
Epigenetic Regulation of MAP3K8 in EBV-Associated Gastric Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
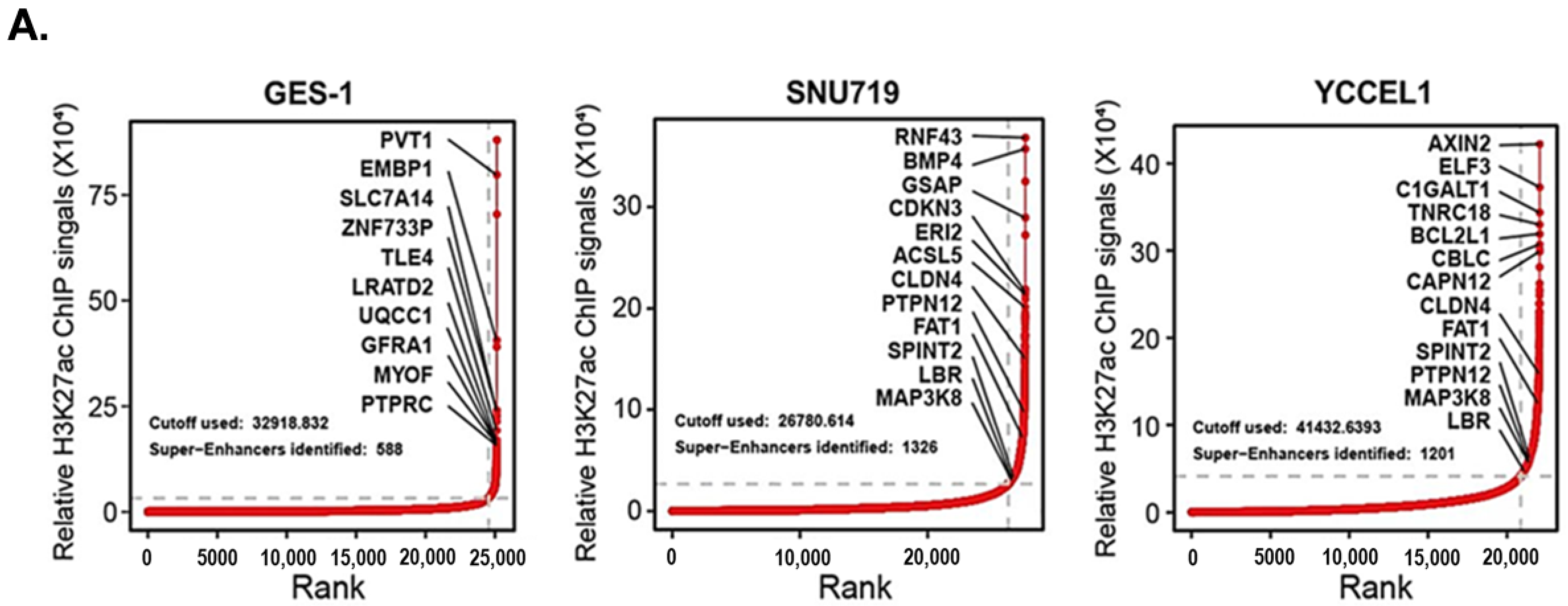
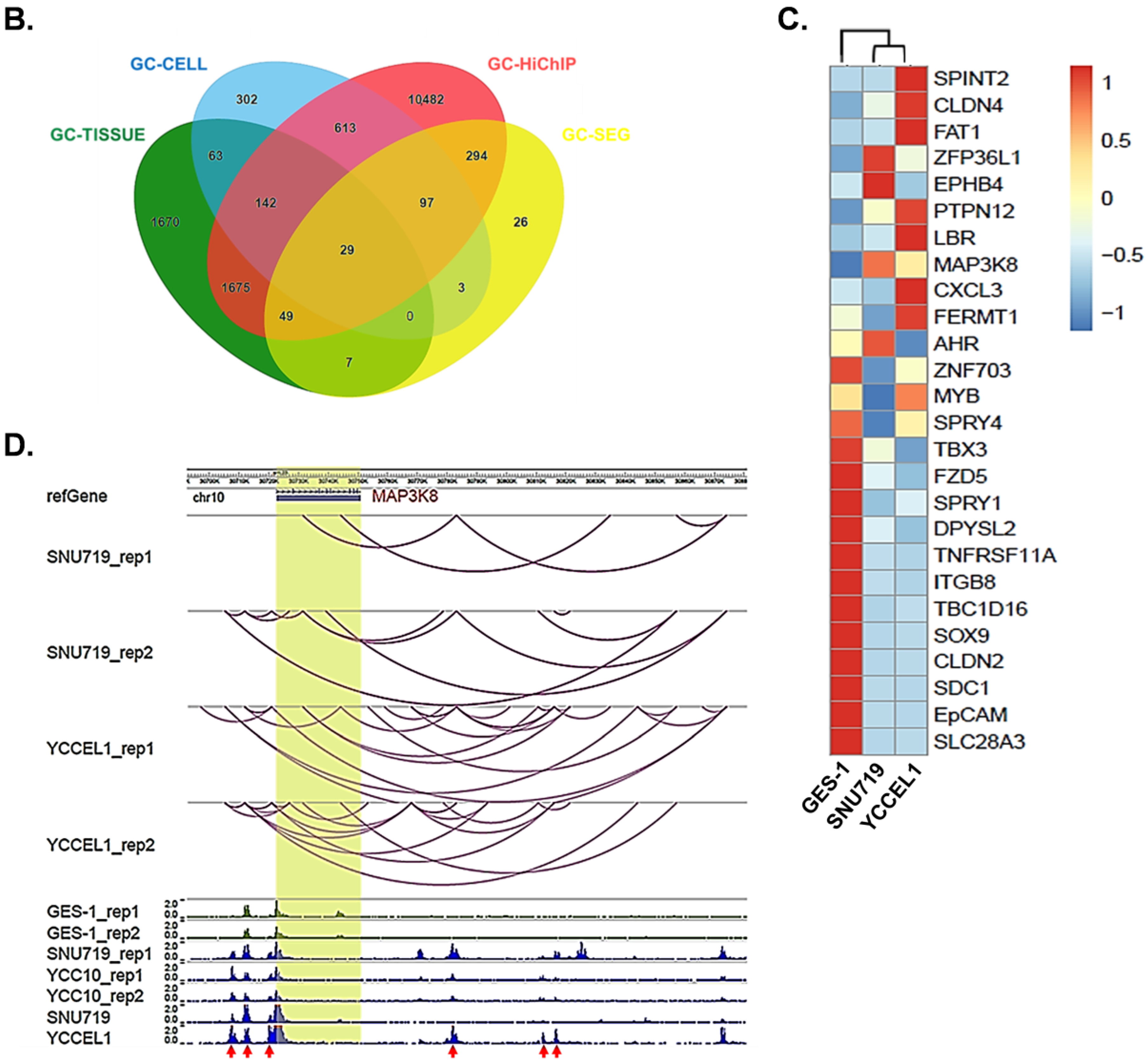
2.1. Profiling Super-Enhancer Landscapes in EBVaGC and Identifying MAP3K8 as an SE-Regulated Gene in EBVaGC
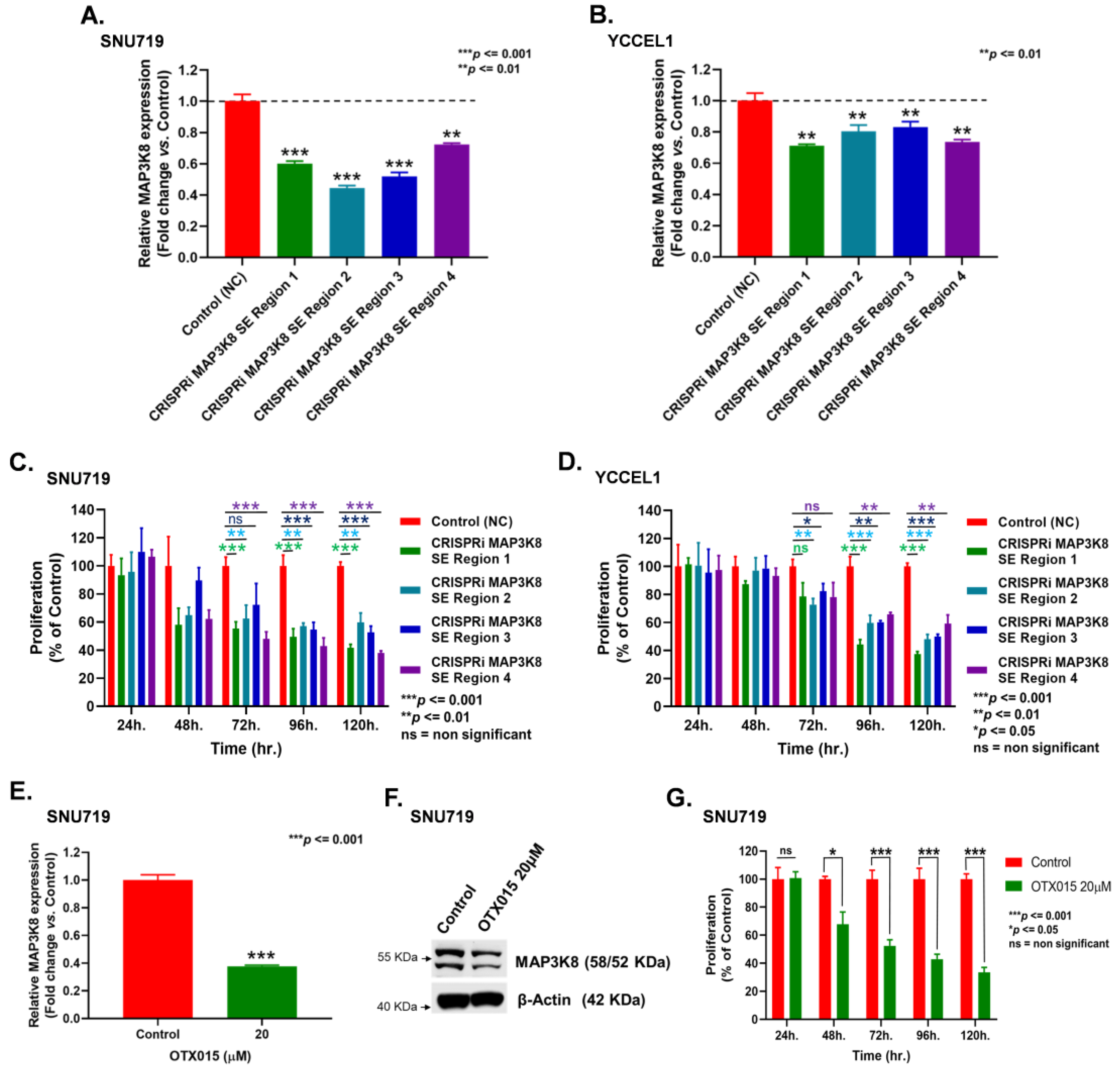
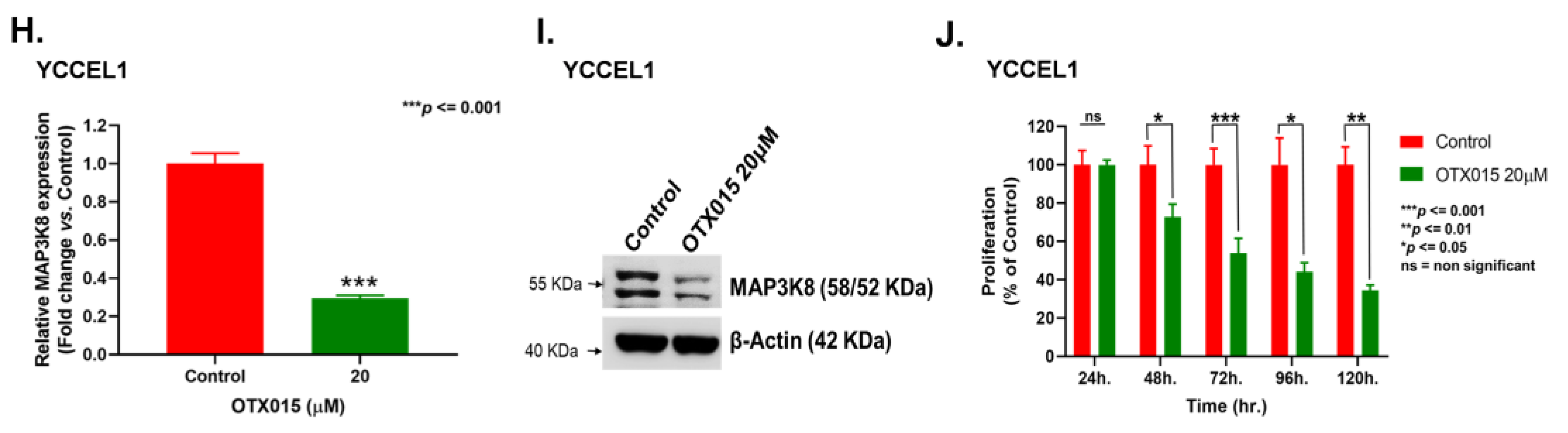
2.2. Super-Enhancers Regulate MAP3K8 Expression in EBVaGC
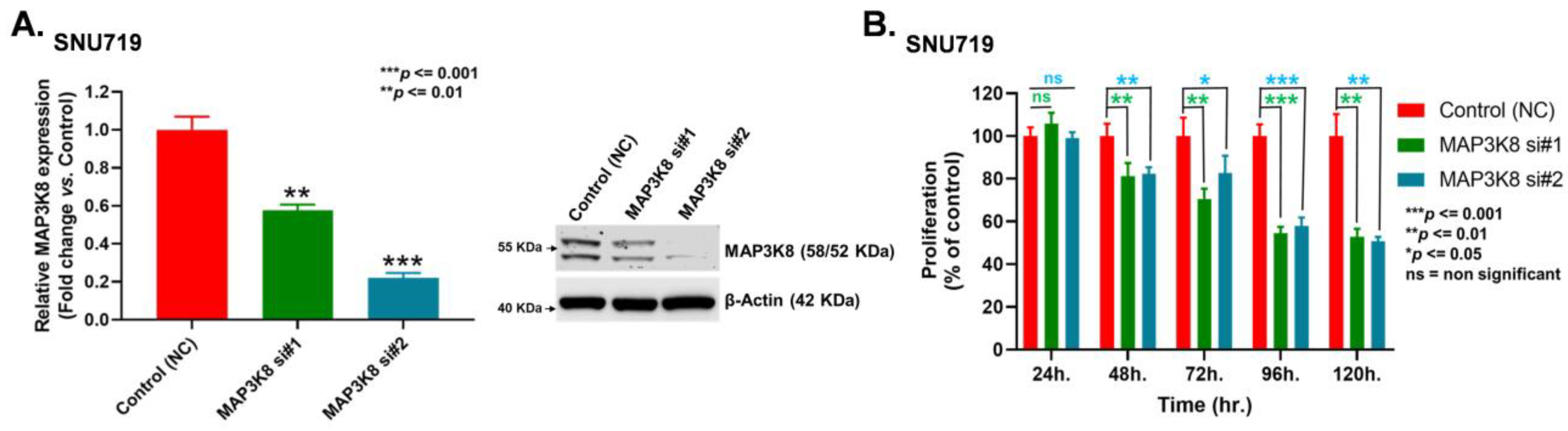
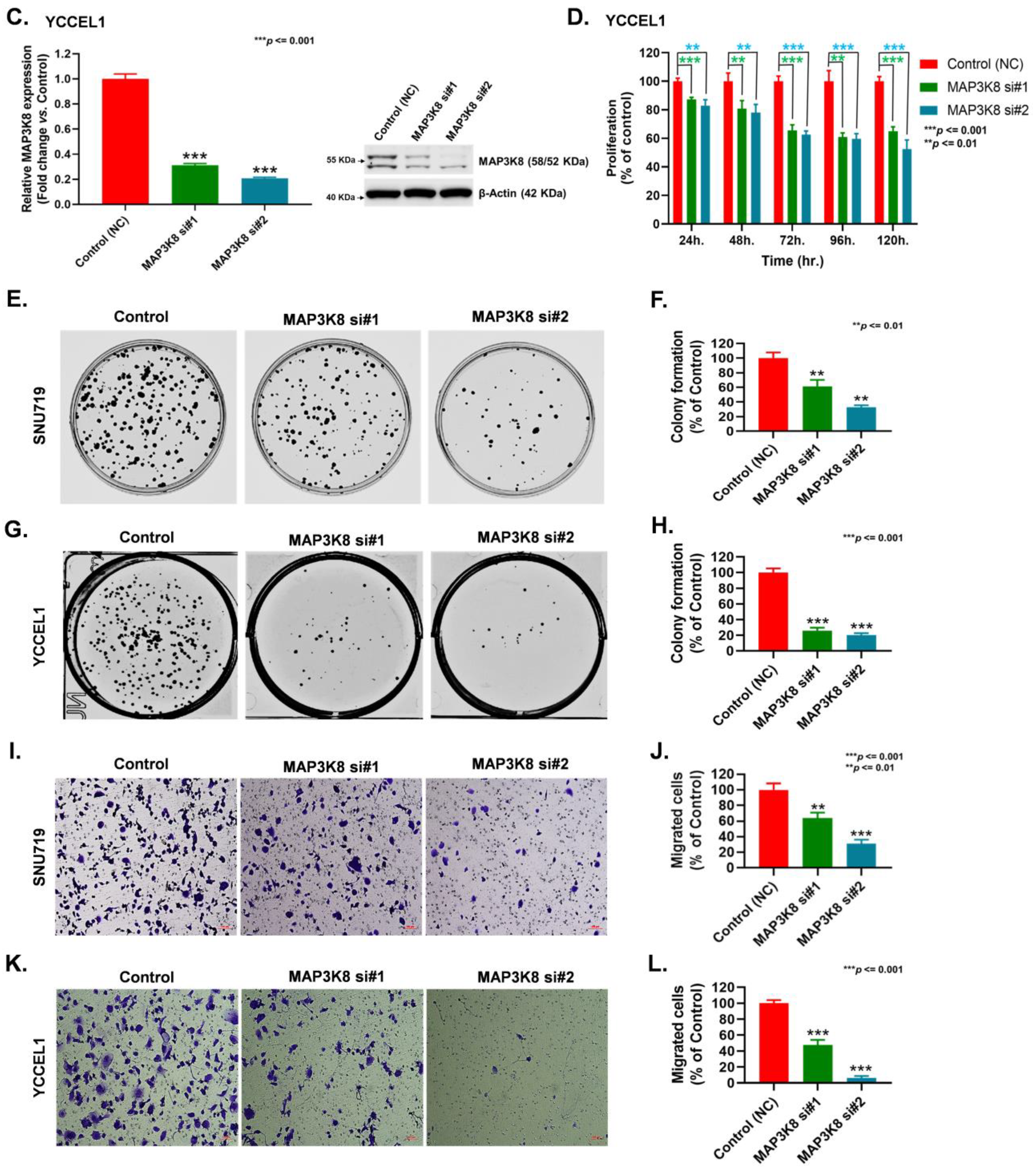
2.3. MAP3K8 Fosters GC Progression by Promoting Cell Proliferation and Migration
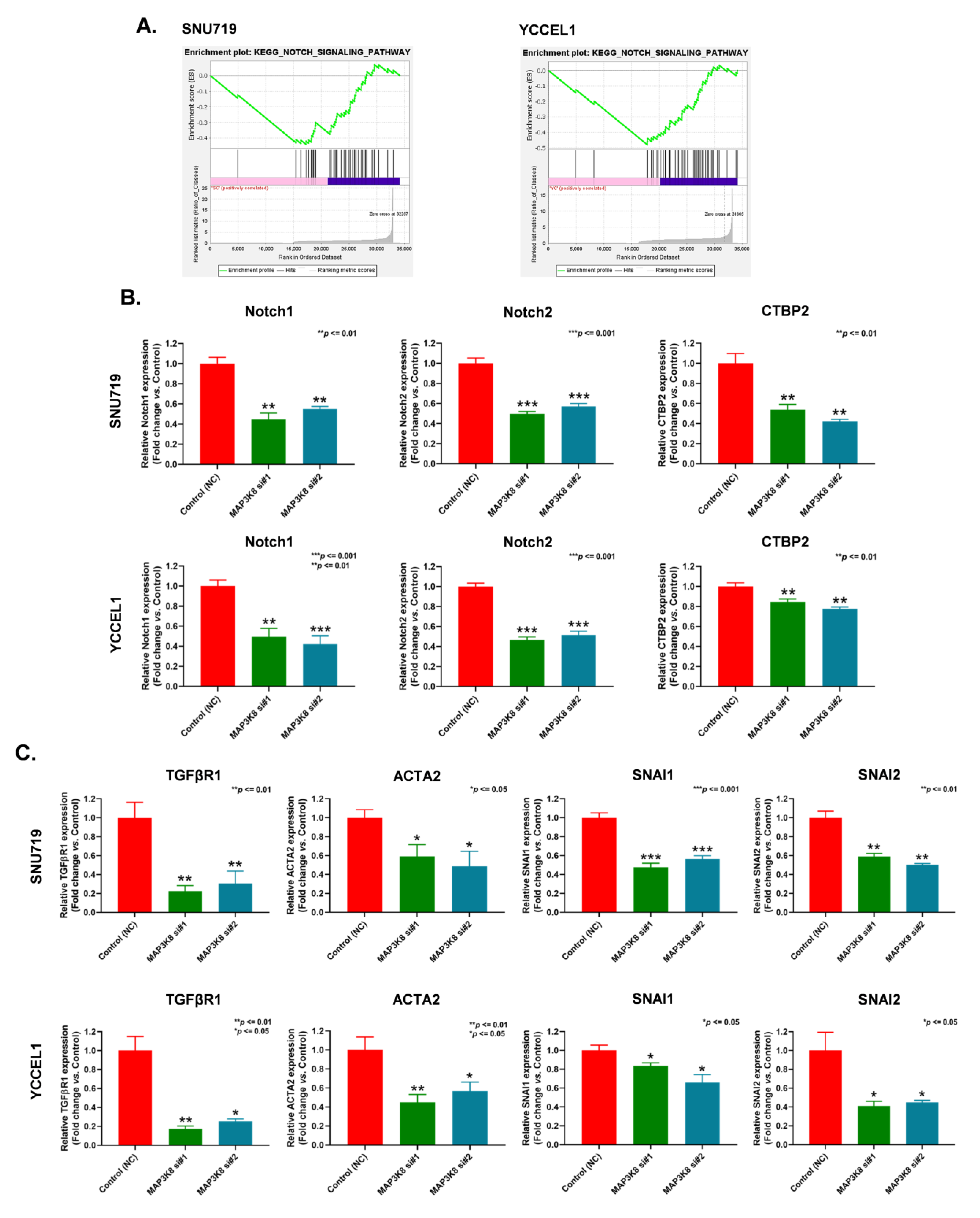
2.4. MAP3K8 Inhibits EBVaGC Progression by Inactivating the Notch Signaling Pathway and Epithelial to Mesenchymal Transition (EMT)
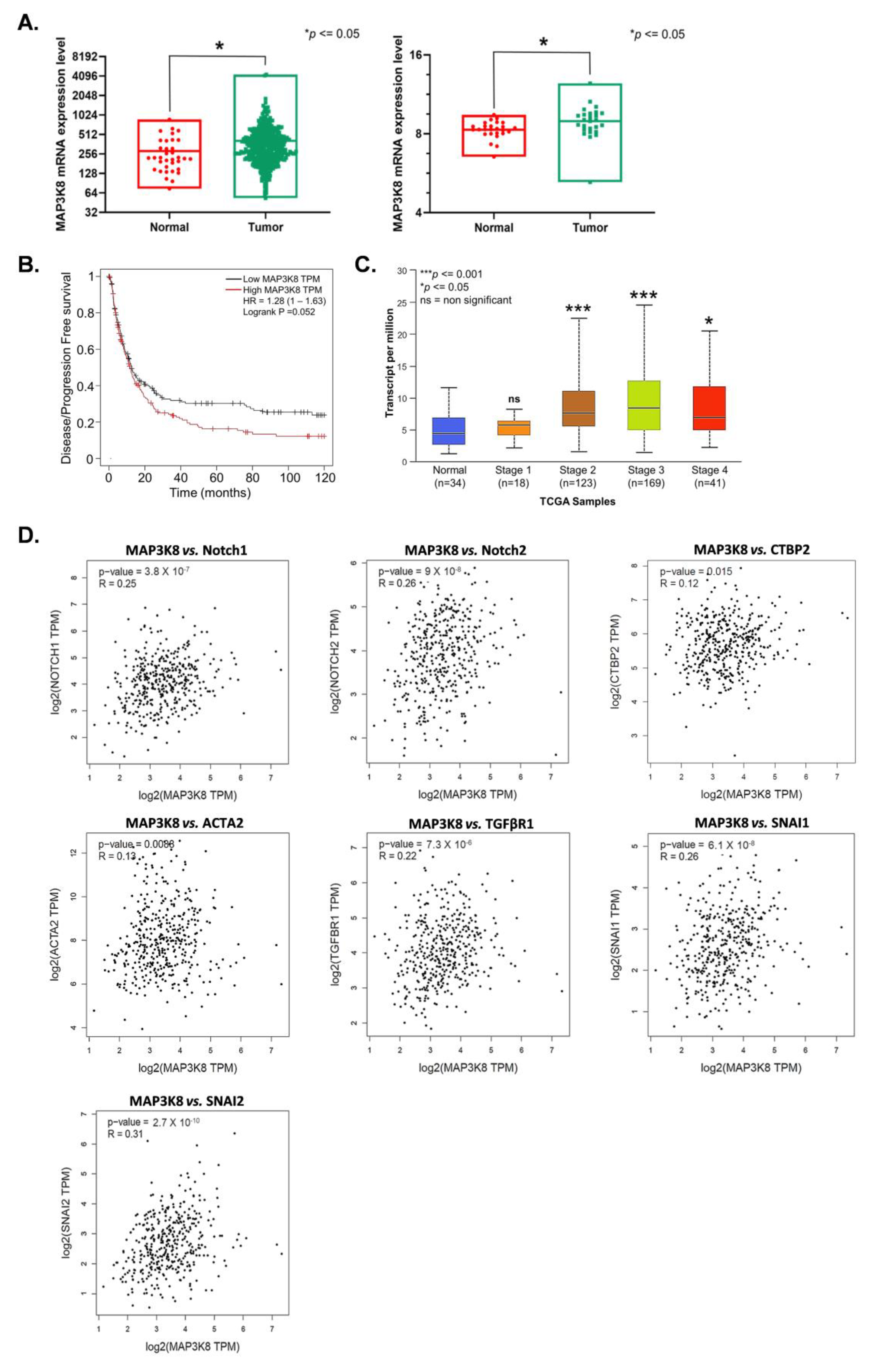
2.5. MAP3K8 Plays a Vital Role in GC Progression
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. H3K27ac ChIP-seq
4.3. H3K27ac HiChIP-seq
4.4. ChIP-seq and HiChIP-seq Data Analysis
4.5. Integrative Analysis
4.6. Plasmid Construction, Transfection, and Establishment of MAP3K8 SE-CRISPRi Stable Cell Lines
4.7. siRNA Transfection
4.8. Birabresib (OTX015) Treatment
4.9. Cell Viability Assay
4.10. Colony Formation Assay
4.11. Boyden Chamber Assisted Cell Migration Assay
4.12. Immunoblotting
4.13. Quantitative Reverse-Transcriptase Polymerase Chain Reaction Analysis (qRT-PCR)
4.14. RNA Sequencing and Pathway Analysis
4.15. Web-Based Analysis of GC Omics Data
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Matsuda, T.; Carlo, V.D.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): Analysis of individual records for 37,513,025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Kono, K. Landscape of EBV-positive gastric cancer. Gastric Cancer 2021, 24, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli, A.; Monavari, S.H.; Mohammadi, F.S.; Kiani, S.J.; Armat, S.; Farahmand, M. Association between Epstein-Barr virus infection and gastric cancer: A systematic review and meta-analysis. BMC Cancer 2020, 20, 493. [Google Scholar] [CrossRef] [PubMed]
- Stanland, L.J.; Luftig, M.A. The role of EBV-induced hypermethylation in gastric cancer tumorigenesis. Viruses 2020, 12, 1222. [Google Scholar] [CrossRef]
- Machlowska, J.; Baj, J.; Sitarz, M.; Maciejewski, R.; Sitarz, R. Gastric Cancer: Epidemiology, risk factors, classification, genomic characteristics and treatment strategies. Int. J. Mol. Sci. 2020, 21, 4012. [Google Scholar] [CrossRef]
