance, J.M.; Ali, S.; Bradley, W.G.; Singer, C.; di monte, D.A. Gene-environment interactions in Parkinson’s and other forms of parkinsonism. Neurotoxicology 2010, 31, 598–602. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Tran, T.; Ruhn, K.A.; Martinez, T.N.; Hong, J.; Marvin, M.; Hartley, M.; Treviño, I.; O’Brien, D.E.; Casey, B.; et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J. Neurosci. 2008, 28, 10825–10834. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef]
- Shyu, W.C.; Lin, S.Z.; Chiang, M.F.; Pang, C.Y.; Chen, S.Y.; Hsin, Y.L.; Thajeb, P.; Lee, Y.J.; Li, H. Early-onset Parkinson’s disease in a Chinese population: 99mTc-TRODAT-1 SPECT, Parkin gene analysis and clinical study. Park. Relat. Disord. 2005, 11, 173–180. [Google Scholar] [CrossRef]
- Pinkaew, D.; Chattopadhyay, A.; King, M.D.; Chunhacha, P.; Liu, Z.; Stevenson, H.L.; Chen, Y.; Sinthujaroen, P.; McDougal, O.M.; Fujise, K. Fortilin binds IRE1α and prevents ER stress from signaling apoptotic cell death. Nat. Commun. 2017, 8, 18. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, L.; Chen, J.; Cao, M.; Wang, J. JS-K as a nitric oxide donor induces apoptosis via the ROS/Ca2+/caspase-mediated mitochondrial pathway in HepG2 cells. Biomed. Pharmacother. 2018, 107, 1385–1392. [Google Scholar] [CrossRef]
- Qiu, M.; Chen, L.; Tan, G.; Ke, L.; Zhang, S.; Chen, H.; Liu, J. A reactive oxygen species activation mechanism contributes to JS-K-induced apoptosis in human bladder cancer cells. Sci. Rep. 2015, 5, 15104. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Kawahara, K.; Oyadomari, S.; Gotoh, T.; Kohsaka, S.; Nakayama, H.; Mori, M. Induction of CHOP and apoptosis by nitric oxide in p53-deficient microglial cells. FEBS Lett. 2001, 506, 135–139. [Google Scholar]
- Oyadomari, S.; Takeda, K.; Takiguchi, M.; Gotoh, T.; Matsumoto, M.; Wada, I.; Akira, S.; Araki, E.; Mori, M. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 10845–10850. [Google Scholar] [CrossRef]
- Takada, K.; Hirose, J.; Yamabe, S.; Uehara, Y.; Mizuta, H. Endoplasmic reticulum stress mediates nitric oxide-induced chondrocyte apoptosis. Biomed. Rep. 2013, 1, 315–319. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Duplan, E.; Giaime, E.; Viotti, J.; Sévalle, J.; Corti, O.; Brice, A.; Ariga, H.; Qi, L.; Checler, F.; Alves da Costa, C. ER-stress-associated functional link between Parkin and DJ-1 via a transcriptional cascade involving the tumor suppressor p53 and the spliced X-box binding protein XBP-1. J. Cell Sci. 2013, 126, 2124–2133. [Google Scholar] [CrossRef]
- Liu, Y.; Adachi, M.; Zhao, S.; Hareyama, M.; Koong, A.C.; Luo, D.; Rando, T.A.; Imai, K.; Shinomura, Y. Preventing oxidative stress: A new role for XBP1. Cell Death Differ. 2009, 16, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Deepti, A.; Deegan, S.; Lisbona, F.; Hetz, C.; Samali, A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1α–XBP1 signaling through a physical interaction. PLoS Biol. 2010, 8, e1000410. [Google Scholar] [CrossRef] [PubMed]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-β neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160. [Google Scholar] [CrossRef]
- Zhong, Y.; Li, J.; Wang, J.J.; Chen, C.; Tran, J.T.; Saadi, A.; Yu, Q.; Le, Y.Z.; Mandal, M.N.; Anderson, R.E.; et al. X-box binding protein 1 is essential for the anti-oxidant defense and cell survival in the retinal pigment epithelium. PLoS ONE 2012, 7, e38616. [Google Scholar] [CrossRef] [PubMed]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Denko, N.C.; Olson, M.; van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef]