- Thrift, A.P.; El-Serag, H.B. Burden of gastric cancer. Clin. Gastroenterol. Hepatol. 2020, 18, 534–542. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Wang, D.; Feng, M.; Wu, X. Epigenetically regulated gene expression profiles reveal four molecular subtypes with prognostic and therapeutic implications in colorectal cancer. Brief. Bioinform. 2021, 22, bbaa309. [Google Scholar] [CrossRef]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 2021, 12, 1786. [Google Scholar] [CrossRef]
- Grady, W.M.; Yu, M.; Markowitz, S.D. Epigenetic alterations in the gastrointestinal tract: Current and emerging use for biomarkers of cancer. Gastroenterology 2021, 160, 690–709. [Google Scholar] [CrossRef] [PubMed]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E. Epigenetic therapies for cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-H.; Qu, Q.; Qi, T.-T.; Teng, X.-Q.; Zhu, H.-H.; Wang, J.-J.; Lu, Q.; Qu, J. Super-enhancers: A new frontier for epigenetic modifiers in cancer chemoresistance. J. Exp. Clin. Cancer Res. 2021, 40, 174. [Google Scholar] [CrossRef] [PubMed]
- Dębek, S.; Juszczyński, P. Super enhancers as master gene regulators in the pathogenesis of hematologic malignancies. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188697. [Google Scholar] [CrossRef] [PubMed]
- Thandapani, P. Super-enhancers in cancer. Pharmacol. Ther. 2019, 99, 129–138. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, T.; Zhang, Y.; Chen, H.; Shen, J.; Jin, X.; Wei, J.; Zhang, E.; Xiao, M.; Fan, Y.; et al. A super-enhancer controls TGF-β signaling in pancreatic cancer through downregulation of TGFBR2. Cell. Signal. 2020, 66, 109470. [Google Scholar] [CrossRef]
- Yu, D.; Yang, X.; Lin, J.; Cao, Z.; Lu, C.; Yang, Z.; Zheng, M.; Pan, R.; Cai, W. Super-enhancer induced IL-20RA promotes proliferation/metastasis and immune evasion in colorectal cancer. Front Oncol. 2021, 11, 724655. [Google Scholar] [CrossRef]
- Zhang, C.; Wei, S.; Sun, W.-P.; Teng, K.; Dai, M.-M.; Wang, F.-W.; Chen, J.-W.; Ling, H.; Ma, X.-D.; Feng, Z.-H.; et al. Super-enhancer-driven AJUBA is activated by TCF4 and involved in epithelial-mesenchymal transition in the progression of hepatocellular carcinoma. Theranostics 2020, 10, 9066–9082. [Google Scholar] [CrossRef]
- Deng, R.; Huang, J.-H.; Wang, Y.; Zhou, L.-H.; Wang, Z.-F.; Hu, B.-X.; Chen, Y.-H.; Dong, Y.; Mai, J.; Li, Z.-L.; et al. Disruption of super-enhancer-driven tumor suppressor gene RCAN1.4 expression promotes the malignancy of breast carcinoma. Mol. Cancer 2020, 19, 122. [Google Scholar] [CrossRef]
- Ooi, W.F.; Xing, M.; Xu, C.; Yao, X.; Ramlee, M.D.; Lim, M.C.; Cao, F.; Lim, K.; Babu, D.; Poon, L.-F.; et al. Epigenomic profiling of primary gastric adenocarcinoma reveals super-enhancer heterogeneity. Nat. Commun. 2016, 7, 12983. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Chai, P.; Zhang, H.; Fan, X. Novel insights into chromosomal conformations in cancer. Mol. Cancer 2017, 16, 173. [Google Scholar] [CrossRef] [PubMed]
- Maass, P.G.; Barutcu, A.R.; Rinn, J.L. Interchromosomal interactions: A genomic love story of kissing chromosomes. J. Cell Biol. 2019, 218, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.D.; Tanimoto, K. Looping, linking, and chromatin activity: New insights into beta-globin locus regulation. Cell 2000, 100, 499–502. [Google Scholar] [CrossRef]
- Chu, Z.; Gu, L.; Hu, Y.; Zhang, X.; Li, M.; Chen, J.; Teng, D.; Huang, M.; Shen, C.-H.; Cai, L.; et al. STAG2 regulates interferon signaling in melanoma via enhancer loop reprogramming. Nat. Commun. 2022, 13, 1859. [Google Scholar] [CrossRef]
- Giambartolomei, C.; Seo, J.-H.; Schwarz, T.; Freund, M.K.; Johnson, R.D.; Spisak, S.; Baca, S.C.; Gusev, A.; Mancuso, N.; Pasaniuc, B.; et al. H3K27ac HiChIP in prostate cell lines identifies risk genes for prostate cancer susceptibility. Am. J. Hum. Genet. 2021, 108, 2284–2300. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting alterations in the RAF-MEK pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef]
- Hebron, E.; Hope, C.; Kim, J.; Jensen, J.L.; Flanagan, C.; Bhatia, N.; Maroulakou, I.; Mitsiades, C.; Miyamoto, S.; Callander, N.; et al. MAP3K8 kinase regulates myeloma growth by cell-autonomous and non-autonomous mechanisms involving myeloma-associated monocytes/macrophages. Br. J. Haematol. 2013, 160, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Kwon, W.; Choi, S.-K.; Kim, D.; Kim, H.-G.; Park, J.-K.; Han, J.E.; Cho, G.-J.; Yun, S.; Yu, W.; Han, S.-H.; et al. ZNF507 affects TGF-β signaling via TGFBR1 and MAP3K8 activation in the progression of prostate cancer to an aggressive state. J. Exp. Clin. Cancer Res. 2021, 40, 291. [Google Scholar] [CrossRef]
- Hao, J.; Cao, Y.; Yu, H.; Zong, L.; An, R.; Xue, Y. Effect of MAP3K8 on prognosis and tumor-related inflammation in renal clear cell carcinoma. Front. Genet. 2021, 12, 674613. [Google Scholar] [CrossRef]