- Wang, L.; Perera, B.G.; Hari, S.B.; Bhhatarai, B.; Backes, B.J.; Seeliger, M.A.; Schürer, S.C.; Oakes, S.A.; Papa, F.R.; Maly, D.J. Divergent allosteric control of the IRE1α endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012, 8, 982–989. [Google Scholar] [CrossRef]
- Shao, D.; Liu, J.; Ni, J.; Wang, Z.; Shen, Y.; Zhou, L.; Huang, Y.; Wang, J.; Xue, H.; Zhang, W.; et al. Suppression of XBP1s mediates high glucose-induced oxidative stress and extracellular matrix synthesis in renal mesangial cell and kidney of diabetic rats. PLoS ONE 2013, 8, e56124. [Google Scholar] [CrossRef]
- Ali, M.M.; Bagratuni, T.; Davenport, E.L.; Nowak, P.R.; Silva-Santisteban, M.C.; Hardcastle, A.; McAndrews, C.; Rowlands, M.G.; Morgan, G.J.; Aherne, W.; et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011, 30, 894–905. [Google Scholar] [CrossRef]
- Itzhak, D.; Bright, M.; McAndrew, P.; Mirza, A.; Newbatt, Y.; Strover, J.; Widya, M.; Thompson, A.; Morgan, G.; Collins, I.; et al. Multiple autophosphorylations significantly enhance the endoribonuclease activity of human inositol requiring enzyme 1α. BMC Biochem. 2014, 15, 3. [Google Scholar] [CrossRef]
- Prischi, F.; Nowak, P.R.; Carrara, M.; Ali, M.M. Phosphoregulation of Ire1 RNase splicing activity. Nat. Commun. 2014, 5, 3554. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; He, Y.; Zhang, H.; Yu, L.; Chen, H.; Xu, Z.; Tang, S.; Urano, F.; Min, W. AIP1 is critical in transducing IRE1-mediated endoplasmic reticulum stress response. J. Biol. Chem. 2008, 283, 11905–11912. [Google Scholar] [CrossRef]
- He, Y.; Beatty, A.; Han, X.; Ji, Y.; Ma, X.; Adelstein, R.S.; Yates, J.R., 3rd; Kemphues, K.; Qi, L. Nonmuscle myosin IIB links cytoskeleton to IRE1α signaling during ER stress. Dev. Cell 2012, 23, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Villalta, S.A.; Feldman, H.C.; Register, A.C.; Rosenthal, W.; Hoffmann-Petersen, I.T.; Mehdizadeh, M.; Ghosh, R.; Wang, L.; Colon-Negron, K.; et al. Targeting ABL-IRE1α signaling spares ER-stressed pancreatic β cells to reverse autoimmune diabetes. Cell Metab. 2017, 25, 883–897. [Google Scholar] [CrossRef]
- Gao, H.M.; Hong, J.S.; Zhang, W.; Liu, B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J. Neurosci. 2002, 22, 782–790. [Google Scholar] [CrossRef]
- Huang, M.; Li, Y.; Tian, T.; Wang, K.; Wang, Y.; Yan, W.; Yang, H. Knockdown of TLR4 Represses the Paraquat-Induced Neuroinflammation and Microglial M1 Polarization. Neurotox. Res. 2020, 38, 741–750. [Google Scholar] [CrossRef]
- Javed, H.; Thangavel, R.; Selvakumar, G.P.; Dubova, I.; Schwartz, N.; Ahmed, M.E.; Zaheer, S.; Kempuraj, D.; Iyer, S.; Zaheer, A.; et al. NLRP3 inflammasome and glia maturation factor coordinately regulate neuroinflammation and neuronal loss in MPTP mouse model of Parkinson’s disease. Int. Immunopharmacol. 2020, 83, 106441. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric oxide and neuronal death. Nitric Oxide 2010, 23, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Mayasi, Y.; Hannoun, A.; Eslami, S.M.; Carandang, R. Nitric oxide and mitochondrial function in neurological diseases. Neuroscience 2018, 376, 48–71. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Borutaite, V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. Biophys. Acta 2004, 1658, 44–49. [Google Scholar] [CrossRef]
- Hyun, D.H.; Lee, M.; Hattori, N.; Kubo, S.; Mizuno, Y.; Halliwell, B.; Jenner, P. Effect of wild-type or mutant Parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J. Biol. Chem. 2002, 277, 28572–28577. [Google Scholar] [CrossRef]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef]