- Ren, J.; Xu, Y.; Liu, J.; Wu, S.; Zhang, R.; Cao, H.; Sun, J. MAP3K8 is a prognostic biomarker and correlated with immune response in glioma. Front. Mol. Biosci. 2021, 8, 779290. [Google Scholar] [CrossRef]
- Gruosso, T.; Garnier, C.; Abelanet, S.; Kieffer, Y.; Lemesre, V.; Bellanger, D.; Bieche, I.; Marangoni, E.; Sastre-Garau, X.; Mieulet, V.; et al. MAP3K8/TPL-2/COT is a potential predictive marker for MEK inhibitor treatment in high-grade serous ovarian carcinomas. Nat. Commun. 2015, 6, 8583. [Google Scholar] [CrossRef] [PubMed]
- Gianì, F.; Russo, G.; Pennisi, M.; Sciacca, L.; Frasca, F.; Pappalardo, F. Computational modeling reveals MAP3K8 as mediator of resistance to vemurafenib in thyroid cancer stem cells. Bioinformatics 2019, 35, 2267–2275. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A comprehensive review on MAPK: A promising therapeutic target in cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed]
- Njunge, L.W.; Estania, A.P.; Guo, Y.; Liu, W.; Yang, L. Tumor progression locus 2 (TPL2) in tumor-promoting inflammation, tumorigenesis and tumor immunity. Theranostics 2020, 10, 8343–8364. [Google Scholar] [CrossRef]
- Nanou, A.; Bourbouli, M.; Vetrano, S.; Schaeper, U.; Ley, S.; Kollias, G. Endothelial Tpl2 regulates vascular barrier function via JNK-mediated degradation of claudin-5 promoting neuroinflammation or tumor metastasis. Cell Rep. 2021, 35, 109168. [Google Scholar] [CrossRef]
- You, Y.; Wen, D.; Zeng, L.; Lu, J.; Xiao, X.; Chen, Y.; Song, H.; Liu, Z. ALKBH5/MAP3K8 axis regulates PD-L1+ macrophage infiltration and promotes hepatocellular carcinoma progression. Int. J. Biol. Sci. 2022, 18, 5001–5018. [Google Scholar] [CrossRef] [PubMed]
- Okabe, A.; Huang, K.K.; Matsusaka, K.; Fukuyo, M.; Xing, M.; Ong, X.; Hoshii, T.; Usui, G.; Seki, M.; Mano, Y.; et al. Cross-species chromatin interactions drive transcriptional rewiring in Epstein-Barr virus-positive gastric adenocarcinoma. Nat. Genet. 2020, 52, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Cantelli, G.; Crosas-Molist, E.; Georgouli, M.; Sanz-Moreno, V. TGFΒ-induced transcription in cancer. Semin. Cancer Biol. 2017, 42, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Bhatia, A.; Toth, Z.; Oh, S.; Inn, K.-S.; Liao, C.-P.; Roy-Burman, P.; Melamed, J.; Coetzee, G.A.; Jung, J.U. TPL2/COT/MAP3K8 (TPL2) activation promotes androgen depletion-independent (ADI) prostate cancer growth. PLoS ONE 2011, 6, e16205. [Google Scholar] [CrossRef] [PubMed]