- Su, Y.; Duan, J.; Ying, Z.; Hou, Y.; Zhang, Y.; Wang, R.; Deng, Y. Increased vulnerability of parkin knock down PC12 cells to hydrogen peroxide toxicity: The role of salsolinol and NM-salsolinol. Neuroscience 2013, 233, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Tokarew, J.M.; El-Kodsi, D.N.; Lengacher, N.A.; Fehr, T.K.; Nguyen, A.P.; Shutinoski, B.; O’Nuallain, B.; Jin, M.; Khan, J.M.; Ng, A.C.H.; et al. Age-associated insolubility of parkin in human midbrain is linked to redox balance and sequestration of reactive dopamine metabolites. Acta Neuropathol. 2021, 141, 725–754. [Google Scholar] [CrossRef]
- Abdullah, A.; Ravanan, P. The unknown face of IRE1alpha—Beyond ER stress. Eur. J. Cell Biol. 2018, 97, 359–368. [Google Scholar] [CrossRef]
- Sado, M.; Yamasaki, Y.; Iwanaga, T.; Onaka, Y.; Ibuki, T.; Nishihara, S.; Mizuguchi, H.; Momota, H.; Kishibuchi, R.; Hashimoto, T.; et al. Protective effect against Parkinson’s disease-related insults through the activation of XBP1. Brain Res. 2009, 1257, 16–24. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Liang, Y.; Yu, H.; Chen, X.; Zheng, T.; Zheng, B.; Wang, L.; Zhao, L.; Shi, C.; et al. Targeting X box-binding protein-1 (XBP1) enhances sensitivity of glioma cells to oxidative stress. Neuropathol. Appl. Neurobiol. 2011, 37, 395–405. [Google Scholar] [CrossRef]
- McLaughlin, T.; Falkowski, M.; Park, J.W.; Keegan, S.; Elliott, M.; Wang, J.J.; Zhang, S.X. Loss of XBP1 accelerates age-related decline in retinal function and neurodegeneration. Mol. Neurodegener. 2018, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Zhang, S.; Rentas, C.; Caldwell, K.A.; Caldwell, G.A. RTCB-1 mediates neuroprotection via XBP-1 mRNA splicing in the unfolded protein response pathway. J. Neurosci. 2014, 34, 16076–16085. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Dis. 2020, 136, 104717. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, J.J.; Ma, J.H.; Jin, C.; Yu, Q.; Zhang, S.X. Activation of the UPR protects against cigarette smoke-induced RPE apoptosis through up-regulation of Nrf2. J. Biol. Chem. 2015, 290, 5367–5380. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, T.-L.; Huang, H.-Y.; Chang, H.-F.; Wu, H.-R.; Wang, M.-J. Enhanced IRE1α Phosphorylation/Oligomerization-Triggered XBP1 Splicing Contributes to Parkin-Mediated Prevention of SH-SY5Y Cell Death under Nitrosative Stress. Int. J. Mol. Sci. 2023, 24, 2017. https://doi.org/10.3390/ijms24032017
Chiu T-L, Huang H-Y, Chang H-F, Wu H-R, Wang M-J. Enhanced IRE1α Phosphorylation/Oligomerization-Triggered XBP1 Splicing Contributes to Parkin-Mediated Prevention of SH-SY5Y Cell Death under Nitrosative Stress. International Journal of Molecular Sciences. 2023; 24(3):2017. https://doi.org/10.3390/ijms24032017
Chicago/Turabian StyleChiu, Tsung-Lang, Hsin-Yi Huang, Hui-Fen Chang, Hsin-Rong Wu, and Mei-Jen Wang. 2023. "Enhanced IRE1α Phosphorylation/Oligomerization-Triggered XBP1 Splicing Contributes to Parkin-Mediated Prevention of SH-SY5Y Cell Death under Nitrosative Stress" International Journal of Molecular Sciences 24, no. 3: 2017. https://doi.org/10.3390/ijms24032017
APA StyleChiu, T.-L., Huang, H.-Y., Chang, H.-F., Wu, H.-R., & Wang, M.-J. (2023). Enhanced IRE1α Phosphorylation/Oligomerization-Triggered XBP1 Splicing Contributes to Parkin-Mediated Prevention of SH-SY5Y Cell Death under Nitrosative Stress. International Journal of Molecular Sciences, 24(3), 2017. https://doi.org/10.3390/ijms24032017