- Tunca, B.; Tezcan, G.; Cecener, G.; Egeli, U.; Zorluoglu, A.; Yilmazlar, T.; Ak, S.; Yerci, O.; Ozturk, E.; Umut, G.; et al. Overexpression of CK20, MAP3K8 and EIF5A correlates with poor prognosis in early-onset colorectal cancer patients. J. Cancer Res. Clin. Oncol. 2013, 139, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M.; Reynolds, S.H.; Anderson, M.; Wiest, J.W. Mutational activation of the MAP3K8 protooncogene in lung cancer. Genes Chromosomes Cancer 2004, 41, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Davies, C.; Blake, S.S.M.; Murray, P.; Najafipour, S.; Tsichlis, P.N.; Young, L.S. The oncogenic protein kinase Tpl-2/Cot contributes to Epstein-Barr virus-encoded latent infection membrane protein 1-induced NF-kappaB signaling downstream of TRAF2. J. Virol. 2002, 76, 4567–4579. [Google Scholar] [CrossRef]
- Sourvinos, G.; Tsatsanis, C.; Spandidos, D.A. Overexpression of the Tpl-2/Cot oncogene in human breast cancer. Oncogene 1999, 18, 4968–4973. [Google Scholar] [CrossRef]
- Chorzalska, A.; Ahsan, N.; Rao, R.S.P.; Roder, K.; Yu, X.; Morgan, J.; Tepper, A.; Hines, S.; Zhang, P.; Treaba, D.O.; et al. Overexpression of Tpl2 is linked to imatinib resistance and activation of MEK-ERK and NF-κB pathways in a model of chronic myeloid leukemia. Mol. Oncol. 2018, 12, 630–647. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Cho, H.J.; Lee, S.J.; Song, H.J.; Cho, H.J.; Park, M.C.; Seol, H.J.; Lee, J.-I.; Kim, S.; Lee, H.M.; et al. Tpl2 induces castration resistant prostate cancer progression and metastasis. Int. J. Cancer 2015, 136, 2065–2077. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.-H.; Hao, J.-H.; Zhang, X.-G.; Shen, C.-C.; Zhang, D.-J.; Zhang, K.-S.; Zheng, H.-X.; Liu, X.-T. Advancement in TPL2-regulated innate immune response. Immunobiology 2019, 224, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ji, G.; Xiao, X.; Chen, X.; Qin, W.-W.; Yang, F.; Li, Y.-F.; Fan, L.-N.; Xi, W.-J.; Huo, Y.; et al. Epigenetically regulated miR-145 suppresses colon cancer invasion and metastasis by targeting LASP1. Oncotarget 2016, 7, 68674–68687. [Google Scholar] [CrossRef]
- Loss, L.A.; Sadanandam, A.; Durinck, S.; Nautiyal, S.; Flaucher, D.; Carlton, V.E.H.; Moorhead, M.; Lu, Y.; Gray, J.W.; Faham, M.; et al. Prediction of epigenetically regulated genes in breast cancer cell lines. BMC Bioinform. 2010, 11, 305. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Nair, V.S.; Elkord, E. PD-L1 expression in human breast cancer stem cells is epigenetically regulated through posttranslational histone modifications. J. Oncol. 2019, 2019, 3958908. [Google Scholar] [CrossRef]
- Yang, S.; Lee, J.-Y.; Hur, H.; Oh, J.H.; Kim, M.H. Up-regulation of HOXB cluster genes are epigenetically regulated in tamoxifen-resistant MCF7 breast cancer cells. BMB Rep. 2018, 51, 450–455. [Google Scholar] [CrossRef]
- Li, X.; Cowell, J.K.; Sossey-Alaoui, K. CLCA2 tumour suppressor gene in 1p31 is epigenetically regulated in breast cancer. Oncogene 2004, 23, 1474–1480. [Google Scholar] [CrossRef]
- Li, J.; Hu, W.-X.; Luo, S.-Q.; Xiong, D.-H.; Sun, S.; Wang, Y.-P.; Bu, X.-F.; Liu, J.; Hu, J. Promoter methylation induced epigenetic silencing of DAZAP2, a downstream effector of p38/MAPK pathway, in multiple myeloma cells. Cell Signal. 2019, 60, 136–145. [Google Scholar] [CrossRef]
- Raedt, T.D.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016, 16, 2829–2837. [Google Scholar] [CrossRef]
- Wen, D.; Wang, Y.; Zhu, Z.; Huang, Z.; Cui, L.; Wu, T.; Liu, C.-Y. Bromodomain and Extraterminal (BET) protein inhibition suppresses tumor progression and inhibits HGF-MET signaling through targeting cancer-associated fibroblasts in colorectal cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165923. [Google Scholar] [CrossRef]
- Masliah-Planchon, J.; Garinet, S.; Pasmant, E. RAS-MAPK pathway epigenetic activation in cancer: miRNAs in action. Oncotarget 2016, 7, 38892–38907. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Calore, F.; Paone, A.; Galli, R.; Calin, G.A. Epigenetic regulation of miRNAs in cancer. Adv. Exp. Med. Biol. 2013, 754, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, K.-T.; Jang, H.-R.; Kim, J.-H.; Noh, S.-M.; Song, K.-S.; Cho, J.-S.; Jeong, H.-Y.; Kim, S.-Y.; Yoo, H.-S.; et al. Epigenetic down-regulation and suppressive role of DCBLD2 in gastric cancer cell proliferation and invasion. Mol. Cancer Res. 2008, 6, 222–230. [Google Scholar] [CrossRef]
- Kim, M.; Jang, H.-R.; Kim, J.-H.; Noh, S.-M.; Song, K.-Y.; Cho, J.-S.; Jeong, H.-Y.; Norman, J.C.; Caswell, P.T.; Kang, G.H.; et al. Epigenetic inactivation of protein kinase D1 in gastric cancer and its role in gastric cancer cell migration and invasion. Carcinogenesis 2008, 29, 629–637. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Z.; Wang, D. Epigenetic regulation of IncRNA KCNKI5-ASI in gastric cancer. Cancer Manag. Res. 2019, 11, 8589–8602. [Google Scholar] [CrossRef]
- Zhou, Z.; Lin, Z.; Pang, X.; Tariq, M.A.; Ao, X.; Li, P.; Wang, J. Epigenetic regulation of long non-coding RNAs in gastric cancer. Oncotarget 2017, 9, 19443–19458. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef]
- Kang, H.-G.; Kim, W.-J.; Noh, M.-G.; Chun, K.-H.; Kim, S.-J. SPON2 is upregulated through notch signaling pathway and promotes tumor progression in gastric cancer. Cancers 2020, 12, 1439. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Cheng, Z.; Wang, Y.-H.; Guo, Z.-H.; Zhang, S.-Q.; Hu, J.-K.; Zhou, Z.-G. Role of notch signaling pathway in gastric cancer: A meta-analysis of the literature. World J. Gastroenterol. 2014, 20, 9191–9199. [Google Scholar] [CrossRef] [PubMed]
- Demitrack, E.S.; Samuelson, L.C. Notch as a driver of gastric epithelial cell proliferation. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, H.; Hu, J.; Xu, Z.; Lin, Y.; Zhou, S. Penicilazaphilone C, a new azaphilone, induces apoptosis in gastric cancer by blocking the notch signaling pathway. Front. Oncol. 2020, 10, 116. [Google Scholar] [CrossRef]
- Dai, F.; Xuan, Y.; Jin, J.-J.; Yu, S.; Long, Z.-W.; Cai, H.; Liu, X.-W.; Zhou, Y.; Wang, Y.-N.; Chen, Z.; et al. CtBP2 overexpression promotes tumor cell proliferation and invasion in gastric cancer and is associated with poor prognosis. Oncotarget 2017, 8, 28736–28749. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-mesenchymal transition in cancer: A historical overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Ishimoto, T.; Miyake, K.; Nandi, T.; Yashiro, M.; Onishi, N.; Huang, K.K.; Lin, S.J.; Kalpana, R.; Tay, S.T.; Suzuki, Y.; et al. Activation of transforming growth factor beta 1 signaling in gastric cancer-associated fibroblasts increases their motility, via expression of rhomboid 5 homolog 2, and ability to induce invasiveness of gastric cancer cells. Gastroenterology 2017, 153, 191–204.e16. [Google Scholar] [CrossRef]
- Wu, L.; Zhao, N.; Zhou, Z.; Chen, J.; Han, S.; Zhang, X.; Bao, H.; Yuan, W.; Shu, X. PLAGL2 promotes the proliferation and migration of gastric cancer cells via USP37-mediated deubiquitination of Snail1. Theranostics 2021, 11, 700–714. [Google Scholar] [CrossRef]
- Pajuelo-Lozano, N.; Alcalá, S.; Sainz, B., Jr.; Perona, R.; Sanchez-Perez, I. Targeting MAD2 modulates stemness and tumorigenesis in human gastric cancer cell lines. Theranostics 2020, 10, 9601–9618. [Google Scholar] [CrossRef]
- Anggorowati, N.; Kurniasari, C.R.; Damayanti, K.; Cahyanti, T.; Widodo, I.; Ghozali, A.; Romi, M.M.; Sari, D.C.R.; Arfian, N. Histochemical and immunohistochemical study of α-SMA, collagen, and PCNA in epithelial ovarian neoplasm. Asian Pac. J. Cancer Prev. 2017, 18, 667–671. [Google Scholar] [CrossRef]
- Lee, H.W.; Park, Y.M.; Lee, S.J.; Cho, H.J.; Kim, D.-H.; Lee, J.-I.; Kang, M.-S.; Seol, H.J.; Shim, Y.M.; Nam, D.-H.; et al. Alpha-smooth muscle actin (ACTA2) is required for metastatic potential of human lung adenocarcinoma. Clin. Cancer Res. 2013, 19, 5879–5889. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.; Liu, Z.; Zhang, M.; Guo, T.; Quan, Q.; Huang, L.; Guo, L.; Cao, L.; Zhang, X. Overexpression of B7-H3 in α-SMA-positive fibroblasts is associated with cancer progression and survival in gastric adenocarcinomas. Front. Oncol. 2020, 9, 1466. [Google Scholar] [CrossRef]
- Chen, J.; Yang, P.; Xiao, Y.; Zhang, Y.; Liu, J.; Xie, D.; Cai, M.; Zhang, X. Overexpression of α-sma-positive fibroblasts (CAFs) in nasopharyngeal carcinoma predicts poor prognosis. J. Cancer 2017, 8, 3897–3902. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Zhang, Z.; Shang, D.; Cheng, J.; Yuan, H.; Wu, Y.; Song, X.; Jiang, H. α-Smooth muscle actin-positive myofibroblasts, in association with epithelial-mesenchymal transition and lymphogenesis, is a critical prognostic parameter in patients with oral tongue squamous cell carcinoma. J. Oral Pathol. Med. 2014, 43, 335–343. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, G.; Yang, T.; Liu, S.; Luo, Y.-L.; Liu, Y.; Zhong, Q. Epigenetic Regulation of MAP3K8 in EBV-Associated Gastric Carcinoma. Int. J. Mol. Sci. 2023, 24, 1964. https://doi.org/10.3390/ijms24031964
Roy G, Yang T, Liu S, Luo Y-L, Liu Y, Zhong Q. Epigenetic Regulation of MAP3K8 in EBV-Associated Gastric Carcinoma. International Journal of Molecular Sciences. 2023; 24(3):1964. https://doi.org/10.3390/ijms24031964
Chicago/Turabian StyleRoy, Gaurab, Ting Yang, Shangxin Liu, Yi-Ling Luo, Yuantao Liu, and Qian Zhong. 2023. "Epigenetic Regulation of MAP3K8 in EBV-Associated Gastric Carcinoma" International Journal of Molecular Sciences 24, no. 3: 1964. https://doi.org/10.3390/ijms24031964
APA StyleRoy, G., Yang, T., Liu, S., Luo, Y.-L., Liu, Y., & Zhong, Q. (2023). Epigenetic Regulation of MAP3K8 in EBV-Associated Gastric Carcinoma. International Journal of Molecular Sciences, 24(3), 1964. https://doi.org/10.3390/ijms24031964